Experimental Combined Immunotherapy of Tumours with Major Histocompatibility Complex Class I Downregulation

 , , , , and
, , , , and
Abstract
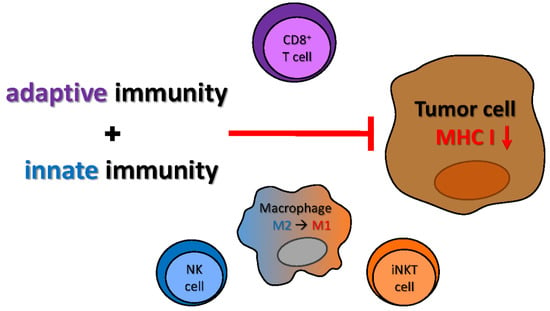
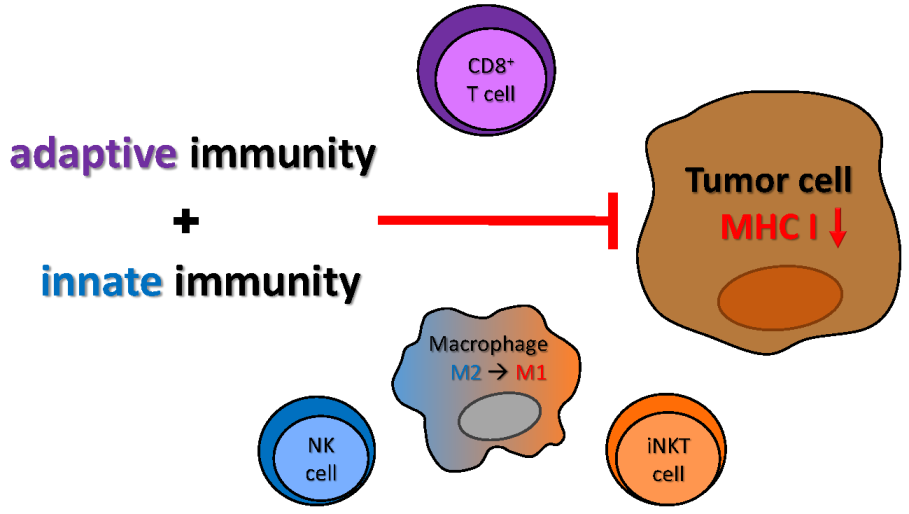
1. Introduction
2. Results
2.1. ODN1826 and α-GalCer Are Effective Vaccine Adjuvants for DNA Immunization against Tumors with Reduced Expression of MHC Class I Molecules
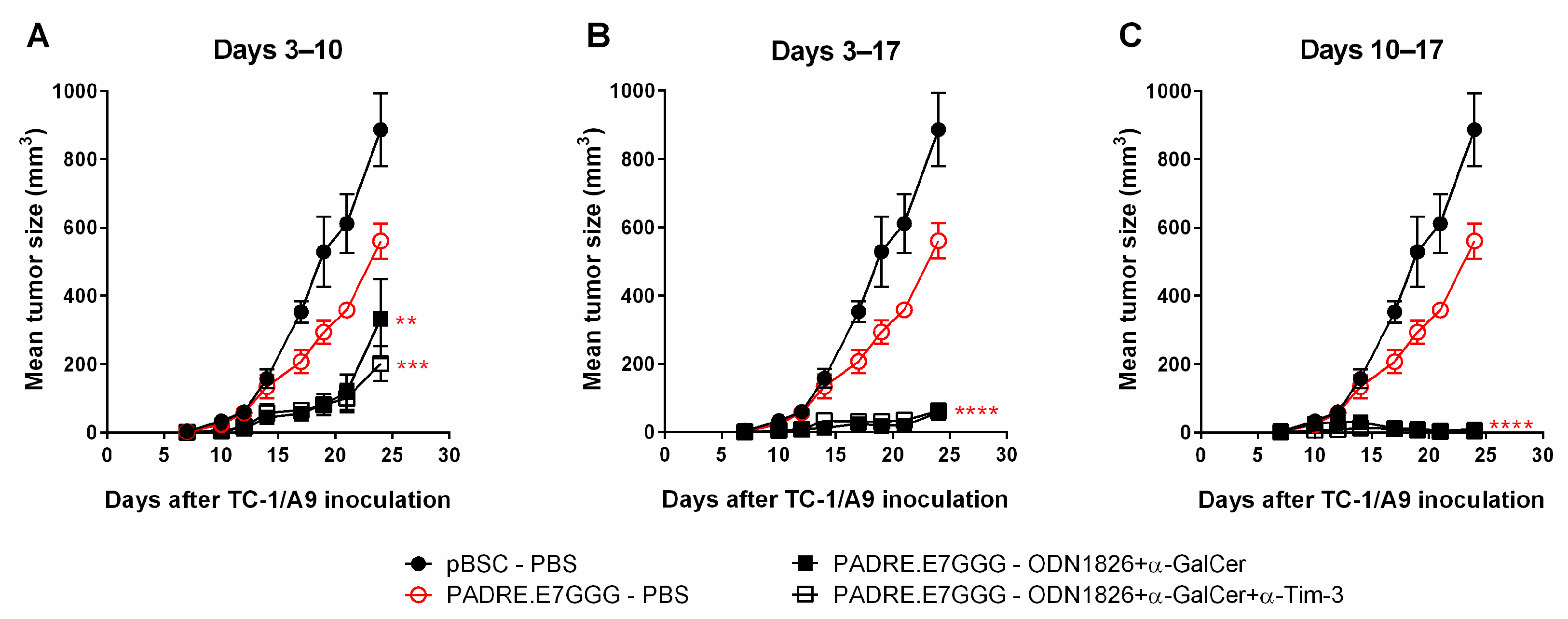
2.2. Delayed Administration of ODN1826 and α-GalCer in Combination Promoted Inhibition of Tumor Growth
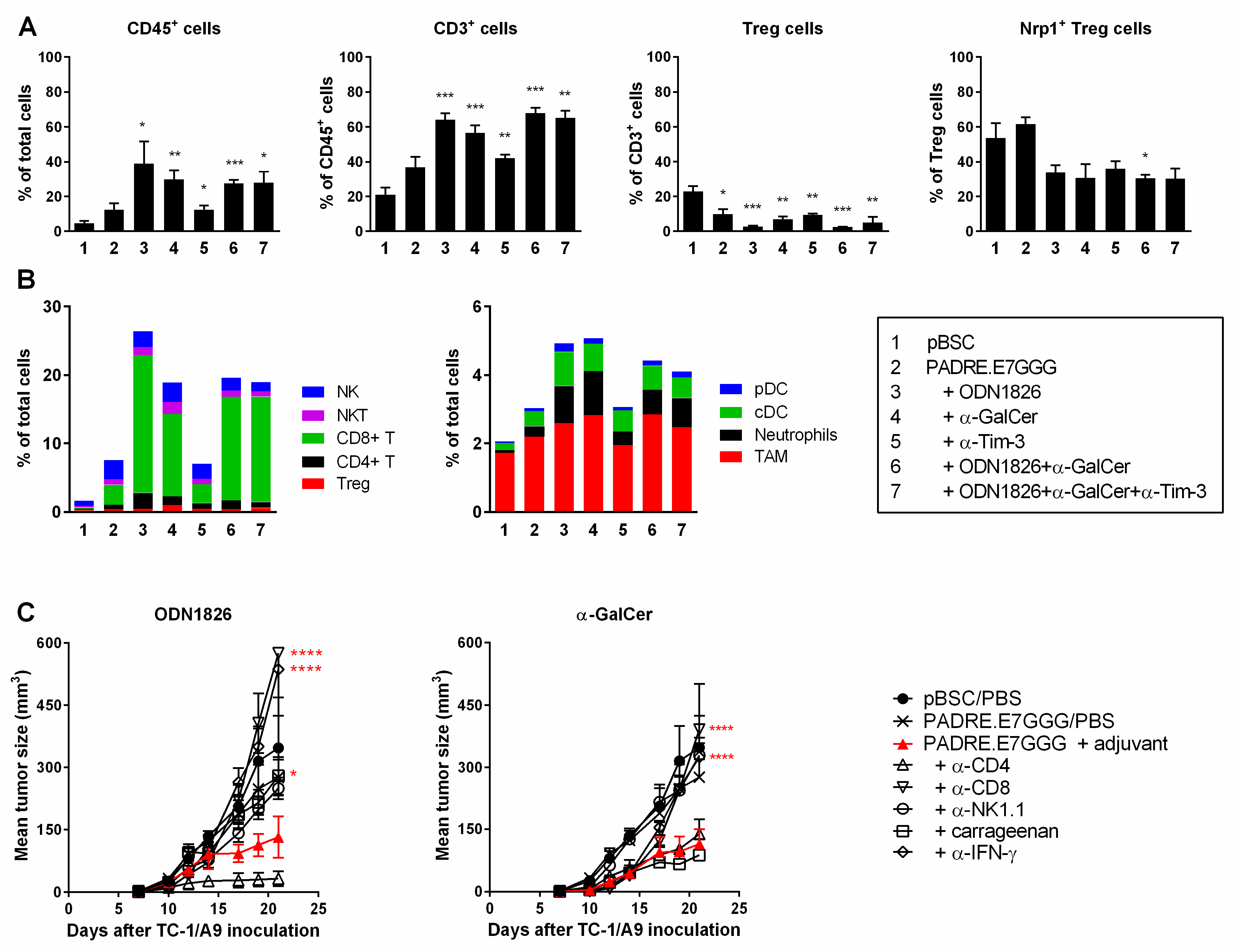
2.3. Immunotherapy Induced Infiltration of Tumors with Various Immune Cells that Differently Affected Tumor Growth
2.4. Combined Immunotherapy Did Not Substantially Enhance Either Systemic or Intratumoral Activation of CD8+ Cells
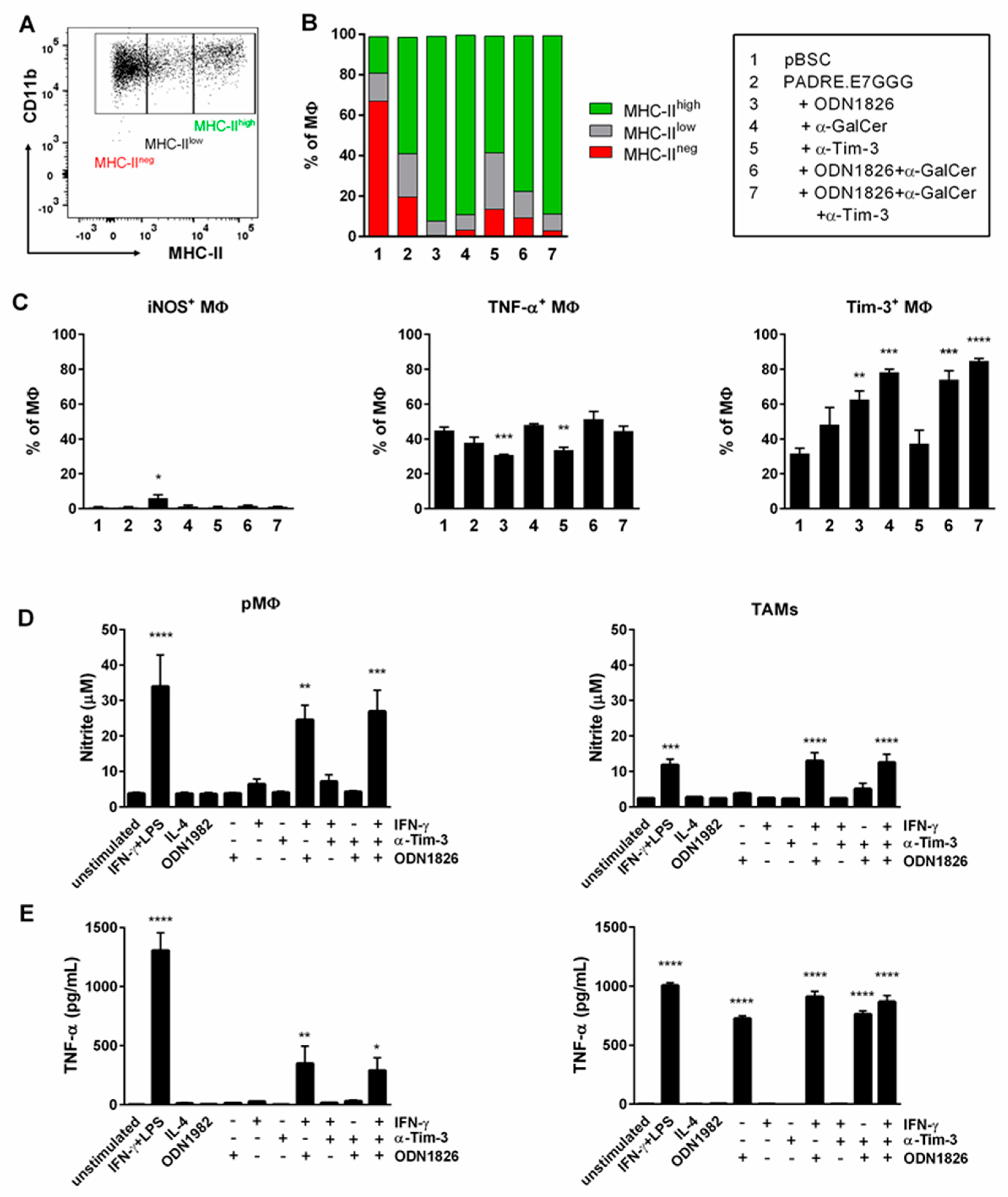
2.5. Immunotherapy Induced Polarization of TAMs into M1 MΦs
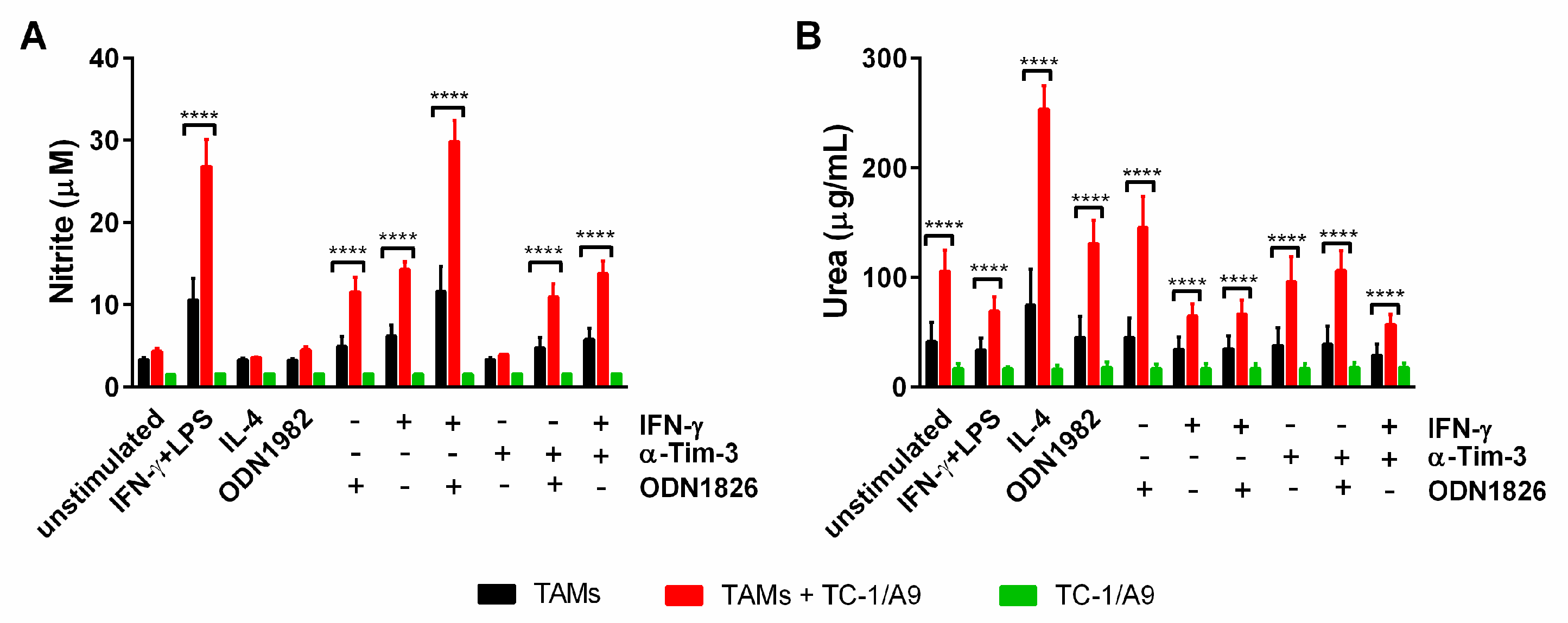
2.6. Co-Culture of TAMs with TC-1/A9 Tumor Cells Enhanced iNOS and Arginase Activity
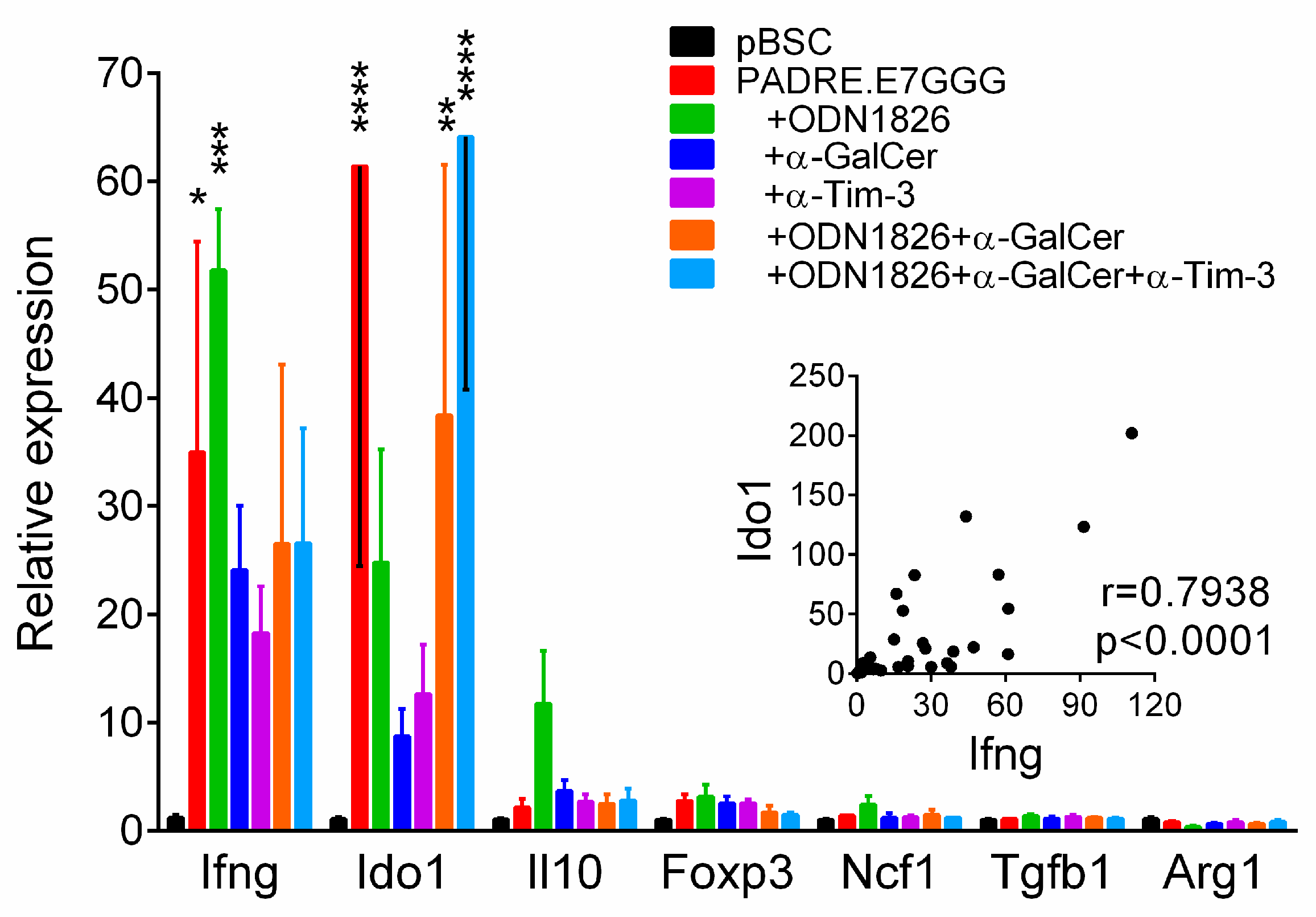
2.7. Enhancement of IFN-γ Expression in Tumors Correlated with Induction of Ido1 Expression
3. Discussion
4. Materials and Methods
4.1. Preparation of Reagents and Media
4.2. Mice
4.3. Tumor Cell Line
4.4. Plasmids
4.5. Combined Immunotherapy Experiments
4.6. Isolation of TAMs
4.7. Isolation of pMΦs
4.8. In Vitro Stimulations of TAMs and pMΦs
4.9. NO Measurement
4.10. TNF-α Enzyme-Linked Immunosorbent Assay (ELISA)
4.11. IFN-γ Enzyme-Linked Immunospot (ELISPOT) Assay
4.12. Co-Culture of TC-1/A9 Tumor Cells with TAMs
4.13. Arginase Microplate Assay
4.14. Flow Cytometry
4.15. Quantification of mRNA Expression by RT-qPCR
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrissey, K.; Yuraszeck, T.; Li, C.; Zhang, Y.; Kasichayanula, S. Immunotherapy and novel combinations in oncology: Current landscape, challenges and opportunities. Clin. Transl. Sci. 2016, 9, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Beyranvand Nejad, E.; Welters, M.J.P.; Arens, R.; van der Burg, S.H. The importance of correctly timing cancer immunotherapy. Expert. Opin. Biol. Ther. 2017, 17, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance and tumor immune escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.-F. DNA vaccine for cancer immunotherapy. Hum. Vaccin. Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Eggermont, A.; Sautès-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology 2013, 2, e25238. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Y.; Lin, Y.-L.; Chiang, B.-L. Levamisole enhances immune response by affecting the activation and maturation of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2008, 151, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.C.; Berzofsky, J.A.; Terabe, M. NKT cell networks in the regulation of tumor immunity. Front. Immunol. 2014, 5, 543. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. Tim-3: An emerging target in the cancer immunotherapy landscape. Cancer Immunol. Res. 2014, 2, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Chen, G.; Shen, B.; Li, Y. Tim-3: An activation marker and activation limiter of innate immune cells. Front. Immunol. 2013, 4, 449. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Ley, K. M1 and M2 macrophages: The chicken and the egg of immunity. J. Innate Immun. 2014, 6, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Ocana-Guzman, R.; Torre-Bouscoulet, L.; Sada-Ovalle, I. TIM-3 regulates distinct functions in macrophages. Front. Immunol. 2016, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Turkowski, K.; Mora, J.; Brune, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Mo, C.; Wang, Y.; Wei, D.; Xiao, H. Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 2013, 138, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Buhtoiarov, I.N.; Sondel, P.M.; Wigginton, J.M.; Buhtoiarova, T.N.; Yanke, E.M.; Mahvi, D.A.; Rakhmilevich, A.L. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology 2011, 132, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Rakhmilevich, A.L.; Baldeshwiler, M.J.; Van De Voort, T.J.; Felder, M.A.R.; Yang, R.K.; Kalogriopoulos, N.A.; Koslov, D.S.; Van Rooijen, N.; Sondel, P.M. Tumor-associated myeloid cells can be activated in vitro and in vivo to mediate antitumor effects. Cancer Immunol. Immunother. 2012, 61, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Buhtoiarov, I.N.; Lum, H.; Berke, G.; Paulnock, D.M.; Sondel, P.M.; Rakhmilevich, A.L. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J. Immunol. 2005, 174, 6013–6022. [Google Scholar] [CrossRef] [PubMed]
- Buhtoiarov, I.N.; Sondel, P.M.; Eickhoff, J.C.; Rakhmilevich, A.L. Macrophages are essential for antitumour effects against weakly immunogenic murine tumours induced by class B CpG-oligodeoxynucleotides. Immunology 2007, 120, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Lum, H.D.; Buhtoiarov, I.N.; Schmidt, B.E.; Berke, G.; Paulnock, D.M.; Sondel, P.M.; Rakhmilevich, A.L. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumour necrosis factor-alpha. Immunology 2006, 118, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.L.; Rakhmilevich, A.; Heninger, E.; Broman, A.T.; Hope, C.; Phan, F.; Miyamoto, S.; Maroulakou, I.; Callander, N.; Hematti, P.; et al. Tumoricidal effects of macrophage-activating immunotherapy in a murine model of relapsed/refractory multiple myeloma. Cancer Immunol. Res. 2015, 3, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Thoreau, M.; Penny, H.L.; Tan, K.; Regnier, F.; Weiss, J.M.; Lee, B.; Johannes, L.; Dransart, E.; Le Bon, A.; Abastado, J.-P.; et al. Vaccine-induced tumor regression requires a dynamic cooperation between T cells and myeloid cells at the tumor site. Oncotarget 2015, 6, 27832–27846. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Woo, S.-R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and function of regulatory T cells is maintained by a neuropilin-1–semaphorin-4a axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. Anatomy of a discovery: M1 and M2 macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2015, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, K.D.; Opel, C.F.; Szeto, G.L.; Tzeng, A.; Zhu, E.F.; Engreitz, J.M.; Williams, R.T.; Rakhra, K.; Zhang, M.H.; Rothschilds, A.M.; et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016, 22, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Aris, M.; Mordoh, J.; Barrio, M.M. Immunomodulatory monoclonal antibodies in combined immunotherapy trials for cutaneous melanoma. Front. Immunol. 2017, 8, 1024. [Google Scholar] [CrossRef] [PubMed]
- Smahel, M.; Sima, P.; Ludvikova, V.; Marinov, I.; Pokorna, D.; Vonka, V. Immunisation with modified HPV16 E7 genes against mouse oncogenic TC-1 cell sublines with downregulated expression of MHC class I molecules. Vaccine 2003, 21, 1125–1136. [Google Scholar] [CrossRef]
- Smahel, M.; Polakova, I.; Sobotkova, E.; Vajdova, E. Systemic administration of CpG oligodeoxynucleotide and levamisole as adjuvants for gene-gun-delivered antitumor DNA vaccines. Clin. Dev. Immunol. 2011, 2011, 176759. [Google Scholar] [CrossRef] [PubMed]
- Kastankova, I.; Polakova, I.; Duskova, M.; Smahel, M. Combined cancer immunotherapy against aurora kinase A. J. Immunother. 2016, 39, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Reinis, M.; Simova, J.; Bubenik, J. Inhibitory effects of unmethylated CpG oligodeoxynucleotides on MHC class I-deficient and -proficient HPV16-associated tumours. Int. J. Cancer 2006, 118, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Simova, J.; Indrova, M.; Bieblova, J.; Mikyskova, R.; Bubenik, J.; Reinis, M. Therapy for minimal residual tumor disease: β-galactosylceramide inhibits the growth of recurrent HPV16-associated neoplasms after surgery and chemotherapy. Int. J. Cancer 2010, 126, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed]
- Lepique, A.P.; Daghastanli, K.R.P.; Cuccovia, I.M.; Villa, L.L. HPV16 tumor associated macrophages suppress antitumor T cell responses. Clin. Cancer Res. 2009, 15, 4391–4400. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, Y.; Katae, M.; Takeda, K.; Yagita, H.; Okumura, K.; Kobayashi, S.; Takeuchi, T.; Kamiyama, T.; Fukuchi, Y.; Aoki, T. Activation of natural killer T cells by α-galactosylceramide impairs DNA vaccine-induced protective immunity against Trypanosoma cruzi. Infect. Immun. 2003, 71, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hung, C.-F.; Wu, T.-C.; Park, Y.-M. DNA vaccine with α-galactosylceramide at prime phase enhances anti-tumor immunity after boosting with antigen-expressing dendritic cells. Vaccine 2010, 28, 7297–7305. [Google Scholar] [CrossRef] [PubMed]
- Bercovici, N.; Trautmann, A. Revisiting the role of T cells in tumor regression. Oncoimmunology 2012, 1, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, T.C.; Sluijter, M.; van Duikeren, S.; West, B.L.; Melief, C.J.M.; Arens, R.; van der Burg, S.H.; van Hall, T. Therapeutic peptide vaccine-induced CD8 T cells strongly modulate intratumoral macrophages required for tumor regression. Cancer. Immunol. Res. 2015, 3, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.W.; Heldwein, K.A.; Means, T.K.; Saukkonen, J.J.; Fenton, M.J. Differential roles of Toll-like receptors in the elicitation of proinflammatory responses by macrophages. Ann. Rheum. Dis. 2001, 60 (Suppl. S3), iii6–iii12. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Øynebråten, I.; Corthay, A. Toll-like receptor ligands and interferon-γ synergize for induction of antitumor M1 macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef] [PubMed]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; de Velde, L.-A.V.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Raymond, D.P.; Crabtree, T.D.; Pelletier, S.J.; Houlgrave, C.W.; Pruett, T.L.; Sawyer, R.G. Enhanced murine macrophage TNF receptor shedding by cytosine-guanine sequences in oligodeoxynucleotides. J. Immunol. 2000, 165, 5153–5160. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.I.; Liao, J.C.; Kuo, L. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. 1998, 274, H342–H348. [Google Scholar] [CrossRef] [PubMed]
- Liscovsky, M.V.; Ranocchia, R.P.; Gorlino, C.V.; Alignani, D.O.; Morón, G.; Maletto, B.A.; Pistoresi-Palencia, M.C. Interferon-γ priming is involved in the activation of arginase by oligodeoxinucleotides containing CpG motifs in murine macrophages. Immunology 2009, 128, e159–e169. [Google Scholar] [CrossRef] [PubMed]
- Calorini, L.; Bianchini, F.; Mannini, A.; Mugnai, G.; Ruggieri, S. Enhancement of nitric oxide release in mouse inflammatory macrophages co-cultivated with tumor cells of a different origin. Clin. Exp. Metastasis 2005, 22, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, T.; Kadonosono, T.; Sakurai, S.; Shiozawa, T.; Goto, T.; Sakai, S.; Kuchimaru, T.; Sakamoto, T.; Watanabe, H.; Kondoh, G.; et al. Novel adherent CD11b+ Gr-1+ tumor-infiltrating cells initiate an immunosuppressive tumor microenvironment. Oncotarget 2018, 9, 11209–11226. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar] [PubMed]
- Smahel, M.; Sima, P.; Ludvikova, V.; Vonka, V. Modified HPV16 E7 genes as DNA vaccine against E7-containing oncogenic cells. Virology 2001, 281, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Smahel, M.; Polakova, I.; Duskova, M.; Ludvikova, V.; Kastankova, I. The effect of helper epitopes and cellular localization of an antigen on the outcome of gene gun DNA immunization. Gene Ther. 2014, 21, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Sidney, J.; Southwood, S.; Ruppert, J.; Oseroff, C.; Maewal, A.; Snoke, K.; Serra, H.M.; Kubo, R.T.; Sette, A. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity 1994, 1, 751–761. [Google Scholar] [CrossRef]
- Ishizaka, S.; Kuriyama, S.; Tsujii, T. In vivo depletion of macrophages by desulfated iota-carrageenan in mice. J. Immunol. Methods 1989, 124, 17–24. [Google Scholar] [CrossRef]
- Corraliza, I.M.; Campo, M.L.; Soler, G.; Modolell, M. Determination of arginase activity in macrophages: A micromethod. J. Immunol. Methods 1994, 174, 231–235. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Conjugate | Clone | Source | Staining | Panels | |||
|---|---|---|---|---|---|---|---|---|
| CD11b | BV421 | M1/70 | BioLegend, 101251 | Surface | ● | ● | ||
| CD11c | BV650 | N418 | BioLegend, 117339 | Surface | ● | |||
| CD25 | APC | PC61.5 | eBiosciences, 17-0251-81 | Surface | ● | |||
| CD3 | APC-Cy7 | 145-2C11 | BioLegend, 100330 | Surface | ● | ● | ● | ● |
| CD317 | APC | 927 | BioLegend, 127015 | Surface | ||||
| CD4 | BV510 | RM4-5 | BioLegend, 100559 | Surface | ● | ● | ||
| CD45 | Alexa Fluor 700 | 30-F11 | BioLegend, 103128 | Surface | ● | ● | ● | ● |
| CD8 | FITC | 53-6.7 | BD Biosciences, 553031 | Surface | ● | ● | ● | |
| F4/80 | BV510 | BM8 | BioLegend, 123135 | Surface | ● | |||
| Foxp3 | PE | FJK-16s | eBiosciences, 12-5773-82 | Nuclear | ● | |||
| Gr-1 | PE/BV786 ♦ | RB6-8C5 | BioLegend, 108407/BD Biosciences, 740850 ♦ | Surface | ● | ♦ | ||
| IFN-γ | BV421 | XMG1.2 | BD Biosciences, 563376 | Intracellular | ● | |||
| MHC-II | PerCP-Cy5.5/PE-Cy7 ♦ | M5/114.15.2 | BioLegend, 107625/BioLegend 107629 ♦ | Surface | ● | ♦ | ||
| NK1.1 | BV650 | PK136 | BioLegend, 108736 | Surface | ● | ● | ||
| iNOS | Alexa Fluor 488 | CXNFT | eBiosciences, 53-5920-80 | Intracellular | ● | |||
| Nrp1 | BV421 | 3E12 | BioLegend, 145209 | Surface | ● | |||
| PD-1 | PE-Cy7 | 29F.1A12 | BioLegend, 135215 | Surface | ● | |||
| Tim-3 | APC | RMT3-23 | BioLegend, 119706 | Surface | ● | ● | ||
| TNF-α | PE-DAZZLE594 | MP6-XT22 | BioLegend, 506346 | Intracellular | ● | ● | ||
| Target Gene | Reference Sequence (NCBI ID) | Forward Primer 5′ → 3′ | Reverse Primer 5′ → 3′ | Amplicon (bp) |
|---|---|---|---|---|
| Ifng | NM_008337.4 | TTCCTCATGGCTGTTTCTGG | CACCATCCTTTTGCCAGTTC | 148 |
| Ido1 | NM_001293690.1 | GTCTGGAGAAAGCCAAGGAA | ATATGCGGAGAACGTGGAAA | 81 |
| Il10 | NM_010548.2 | GGTGAGAAGCTGAAGACCC | ATGGCCTTGTAGACACCTTG | 137 |
| Foxp3 | NM_001199347.1 | ACCTGGCTGGGAAGATG | TCCCGAGGAGCAGACC | 124 |
| Ncf1 | NM_010876.4 | TGTTCCTGGTTAAGTGGCAG | GGTGTGGGATGACTCTGTTC | 144 |
| Tgfb1 | NM_011577.2 | TCAGACATTCGGGAAGCAG | AAGGTAACGCCAGGAATTGT | 135 |
| Arg1 | NM_007482.3 | ATGGAAGAGTCAGTGTGGTG | GGGAGTGTTGATGTCAGTGT | 128 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzelak, A.; Polakova, I.; Smahelova, J.; Vackova, J.; Pekarcikova, L.; Tachezy, R.; Smahel, M. Experimental Combined Immunotherapy of Tumours with Major Histocompatibility Complex Class I Downregulation. Int. J. Mol. Sci. 2018, 19, 3693. https://doi.org/10.3390/ijms19113693
Grzelak A, Polakova I, Smahelova J, Vackova J, Pekarcikova L, Tachezy R, Smahel M. Experimental Combined Immunotherapy of Tumours with Major Histocompatibility Complex Class I Downregulation. International Journal of Molecular Sciences. 2018; 19(11):3693. https://doi.org/10.3390/ijms19113693
Chicago/Turabian StyleGrzelak, Adrianna, Ingrid Polakova, Jana Smahelova, Julie Vackova, Lucie Pekarcikova, Ruth Tachezy, and Michal Smahel. 2018. "Experimental Combined Immunotherapy of Tumours with Major Histocompatibility Complex Class I Downregulation" International Journal of Molecular Sciences 19, no. 11: 3693. https://doi.org/10.3390/ijms19113693
APA StyleGrzelak, A., Polakova, I., Smahelova, J., Vackova, J., Pekarcikova, L., Tachezy, R., & Smahel, M. (2018). Experimental Combined Immunotherapy of Tumours with Major Histocompatibility Complex Class I Downregulation. International Journal of Molecular Sciences, 19(11), 3693. https://doi.org/10.3390/ijms19113693