RNA Sequencing-Based Bulked Segregant Analysis Facilitates Efficient D-genome Marker Development for a Specific Chromosomal Region of Synthetic Hexaploid Wheat

 ,
,
Abstract
1. Introduction
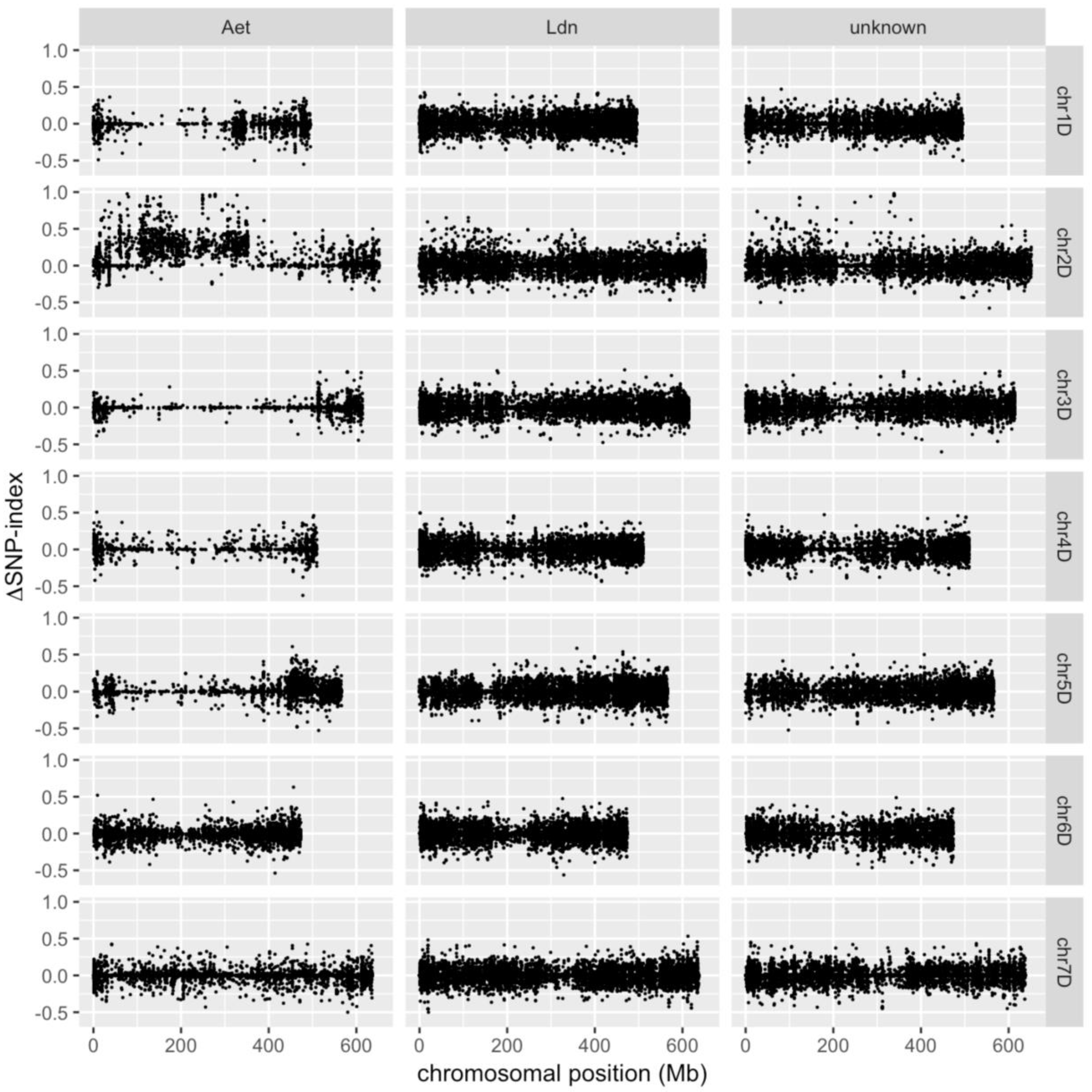
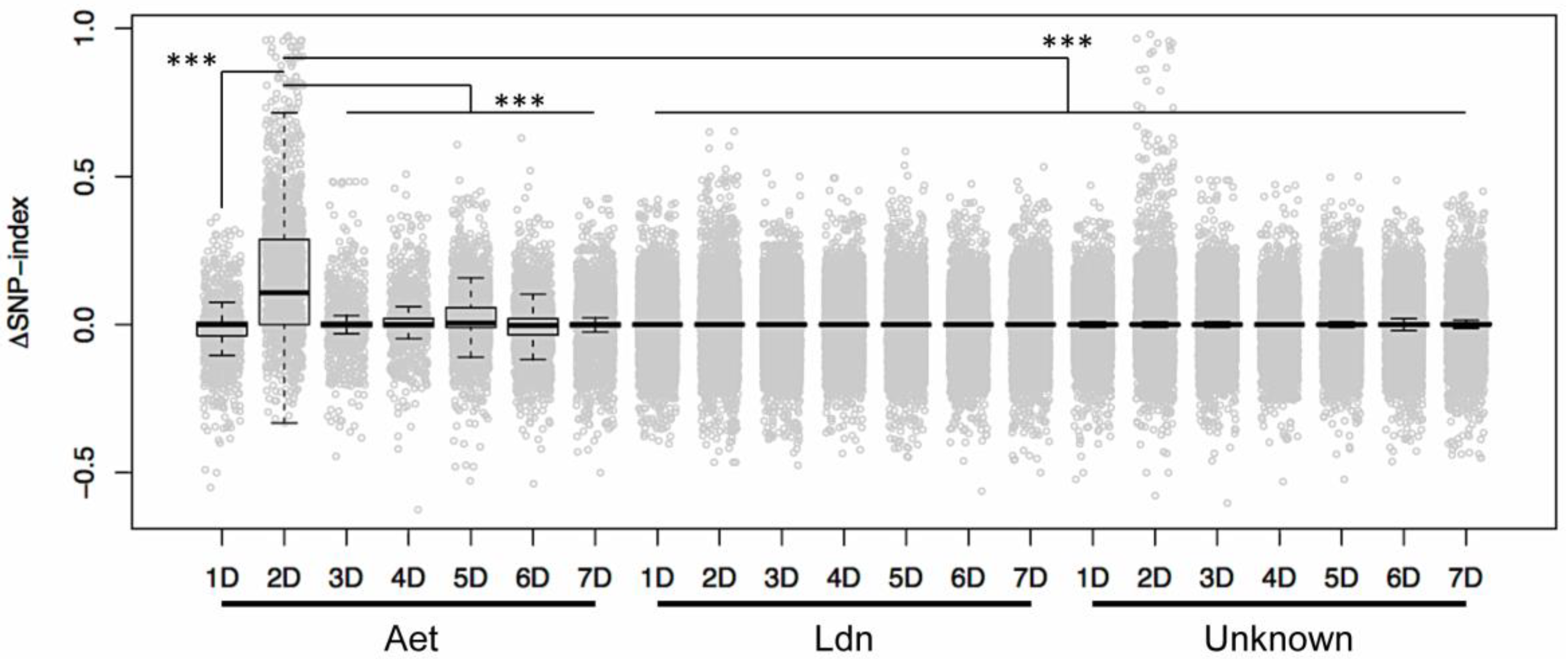
2. Results and Discussion
3. Materials and Methods
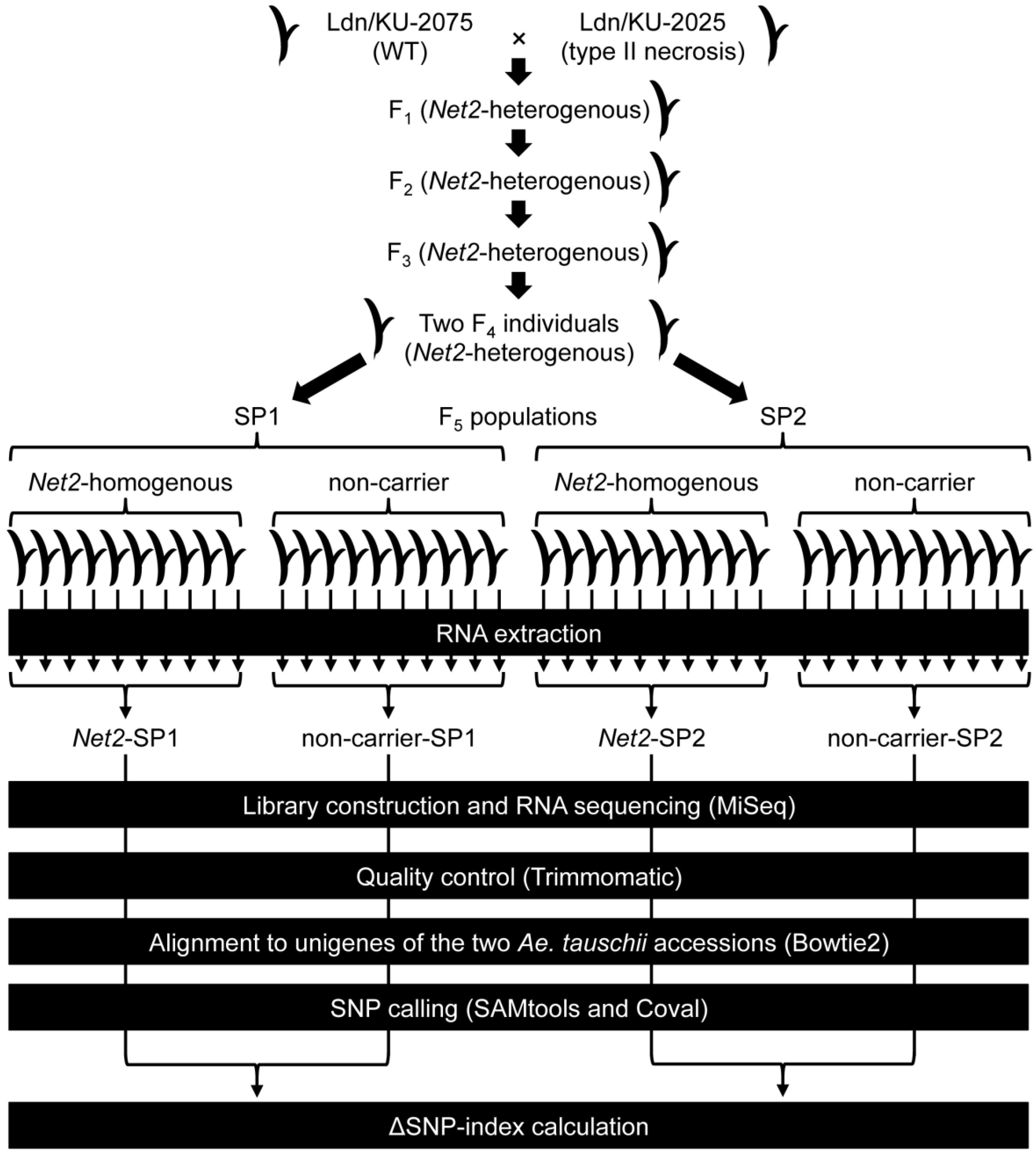
3.1. Plant Materials
3.2. Library Construction and RNA Sequencing
3.3. Alignment of RNA-seq Reads to de Novo Assembled Transcripts of the Parental Ae. tauschii Accessions
3.4. Identification of D-genome Specific SNPs and Calculation of ΔSNP-Index
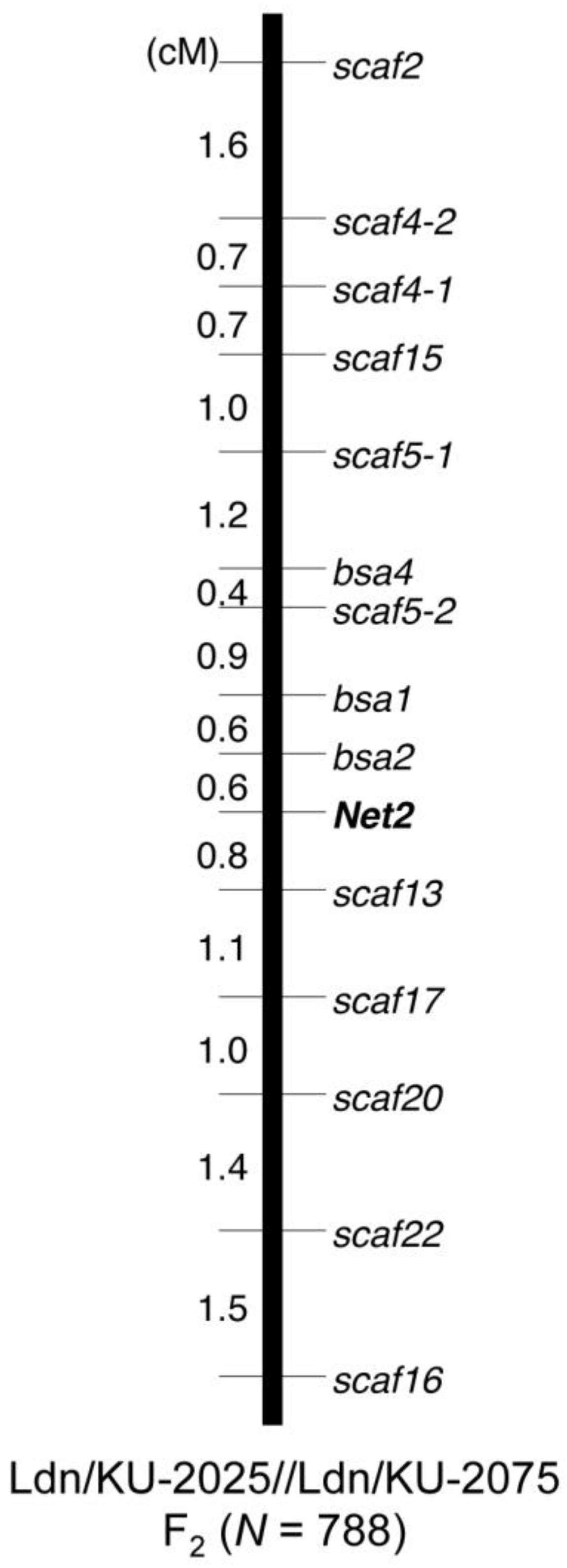
3.5. Molecular Marker Development and Genotyping
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA-seq | RNA sequencing |
| NGS | next-generation sequencing |
| BSA | bulked segregant analysis |
| SNP | single nucleotide polymorphism |
| Indel | insertion/deletion |
| NR | non-redundant |
| SP | Segregating Population |
| dCAPS | derived cleaved amplified polymorphic sequence |
| Ldn | Langdon |
References
- Matsuoka, Y. Evolution of polyploid Triticum wheats under cultivation: The role of domestication, natural hybridization and allopolyploid speciation in their diversification. Plant Cell Physiol. 2011, 52, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, M.C.; Chen, Z.; You, F.M.; Wei, Y.; Zheng, Y.; Dvorak, J. Aegilops tauschii single nucleotide polymorphisms shed light on the origins of wheat D-genome genetic diversity and pinpoint the geographic origin of hexaploid wheat. New Phytol. 2013, 198, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Nasuda, S. Durum wheat as a candidate for the unknown female progenitor of bread wheat: An empirical study with a highly fertile F1 hybrid with Aegilops tauschii Coss. Theor. Appl. Genet. 2004, 109, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Zohary, D.; Harlan, J.R.; Vardi, A. The wild diploid progenitors of wheat and their breeding value. Euphytica 1969, 18, 58–65. [Google Scholar] [CrossRef]
- Mujeeb-Kazi, A.; Rosas, V.; Roldan, S. Conservation of the genetic variation of Triticum tauschii (Coss.) Schmalh. (Aegilops squarrosa auct. non L.) in synthetic hexaploid wheats (T. turgidum L. s.lat. x T. tauschii; 2n=6x=42, AABBDD) and its potential utilization for wheat improvement. Genet. Resour. Crop Evol. 1996, 43, 129–134. [Google Scholar] [CrossRef]
- Jones, H.; Gosman, N.; Horsnell, R.; Rose, G.A.; Everst, L.A.; Bentley, A.R.; Tha, S.; Uauy, C.; Kowalski, A.; Novoselovic, D.; et al. Strategy for exploiting exotic germplasm using genetic, morphological, and environmental diversity: The Aegilops tauschii Coss. example. Theor. Appl. Genet. 2013, 126, 1793–1808. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Yorgancilar, A.; Baenziger, P.S. Genome-wide association study reveals favorable alleles associated with common bunt resistance in synthetic hexaploid wheat. Euphytica 2018, 214, 200. [Google Scholar] [CrossRef]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Baenziger, P.S. Genome-wide association study reveals novel genomic regions for grain yield and yield-related traits in drought-stressed synthetic hexaploid wheat. Int. J. Mol. Sci. 2018, 19, 3011. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, M.; Baenziger, P.S.; Waters, B.M.; Poudel, R.; Belamkar, V.; Poland, J.; Morgounov, A. Genome-wide association study reveals novel genomic regions associated with 10 grain minerals in synthetic hexaploid wheat. Int. J. Mol. Sci. 2018, 19, 3237. [Google Scholar] [CrossRef] [PubMed]
- Gorafi, Y.S.A.; Kim, J.S.; Elbashir, A.A.E.; Tsujimoto, H. A population of wheat multiple synthetic derivatives: An effective platform to explore, harness and utilize genetic diversity of Aegilops tauschii for wheat improvement. Theor. Appl. Genet. 2018, 131, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Dreisigacker, S.; Melchinger, A.E.; Reif, J.C.; Mujeeb-Kazi, A.; Van Ginkel, M.; Hoisington, D.; Warburton, M.L. Quantifying novel sequence variation and selective advantage in synthetic hexaploid wheats and their backcross-derived lines using SSR markers. Mol. Breed. 2005, 15, 1–10. [Google Scholar] [CrossRef]
- Jafarzadeh, J.; Bonnett, D.; Jannink, J.L.; Akdemir, D.; Dreisigacker, S.; Sorrells, M.E. Breeding value of primary synthetic wheat genotypes for grain yield. PLoS ONE 2016, 11, e0162860. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Poland, J.; Baenziger, P.S. Unlocking the novel genetic diversity and population structure of synthetic hexaploid wheat. BMC Genom. 2018, 19, 591. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Mujeeb-Kazi, A.; Ogbonnaya, F.C.; He, Z.; Rajaram, S. Wheat genetic resources in the post-genomics era: Promise and challenges. Ann. Bot. 2018, 121, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Van Slageren, M.W. Wild Wheats: A Monograph of Aegilops L. and Amblyopyrum (Jaub. & Spach) Eig (Poaceae); Wageningen Agricultural University: Wageningen, The Netherlands, 1994; pp. 326–344. ISBN 90-6754-377-2. [Google Scholar]
- Matsuoka, Y.; Takumi, S.; Kawahara, T. Flowering time diversification and dispersal in central Eurasian wild wheat Aegilops tauschii Coss.: Genealogical and ecological framework. PLoS ONE 2008, 3, e3138. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Yamasaki, M.; Matsuoka, Y.; Kawahara, T.; Takumi, S. Population structure of wild wheat D-genome progenitor Aegilops tauschii Coss.: Implications for intraspecific lineage diversification and evolution of common wheat. Mol. Ecol. 2010, 19, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Kawahara, T.; Takumi, S. Intraspecific lineage divergence and its association with reproductive trait change during species range expansion in central Eurasian wild wheat Aegilops tauschii Coss. (Poaceae). BMC Evol. Biol. 2015, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Takumi, S. The role of reproductive isolation in allohexaploid speciation pattern: Empirical insights from the progenitors of common wheat. Sci. Rep. 2017, 7, 16004. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Hosogi, N.; Park, P.; Takumi, S. Hypersensitive response-like reaction is associated with hybrid necrosis in interspecific crosses between tetraploid wheat and Aegilops tauschii Coss. PLoS ONE 2010, 5, e11326. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Shitsukawa, N.; Hosogi, N.; Park, P.; Takumi, S. Autoimmune response and repression of mitotic cell division occur in inter-specific crosses between tetraploid wheat and Aegilops tauschii Coss. that show low temperature-induced hybrid necrosis. Plant J. 2011, 68, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Nishijima, R.; Iehisa, J.C.M.; Takumi, S. Fine mapping and genetic association analysis of Net2, the causative D-genome locus of low temperature-induced hybrid necrosis in interspecific crosses between tetraploid wheat and Aegilops tauschii. Genetica 2016, 144, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Brozynska, M.; Furtado, A.; Henry, R.J. Genomics of crop wild relatives: Expanding the gene pool for crop improvement. Plant Biotechnol. J. 2016, 14, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, G.; Saito, M.; Tanaka, T.; Katayose, Y.; Kanamori, H.; Kurita, K.; Nakamura, T. An efficient approach for the development of genome-specific markers in allohexaploid wheat (Triticum aestivum L.) and its application in the construction of high-density linkage maps of the D genome. DNA Res. 2018, 25, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Iehisa, J.C.M.; Shimizu, A.; Sato, K.; Nasuda, S.; Takumi, S. Discovery of high-confidence single nucleotide polymorphisms from large-scale de novo analysis of leaf transcripts of Aegilops tauschii, a wild wheat progenitor. DNA Res. 2012, 19, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Iehisa, J.C.M.; Shimizu, A.; Sato, K.; Nishijima, R.; Sakaguchi, K.; Matsuda, R.; Nasuda, S.; Takumi, S. Genome-wide marker development for the wheat D genome based on single nucleotide polymorphisms identified from transcripts in the wild wheat progenitor Aegilops tauschii. Theor. Appl. Genet. 2014, 127, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, R.; Yoshida, K.; Motoi, Y.; Sato, K.; Takumi, S. Genome-wide identification of novel genetic markers from RNA sequencing assembly of diverse Aegilops tauschii accessions. Mol. Genet. Genom. 2016, 291, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Schneeberge, K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 2014, 15, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Wang, P.; Xu, Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol. J. 2016, 14, 1941–1955. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yeh, C.T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; Song, W.; Xing, J.; Zhao, Y.; Zhang, R.; Li, C.; Duan, M.; Luo, M.; Shi, Z.; Zhao, J. Identification of genes potentially associated with the fertility instability of S-type cytoplasmic male sterility in maize via bulked segregant RNA-Seq. PLoS ONE 2016, 11, e0163489. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhu, J.; Su, H.; Huang, M.; Wang, H.; Ding, S.; Zhang, B.; Luo, A.; Wei, S.; Tian, X.; et al. Bulked segregant RNA-seq revealed differential expression and SNPs of candidate genes associated with waterlogging tolerance in maize. Front. Plant Sci. 2017, 8, 1022. [Google Scholar] [CrossRef] [PubMed]
- Borrill, P.; Adamski, N.; Uauy, C. Genomics as the key to unlocking the polyploid potential of wheat. New Phytol. 2015, 208, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Adamski, N.M.; Mugford, S.G.; Jiang, C.C.; Febrer, M.; Uauy, C. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol. 2012, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Gonzalez, R.H.; Segovia, V.; Bird, N.; Fenwick, P.; Holdgate, S.; Bery, S.; Jack, P.; Caccamo, M.; Uauy, C. RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. J. 2015, 13, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Xie, J.; Hu, J.; Qiu, D.; Liu, Z.; Li, J.; Li, M.; Zhang, H.; Yang, L.; Liu, H.; et al. Development of molecular markers linked to powdery mildew resistance gene Pm4b by combining SNP discovery from transcriptome sequencing data with bulked segregant analysis (BSR-Seq) in wheat. Front. Plant Sci. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.-C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Ywardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Yoshida, K.; Takumi, S. Hybrid incompatibilities in interspecific crosses between tetraploid wheat and its wild relative Aegilops umbellulata. Plant Mol. Biol. 2017, 95, 625–645. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Yoshida, K.; Mizuno, N.; Nasuda, S.; Sato, K.; Takumi, S. Origin of the wheat B-genome chromosomes conferred by RNA sequencing analysis of leaf transcripts in the section Sitopsis species of Aegilops. DNA Res. 2018. under review. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup 1000 Genome Project Data Processing. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Natsume, S.; Yoshida, K.; MacLean, D.; Cano, L.; Kamoun, S.; Terauchi, R. Coval: Improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS ONE 2013, 8, e75402. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, T.; Murai, K.; Takumi, S. Distinct genetic regulation of flowering time and grain-filling period based on empirical study of D genome diversity in synthetic hexaploid wheat lines. Breed. Sci. 2011, 61, 130–141. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016; Available online: http://www.rstudio.com/ (accessed on 1 November 2016).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 1 November 2016).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Lander, E.S.; Green, P.; Abrahamson, J.; Barlow, A.; Daly, M.J.; Lincoln, S.E.; Newburg, L. MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1987, 1, 174–181. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Read Pairs | Filtered Read Pairs (%) a | Aligned to the Ae. tauschii Transcripts b (%) c | |
|---|---|---|---|---|
| KU-2075 | KU-2025 | |||
| Synthetic hexaploids | ||||
| non-carrier-SP1-1st | 4,202,114 | 2,799,202 (66.61%) | 2,020,405 (72.18%) | 1,930,400.5 (68.96%) |
| non-carrier-SP1-2nd | 4,059,840 | 2,858,956 (70.42%) | 2,062,291 (72.13%) | 1,971,110.5 (68.95%) |
| non-carrier-SP2-1st | 4,492,358 | 2,953,088 (65.74%) | 2,164,656 (73.3%) | 2,083,120 (70.54%) |
| non-carrier-SP2-2nd | 4,115,352 | 2,864,271 (69.60%) | 2,098,383 (73.26%) | 2,020,752.5 (70.55%) |
| Net2-SP1-1st | 4,710,499 | 3,148,652 (66.84%) | 2,208,392 (70.14%) | 2,110,452.5 (67.03%) |
| Net2-SP1-2nd | 4,403,630 | 3,108,568 (70.59%) | 2,178,403 (70.08%) | 2,082,004 (66.98%) |
| Net2-SP2-1st | 4,828,182 | 3,249,596 (67.30%) | 2,420,056 (74.47%) | 2,348,814 (72.28%) |
| Net2-SP2-2nd | 5,216,082 | 3,709,478 (71.12%) | 2,763,471 (74.5%) | 2,684,019 (72.36%) |
| Tetraploid wheat | ||||
| cv. Langdon | 6,316,174 | 4,372,660 (69.23%) | 2,974,277 (68.02%) | 2,661,487 (60.87%) |
| Transcripts a | KU-2075 | KU-2025 | ||
|---|---|---|---|---|
| The Number of SNP | Total | Anchored to the Genome b (%) | Total | Anchored to the Genome b (%) |
| Synthetic hexaploids | ||||
| non-carrier-SP1 | 277,605 | 275,799 (99.35%) | 262,966 | 261,128 (99.30%) |
| non-carrier-SP2 | 276,564 | 274,772 (99.35%) | 269,175 | 267,249 (99.28%) |
| Net2-SP1 | 318,046 | 315,859 (99.31%) | 296,819 | 294,739 (99.30%) |
| Net2-SP2 | 298,496 | 296,419 (99.30%) | 285,798 | 283,684 (99.26%) |
| Tetraploid wheat | ||||
| cv. Langdon | 429,346 | 421,957 (98.28%) | 350,871 | 345,657 (98.51%) |
| Chr. | D-genome-Specific | Homoeologous | Unclassified | Total |
|---|---|---|---|---|
| 1D | 1674 | 29,307 | 11,760 | 42,741 |
| 2D | 2295 | 33,822 | 13,975 | 50,092 |
| 3D | 1611 | 31,932 | 15,532 | 49,075 |
| 4D | 1698 | 28,781 | 11,776 | 42,255 |
| 5D | 2936 | 34,961 | 14,336 | 52,233 |
| 6D | 3730 | 24,534 | 10,966 | 39,230 |
| 7D | 3983 | 28,593 | 11,606 | 44,182 |
| Total | 17,927 | 211,930 | 89,951 | 319,808 |
| Marker Name | Primer Sequence (5′ to 3′) | Restriction Enzyme |
|---|---|---|
| bsa1 | TCATGACCTGCTGGTTTGTT | StyI |
| GATTCCAATGTTATTTCTGAACCCT | ||
| bsa2 | TCACAACATTCGCAGGTCAT | HpaII |
| TGGTTCTGTTGATCTCACTGCC | ||
| bsa4 | ACAAGTCGGATATCGCCAAA | HinfI |
| CAGCTAAAAACTGTTTGCTTGAGA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishijima, R.; Yoshida, K.; Sakaguchi, K.; Yoshimura, S.-i.; Sato, K.; Takumi, S. RNA Sequencing-Based Bulked Segregant Analysis Facilitates Efficient D-genome Marker Development for a Specific Chromosomal Region of Synthetic Hexaploid Wheat. Int. J. Mol. Sci. 2018, 19, 3749. https://doi.org/10.3390/ijms19123749
Nishijima R, Yoshida K, Sakaguchi K, Yoshimura S-i, Sato K, Takumi S. RNA Sequencing-Based Bulked Segregant Analysis Facilitates Efficient D-genome Marker Development for a Specific Chromosomal Region of Synthetic Hexaploid Wheat. International Journal of Molecular Sciences. 2018; 19(12):3749. https://doi.org/10.3390/ijms19123749
Chicago/Turabian StyleNishijima, Ryo, Kentaro Yoshida, Kohei Sakaguchi, Shin-ichi Yoshimura, Kazuhiro Sato, and Shigeo Takumi. 2018. "RNA Sequencing-Based Bulked Segregant Analysis Facilitates Efficient D-genome Marker Development for a Specific Chromosomal Region of Synthetic Hexaploid Wheat" International Journal of Molecular Sciences 19, no. 12: 3749. https://doi.org/10.3390/ijms19123749
APA StyleNishijima, R., Yoshida, K., Sakaguchi, K., Yoshimura, S.-i., Sato, K., & Takumi, S. (2018). RNA Sequencing-Based Bulked Segregant Analysis Facilitates Efficient D-genome Marker Development for a Specific Chromosomal Region of Synthetic Hexaploid Wheat. International Journal of Molecular Sciences, 19(12), 3749. https://doi.org/10.3390/ijms19123749