Temporal Frame of Immune Cell Infiltration during Heart Failure Establishment: Lessons from Animal Models

 , , ,
, , ,
Abstract
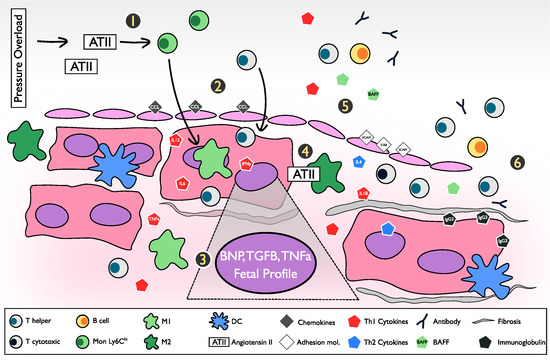
1. Introduction
2. Heart Failure Animal Models
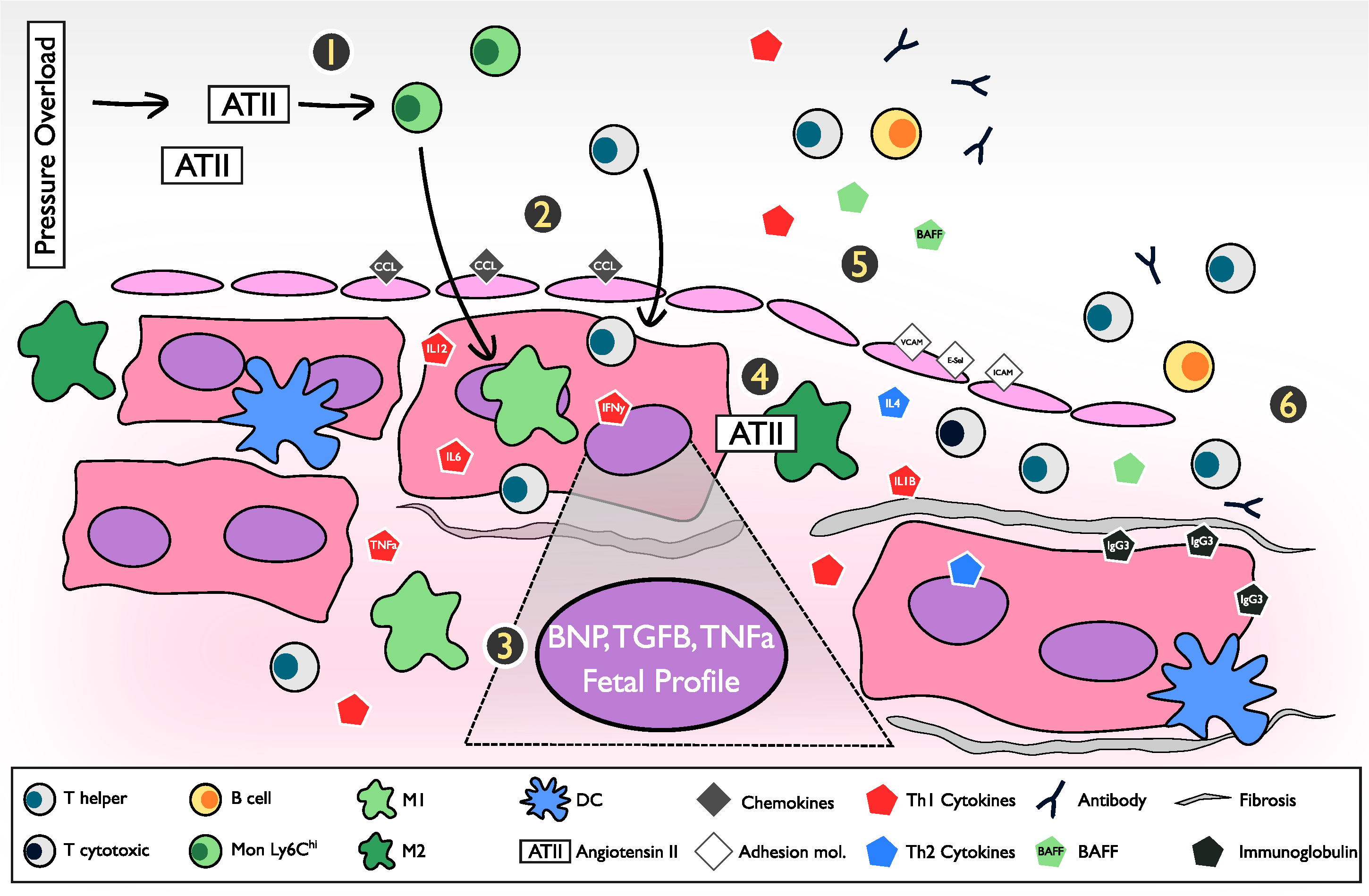
2.1. Ischemic vs. Non-Ischemic (Pressure Overload Model)
2.1.1. Acute Insult vs. Progressive
2.1.2. First to Arrive
2.1.3. Beneficial vs. Pathological Fibrosis
2.2. Pressure Overload
3. Understanding the Different Roles of Immune Response
3.1. T and B Cells
3.2. Mononuclear Phagocytic Cells: Monocytes, Macrophages, and Dendritic Cells
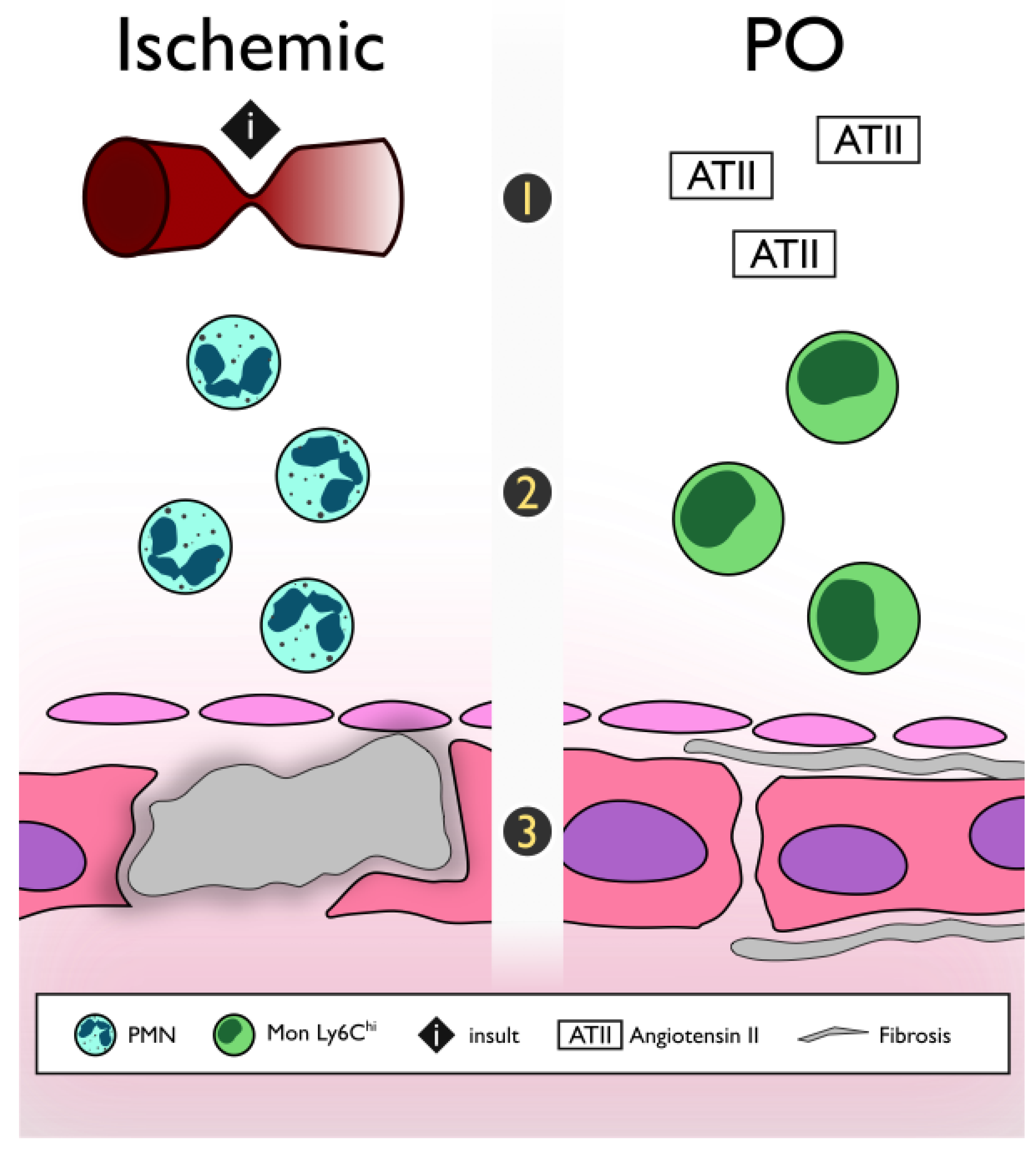
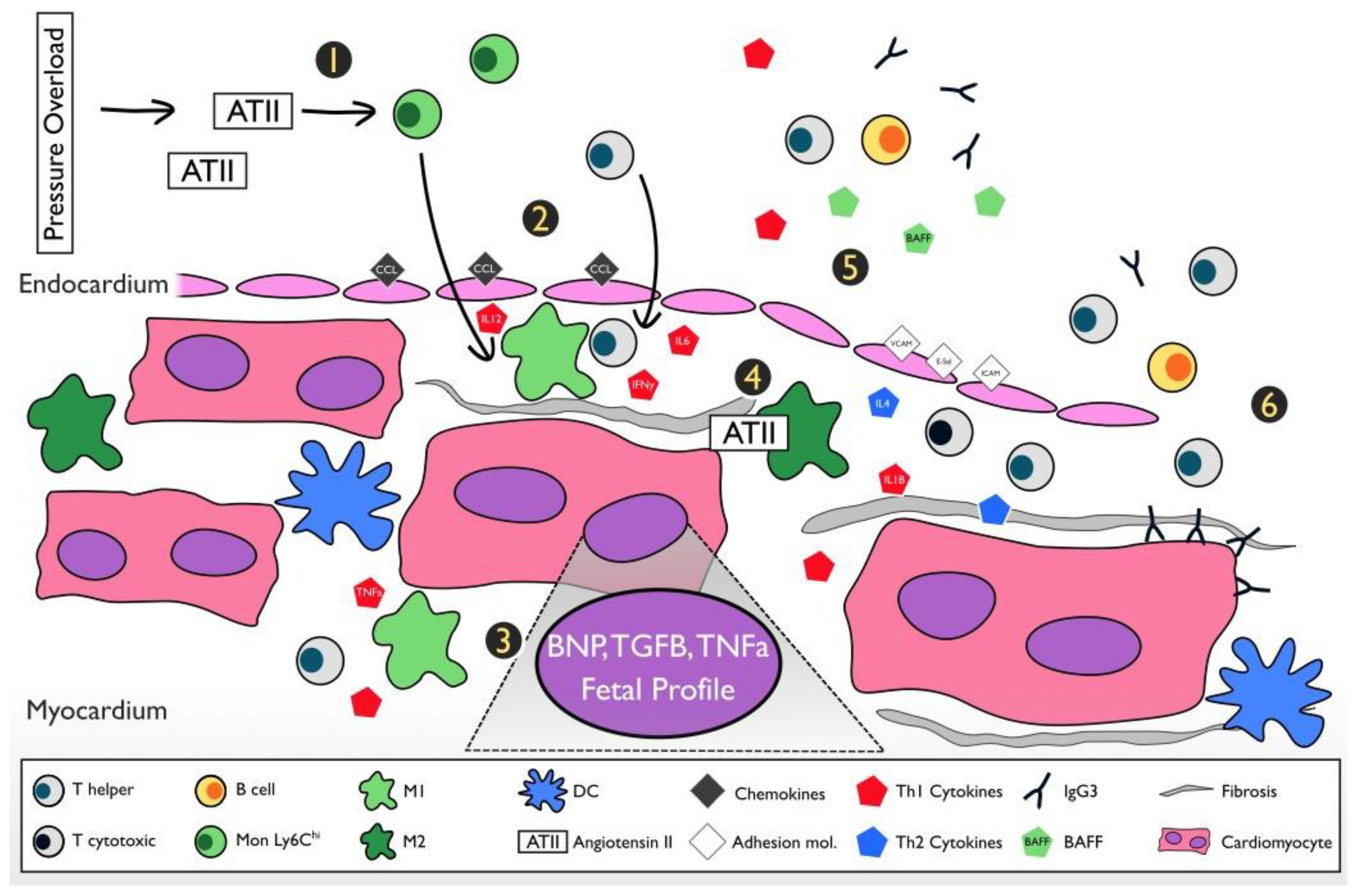
4. The Inflammatory Pathway and Pathogenesis of the Pressure Overload Heart Failure Model
5. Findings in Animal Models and Their Correlation with Human Heart Failure
5.1. Transition from Hypertension to Heart Failure
5.2. Murine Pressure Overload Heart Failure Model and Clinical Pressure Overload Heart Failure
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ADP | Acute decompensated phase |
| ATII | Angiotensin II |
| BNP | Brain natriuretic peptide |
| BP | Blood pressure |
| CHF | Congestive and end-stage heart failure |
| CMP | Cardiomyopathy |
| CP | Compensated phase |
| DCs | Dendritic cell |
| DP-HF | Decompensated phase and heart failure establishment |
| Dsfx | Dysfunction |
| EDV | End-diastolic volume |
| EF | Ejection fraction |
| E-Sel | E-selectin |
| HFmrEF | Heart failure with moderately reduced ejection fraction |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| HTN | Hypertension |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IFN-ɣ | Interferon-ɣ |
| IgG | Immunoglobulin G |
| IL | Interleukin |
| LVEF | Left ventricle ejection fraction |
| M1 | Pro-inflammatory macrophage |
| M2 | Anti-inflammatory macrophage |
| MAP | Mean arterial pressure |
| MCP1 | Monocyte chemoattractant protein-1 |
| MLN | Mediastinal lymph node |
| Mon | Monocyte |
| MnPs | Mononuclear phagocytes |
| NYHA | New York Heart Association |
| PMN | Polymorphonuclear neutrophils |
| PO | Pressure overload |
| PO-HF | Pressure overload heart failure |
| RV | Right ventricle |
| TAC | Transverse aortic constriction |
| TGFβ | Transforming growth factor-β |
| TNFα | Tumor necrosis factor-α |
| Treg | T regulatory cell |
| VCAM-1 | Vascular cell adhesion molecule 1 |
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed]
- Conrad, N.; Judge, A.; Tran, J.; Mohseni, H.; Hedgecott, D.; Crespillo, A.P.; Allison, M.; Hemingway, H.; Cleland, J.G.; McMurray, J.J.V.; et al. Temporal trends and patterns in heart failure incidence: A population-based study of 4 million individuals. Lancet 2018, 391, 572–580. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics--2013 update: A report from the American Heart Association. Circulation 2013, 127, e6–e245. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Kenchaiah, S.; Larson, M.G.; Benjamin, E.J.; Kupka, M.J.; Ho, K.K.L.; Murabito, J.M.; Vasan, R.S. Long-term trends in the incidence of and survival with heart failure. N. Engl. J. Med. 2002, 347, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Nymo, S.H.; Aukrust, P.; Kjekshus, J.; McMurray, J.J.V.; Cleland, J.G.F.; Wikstrand, J.; Muntendam, P.; Wienhues-Thelen, U.; Latini, R.; Askevold, E.T.; et al. Limited Added Value of Circulating Inflammatory Biomarkers in Chronic Heart Failure. JACC Heart Fail 2017, 5, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Maggioni, A.P.; Dahlström, U.; Filippatos, G.; Chioncel, O.; Crespo Leiro, M.; Drozdz, J.; Fruhwald, F.; Gullestad, L.; Logeart, D.; Fabbri, G.; et al. EURObservational Research Programme: Regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESC-HF Pilot). Eur. J. Heart Fail. 2013, 15, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Trujillo, L.; Vázquez-Garza, E.; Castillo, E.C.; García-Rivas, G.; Torre-Amione, G. Role of Adaptive Immunity in the Development and Progression of Heart Failure: New Evidence. Arch. Med. Res. 2017, 48, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Biomarkers in Heart Failure. N. Engl. J. Med. 2008, 358, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Seta, Y.; Shan, K.; Bozkurt, B.; Oral, H.; Mann, D.L. Basic mechanisms in heart failure: The cytokine hypothesis. J. Card. Fail. 1996, 2, 243–249. [Google Scholar] [CrossRef]
- Mann, D.L. Inflammatory mediators and the failing heart: Past, present, and the foreseeable future. Circ. Res. 2002, 91, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sada, E.; Torres-Quintanilla, A.; Silva-Platas, C.; García, N.; Willis, B.C.; Rodríguez-Rodríguez, C.; De la Peña, E.; Bernal-Ramírez, J.; Treviño-Saldaña, N.; Oropeza-Almazán, Y.; et al. Proinflammatory Cytokines Are Soluble Mediators Linked with Ventricular Arrhythmias and Contractile Dysfunction in a Rat Model of Metabolic Syndrome. Available online: https://www.hindawi.com/journals/omcl/2017/7682569/ (accessed on 14 November 2017).
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll. Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Durand, J.B.; Bies, R.D.; Young, J.B.; Mann, D.L. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 1996, 93, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Anti-TNF Therapy Against Congestive Heart Failure Investigators Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, R. Heart failure and mouse models. Disease Models Mech. 2010, 3, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Toischer, K.; Rokita, A.G.; Unsöld, B.; Zhu, W.; Kararigas, G.; Sossalla, S.; Reuter, S.P.; Becker, A.; Teucher, N.; Seidler, T.; et al. Differential cardiac remodeling in preload versus afterload. Circulation 2010, 122, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Yang, X.-P.; Carretero, O.A.; Nakagawa, P.; D’Ambrosio, M.; Leung, P.; Xu, J.; Peterson, E.L.; González, G.E.; Harding, P.; et al. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp. Physiol. 2011, 96, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Gomolak, J.R.; Didion, S.P. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front. Physiol. 2014, 5, 396. [Google Scholar] [CrossRef] [PubMed]
- Valero-Muñoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure with Preserved Ejection Fraction: A “Fishing Expedition”. JACC Basic Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Paradis, P.; Schiffrin, E.L. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 2008, 29, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Riojas-Hernández, A.; Bernal-Ramírez, J.; Rodríguez-Mier, D.; Morales-Marroquín, F.E.; Domínguez-Barragán, E.M.; Borja-Villa, C.; Rivera-Álvarez, I.; García-Rivas, G.; Altamirano, J.; García, N. Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 2015, 141, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.R.Y.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.N.; Dechend, R.; Mervaala, E.M.; Park, J.K.; Schmidt, F.; Fiebeler, A.; Theuer, J.; Breu, V.; Ganten, D.; Haller, H.; et al. NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000, 35, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.R.; Chung, A.C.K.; Yang, F.; Yue, W.; Deng, C.; Lau, C.P.; Tse, H.F.; Lan, H.Y. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension 2010, 55, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Li, Y.; Hua, F.; Ma, J.; Gao, X.; Kelley, J.; Zhao, A.; Haddad, G.E.; Williams, D.L.; William Browder, I.; et al. Reduced cardiac hypertrophy in toll-like receptor 4-deficient mice following pressure overload. Cardiovasc. Res. 2005, 68, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Higashikuni, Y.; Tanaka, K.; Kato, M.; Nureki, O.; Hirata, Y.; Nagai, R.; Komuro, I.; Sata, M. Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1β upregulation via nuclear factor κB activation. J. Am. Heart Assoc. 2013, 2, e000267. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A. Endogenous ligands of Toll-like receptors: Implications for regulating inflammatory and immune responses. Trends Immunol. 2002, 23, 509–512. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Lee, K.; Li, N.; Corbett, D.; Mendoza, L.; Frangogiannis, N.G. Characterization of the Inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem. Cell Biol. 2009, 131, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Frangogiannis, N.G. The Role of Macrophages in Nonischemic Heart Failure. JACC Basic Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Prabhu, S.D. Mononuclear Phagocytes Are Dispensable for Cardiac Remodeling in Established Pressure-Overload Heart Failure. PLoS ONE 2017, 12, e0170781. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-P.; Erskine, J.; Zhang, W.-W.; Zheng, R.-H.; Zhang, L.-H.; Duron, G.; Gendreau, J.; Zhao, Z.-Q. Recruitment of macrophages from the spleen contributes to myocardial fibrosis and hypertension induced by angiotensin II. J. Renin Angiotensin Aldosterone Syst. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Schelbert, E.B.; Díez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Guarda, E. Structural remodeling in hypertensive heart disease and the role of hormones. Hypertension 1994, 23, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.A.; Finato, N.; Rocco, M.; Feruglio, G.A.; Puricelli, C.; Cigola, E.; Quaini, F.; Sonnenblick, E.H.; Olivetti, G.; Anversa, P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 1994, 89, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velázquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ. Heart Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Reyes, A.M.; Youker, K.A.; Trevino, A.R.; Celis, R.; Hamilton, D.J.; Flores-Arredondo, J.H.; Orrego, C.M.; Bhimaraj, A.; Estep, J.D.; Torre-Amione, G. Full Expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Kallikourdis, M.; Martini, E.; Carullo, P.; Sardi, C.; Roselli, G.; Greco, C.M.; Vignali, D.; Riva, F.; Berre, A.M.O.; Stølen, T.O.; et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 2017, 8, 14680. [Google Scholar] [CrossRef] [PubMed]
- Youker, K.A.; Assad-Kottner, C.; Cordero-Reyes, A.M.; Trevino, A.R.; Flores-Arredondo, J.H.; Barrios, R.; Fernandez-Sada, E.; Estep, J.D.; Bhimaraj, A.; Torre-Amione, G. High proportion of patients with end-stage heart failure regardless of aetiology demonstrates anti-cardiac antibody deposition in failing myocardium: Humoral activation, a potential contributor of disease progression. Eur. Heart J. 2014, 35, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, Y.; Jia, L.; Cheng, J.; Qi, Y.; Zhang, H.; Du, J. Reciprocal interaction between macrophages and T cells stimulates IFN-γ and MCP-1 production in Ang II-induced cardiac inflammation and fibrosis. PLoS ONE 2012, 7, e35506. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, G.; Alarcón, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepúlveda, M.; et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef] [PubMed]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Fujiu, K.; Nagai, R. Contributions of cardiomyocyte–cardiac fibroblast–immune cell interactions in heart failure development. Basic Res. Cardiol. 2013, 108. [Google Scholar] [CrossRef] [PubMed]
- Fujiu, K.; Wang, J.; Nagai, R. Cardioprotective function of cardiac macrophages. Cardiovasc. Res. 2014, 102, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Drazner, M.H. The progression of hypertensive heart disease. Circulation 2011, 123, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition from Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail 2017, 5, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Iriarte, M.; Murga, N.; Sagastagoitia, D.; Morillas, M.; Boveda, J.; Molinero, E.; Etxebeste, J.; Salcedo, A.; Rodriguez, E.; Ormaetxe, J.M. Classification of hypertensive cardiomyopathy. Eur. Heart J. 1993, 14 (Suppl. J), 95–101. [Google Scholar] [PubMed]
- Drazner, M.H. The transition from hypertrophy to failure: How certain are we? Circulation 2005, 112, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Arena, R.; Abouzaki, N.; Van Tassell, B.W.; Canada, J.; Shah, K.; Biondi-Zoccai, G.; Voelkel, N.F. Heart failure with preserved ejection fraction: Refocusing on diastole. Int. J. Cardiol. 2015, 179, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Roger, V.L.; Weston, S.A.; Jiang, R.; Redfield, M.M. Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ. Heart Fail. 2012, 5, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Conceição, G.; Heinonen, I.; Lourenço, A.P.; Duncker, D.J.; Falcão-Pires, I. Animal models of heart failure with preserved ejection fraction. Neth. Heart J. 2016, 24, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Ho, K.; Thom, T. Changing epidemiological features of cardiac failure. Br. Heart J. 1994, 72, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ogden, L.G.; Bazzano, L.A.; Vupputuri, S.; Loria, C.; Whelton, P.K. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch. Intern. Med. 2001, 161, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Bonow, R.O. Chronic heart failure in the United States: A manifestation of coronary artery disease. Circulation 1998, 97, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Tsutamoto, T.; Hisanaga, T.; Wada, A.; Maeda, K.; Ohnishi, M.; Fukai, D.; Mabuchi, N.; Sawaki, M.; Kinoshita, M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998, 31, 391–398. [Google Scholar] [CrossRef]
- Phelan, D.; Watson, C.; Martos, R.; Collier, P.; Patle, A.; Donnelly, S.; Ledwidge, M.; Baugh, J.; McDonald, K. Modest elevation in BNP in asymptomatic hypertensive patients reflects sub-clinical cardiac remodeling, inflammation and extracellular matrix changes. PLoS ONE 2012, 7, e49259. [Google Scholar] [CrossRef] [PubMed]
- Collier, P.; Watson, C.J.; Voon, V.; Phelan, D.; Jan, A.; Mak, G.; Martos, R.; Baugh, J.A.; Ledwidge, M.T.; McDonald, K.M. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur. J. Heart Fail. 2011, 13, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [PubMed]
- López, B.; González, A.; Querejeta, R.; Larman, M.; Díez, J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J. Am. Coll. Cardiol. 2006, 48, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Foronjy, R.F.; Sun, J.; Lemaitre, V.; D’Armiento, J.M. Transgenic expression of matrix metalloproteinase-1 inhibits myocardial fibrosis and prevents the transition to heart failure in a pressure overload mouse model. Hypertens. Res. 2008, 31, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Jekell, A.; Malmqvist, K.; Wallén, N.H.; Mörtsell, D.; Kahan, T. Markers of Inflammation, Endothelial Activation, and Arterial Stiffness in Hypertensive Heart Disease and the Effects of Treatment: Results from the SILVHIA Study. J. Cardiovasc. Pharmacol. 2013, 62, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Greene, S.J.; Butler, J.; Sabbah, H.N.; Shantsila, E.; Lip, G.Y.H.; Gheorghiade, M. The immunological axis in heart failure: Importance of the leukocyte differential. Heart Fail. Rev. 2013, 18, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Huehnergarth, K.V.; Mozaffarian, D.; Sullivan, M.D.; Crane, B.A.; Wilkinson, C.W.; Lawler, R.L.; McDonald, G.B.; Fishbein, D.P.; Levy, W.C. Usefulness of relative lymphocyte count as an independent predictor of death/urgent transplant in heart failure. Am. J. Cardiol. 2005, 95, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Sakatani, T.; Hadase, M.; Kawasaki, T.; Kamitani, T.; Kawasaki, S.; Sugihara, H. Usefulness of the Percentage of Plasma Lymphocytes as a Prognostic Marker in Patients with Congestive Heart Failure. Jpn. Heart J. 2004, 45, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Hodge, D.O.; Rodeheffer, R.J.; McGregor, C.G.; Thomson, S.P.; Gibbons, R.J. Predictive power of the relative lymphocyte concentration in patients with advanced heart failure. Circulation 1998, 97, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Acanfora, D.; Gheorghiade, M.; Trojano, L.; Furgi, G.; Pasini, E.; Picone, C.; Papa, A.; Iannuzzi, G.L.; Bonow, R.O.; Rengo, F. Relative lymphocyte count: A prognostic indicator of mortality in elderly patients with congestive heart failure. Am. Heart J. 2001, 142, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.-T. Differential white blood cell count and incident heart failure in men and women in the EPIC-Norfolk study. Eur. Heart J. 2012, 33, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Bekwelem, W.; Lutsey, P.L.; Loehr, L.R.; Agarwal, S.K.; Astor, B.C.; Guild, C.; Ballantyne, C.M.; Folsom, A.R. White blood cell count, C-reactive protein, and incident heart failure in the Atherosclerosis Risk in Communities (ARIC) Study. Ann. Epidemiol. 2011, 21, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Khan, M.A.F.; Mentz, R.J.; O’Connor, C.M.; Metra, M.; Dittrich, H.C.; Ponikowski, P.; Teerlink, J.R.; Cotter, G.; Davison, B.; et al. Biomarker Profiles of Acute Heart Failure Patients With a Mid-Range Ejection Fraction. JACC Heart Fail. 2017, 5, 507–517. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; Lok, D.J.A.; Jaarsma, T.; van der Meer, P.; Voors, A.A.; Hillege, H.L.; van Veldhuisen, D.J. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann. Med. 2011, 43, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Van Kimmenade, R.R.; Januzzi, J.L.; Ellinor, P.T.; Sharma, U.C.; Bakker, J.A.; Low, A.F.; Martinez, A.; Crijns, H.J.; MacRae, C.A.; Menheere, P.P.; et al. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J. Am. Coll. Cardiol. 2006, 48, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Canepa, M.; Fonseca, C.; Chioncel, O.; Laroche, C.; Crespo-Leiro, M.G.; Coats, A.J.S.; Mebazaa, A.; Piepoli, M.F.; Tavazzi, L.; Maggioni, A.P.; et al. Performance of Prognostic Risk Scores in Chronic Heart Failure Patients Enrolled in the European Society of Cardiology Heart Failure Long-Term Registry. JACC Heart Fail 2018, 6, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Ketchum, E.S.; Levy, W.C. Establishing prognosis in heart failure: A multimarker approach. Prog. Cardiovasc. Dis. 2011, 54, 86–96. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | TAC | ATII-Infusion |
|---|---|---|
| LV structure | Concentric → dilated | Concentric → dilated |
| Systolic function | Preserved → reduced | Preserved* → reduced* |
| Diastolic function | Dfxn | Dfxn |
| Cardiomyocyte hypertrophy | Yes | Yes |
| Myocardial fibrosis | Yes | Yes |
| Increased BNP expression | Yes | Yes |
| Can it be used as a HFpEF model? | No | Yes |
| Phase | 1. ADP | 2. CP | 3. DP-HF | 4. CHF | ||
|---|---|---|---|---|---|---|
| Time | 3rd day | 1st week | 2nd week | 4th week | 6th week | 8th week |
| Hemodynamic & Structural parameters | ||||||
| LVEF [40,50,51] | ↓ | ↔ | ↔ | ↓ | ↓+ | |
| FS [38,47,49] | ↔ | ↔ | ↓ | ↓ | ↓ | |
| EDV [40] | ↔ | ↔ | ↔ | ↑ | ↑ | ↑+ |
| BP [50] | ↑ | ↑ | ↑ | |||
| LV hypertrophic response [38] | concentric | eccentric | eccentric | |||
| Fibrotic response [38,48] | ↑ | ↑+ | ↑ | ↑ | ↑ | |
| RV hypertrophic response [40] | ↑ | |||||
| Immune cell infiltration | ||||||
| CD45+ [38,40] | ↔ | ↑ | ↔/↑ | |||
| CD3+ [47,49] | ↔ | ↑ | ↑ | ↑ | ↑ | ↑ |
| CD3+, CD4+ [47,49] | ↑ | ↑ | ↑ | |||
| CD3+, CD8+ [48,49] | ↑ | ↔ | ||||
| Treg [48] | ↑ | |||||
| M [38,40] | ↔ | ↑ | ↔ | ↔ | ||
| M1 [48] | ↔ | ↑ | ↔ | |||
| M2 [48] | ↔ | ↑ | ↔ | |||
| Gr1+ [47] | ↔ | ↔ | ||||
| Classical DCs [40] | ↔ | ↑ | ↔ | ↑ | ||
| Peripheral circulating immune cells | ||||||
| Mon Ly6Chi [40] | ↔ | ↑ | ↔ | |||
| Mon Ly6Clo [40] | ↔ | ↔ | ↔ | |||
| Cytokines: | ||||||
| BAFF (B cell activating factor) [50] | ↑ | |||||
| IL-1β, IFN-ɣ, TNFα, IL-6, IL-10 [50] | ↔ | |||||
| Mediastinic Lymph Node | ||||||
| CD3+ [47] | ↑ | ↑ | ↑ | ↑ | ||
| CD3+, CD4+ [47] | ↑ | ↑ | ↑ | |||
| CD3+, CD8+ [47] | ↑ | ↔ | ||||
| Gene Expression | ||||||
| BNP [48] | ↑ | ↑ | ||||
| TGF-β [38] | ↑ | ↑ | ||||
| Chemokines: | ||||||
| CCL4, CCL5, CXCL11 [51] | ↑ | ↔ | ||||
| MCP-1(CCL2) [38,51] | ↑ | ↔ | ||||
| CCL7, CCL12 [48] | ↑ | |||||
| CXCL10, CX3CL1, CXCL16, CCL17 [49] | ↑ | |||||
| CD3e [51] | ↑ | |||||
| Cytokines: | ||||||
| TNFα, IL-6, IFN-ɣ [47,48,49,50,51] | ↑ | ↑ | ||||
| IL-1β, IL-8, BAFF [47,50,51] | ↑ | |||||
| IL-4 [48,51] | ↓ | ↑ | ||||
| IL-10 [48,50] | ↔ | ↓ | ||||
| RORɣt [47] | ↑ | |||||
| Foxp3 [47,51] | ↔ | |||||
| Adhesion molecules | ||||||
| VCAM-1, E-Sel, ICAM-1 [47] | ↑ | |||||
| Heart tissue immunostaining | ||||||
| IgG1, IgG2, IgG4 [50] | ↓ | |||||
| IgG3 [50] | ↑ | |||||
| BAX [50] | ↑ | |||||
| Anti-ssDNA [50] | ↑ | |||||
| Human Progression of HDD [62,63] | Murine Progression of PO-HF [38,40] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Degree | Dfxn | LV | EF | Peripheral | ↑Local markers | Phase | Dfxn | LV | EF | Peripheral | ↑ Local markers | ||
| ↑Cytokines | ↑ Cells | ↑Cytokines | ↑ Cells | ||||||||||
| I | D | P | P | IL6, CRP, MMP9, BNP [56,69,71,75] | Mon [56] | ADP | P | Preserved | R | ||||
| II | D | C-LVH | P | CP | P | C-LVH | P | Mon [40] | T cell, macrophages, DCs [40,47,48,49] BNP, TGFB, MCP1, CCLs, TNFa, IL6, IFN-ɣ [38,47,48,49,52] | ||||
| III | D | C-LVH | P | TNFα, MCP1, IL6, IL12, IL8, MMP9, BNP [56,71] | Mon [56] | Col-I, Col-3, Col-1/Col-3 TGF-β, VCAM-1, TIMP1, MMP2, ↓MMP1, Lukocytes, T-cells, CD11a+ cells [72] | DP-HF | S | Dilation | P | BAFF [50] | T cell [47,48,49] BNP, TGFB, MCP1, TNFa, IL6, IL8, IFN-ɣ. IL1B, IL4, ↓IL10, VCAM-1, ICAM-1, E-Sel [38,47,48,49,50,51] | |
| IV | S | Dilation | R | TNF α, MMP1/TIMP1, BNP [16,73] | T cells, TNFα, ↓TNFR [16,47] | CHF | S | Dilation | R | T cell, DCs [40,47,49] | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenes-Castro, D.; Castillo, E.C.; Vázquez-Garza, E.; Torre-Amione, G.; García-Rivas, G. Temporal Frame of Immune Cell Infiltration during Heart Failure Establishment: Lessons from Animal Models. Int. J. Mol. Sci. 2018, 19, 3719. https://doi.org/10.3390/ijms19123719
Brenes-Castro D, Castillo EC, Vázquez-Garza E, Torre-Amione G, García-Rivas G. Temporal Frame of Immune Cell Infiltration during Heart Failure Establishment: Lessons from Animal Models. International Journal of Molecular Sciences. 2018; 19(12):3719. https://doi.org/10.3390/ijms19123719
Chicago/Turabian StyleBrenes-Castro, David, Elena C. Castillo, Eduardo Vázquez-Garza, Guillermo Torre-Amione, and Gerardo García-Rivas. 2018. "Temporal Frame of Immune Cell Infiltration during Heart Failure Establishment: Lessons from Animal Models" International Journal of Molecular Sciences 19, no. 12: 3719. https://doi.org/10.3390/ijms19123719
APA StyleBrenes-Castro, D., Castillo, E. C., Vázquez-Garza, E., Torre-Amione, G., & García-Rivas, G. (2018). Temporal Frame of Immune Cell Infiltration during Heart Failure Establishment: Lessons from Animal Models. International Journal of Molecular Sciences, 19(12), 3719. https://doi.org/10.3390/ijms19123719