Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β


Abstract
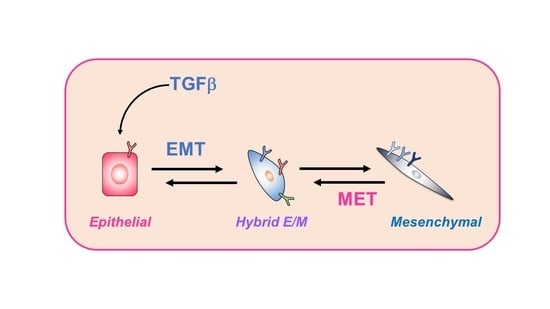
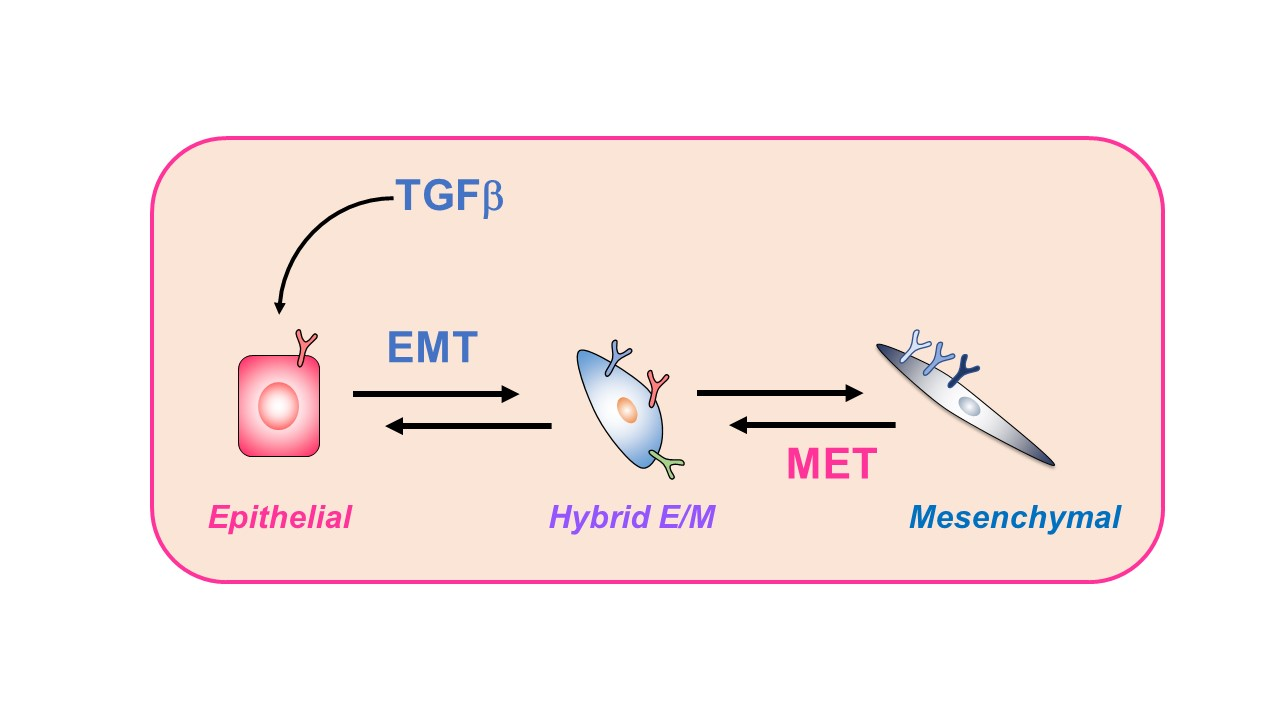{kind=link}
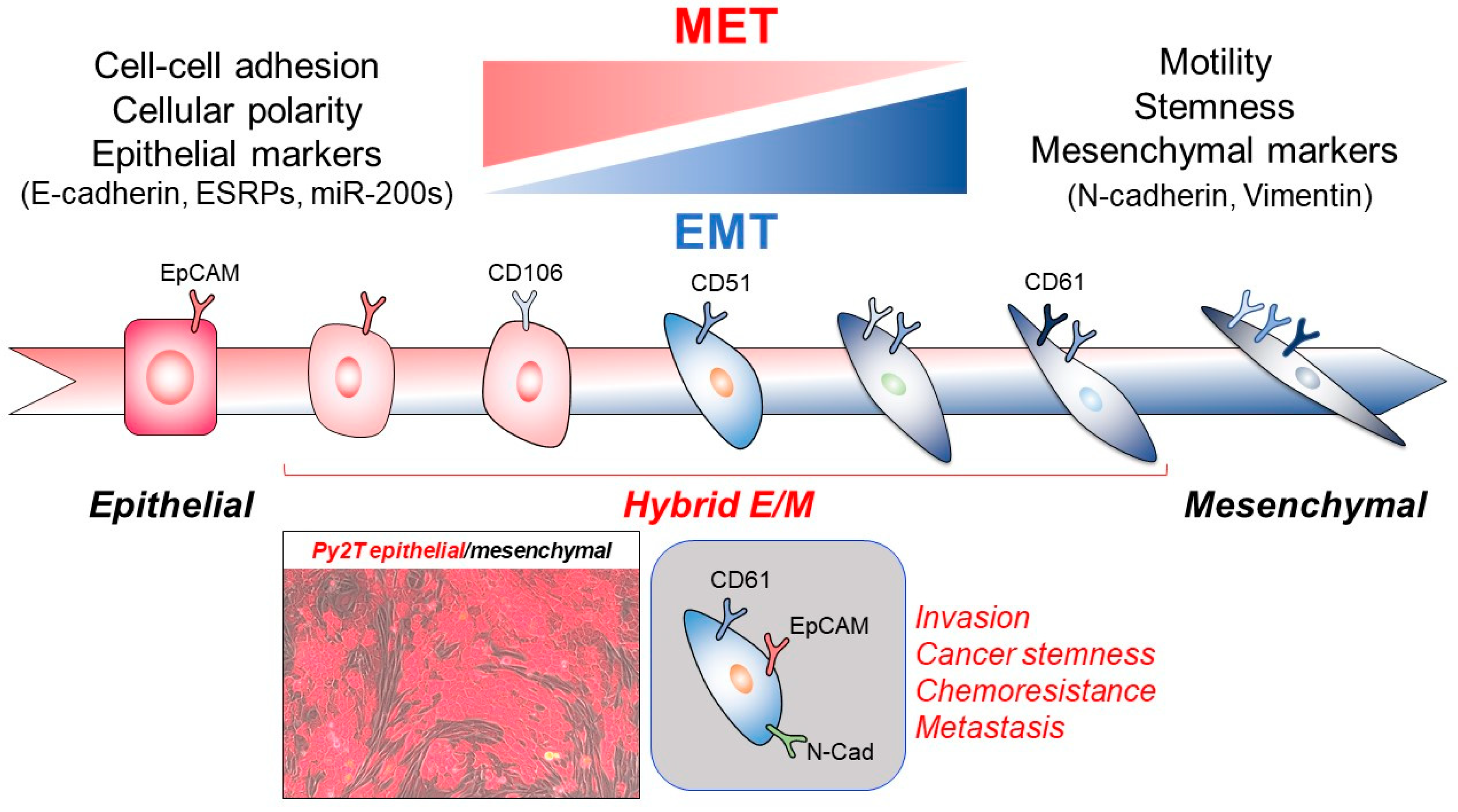{kind=link}
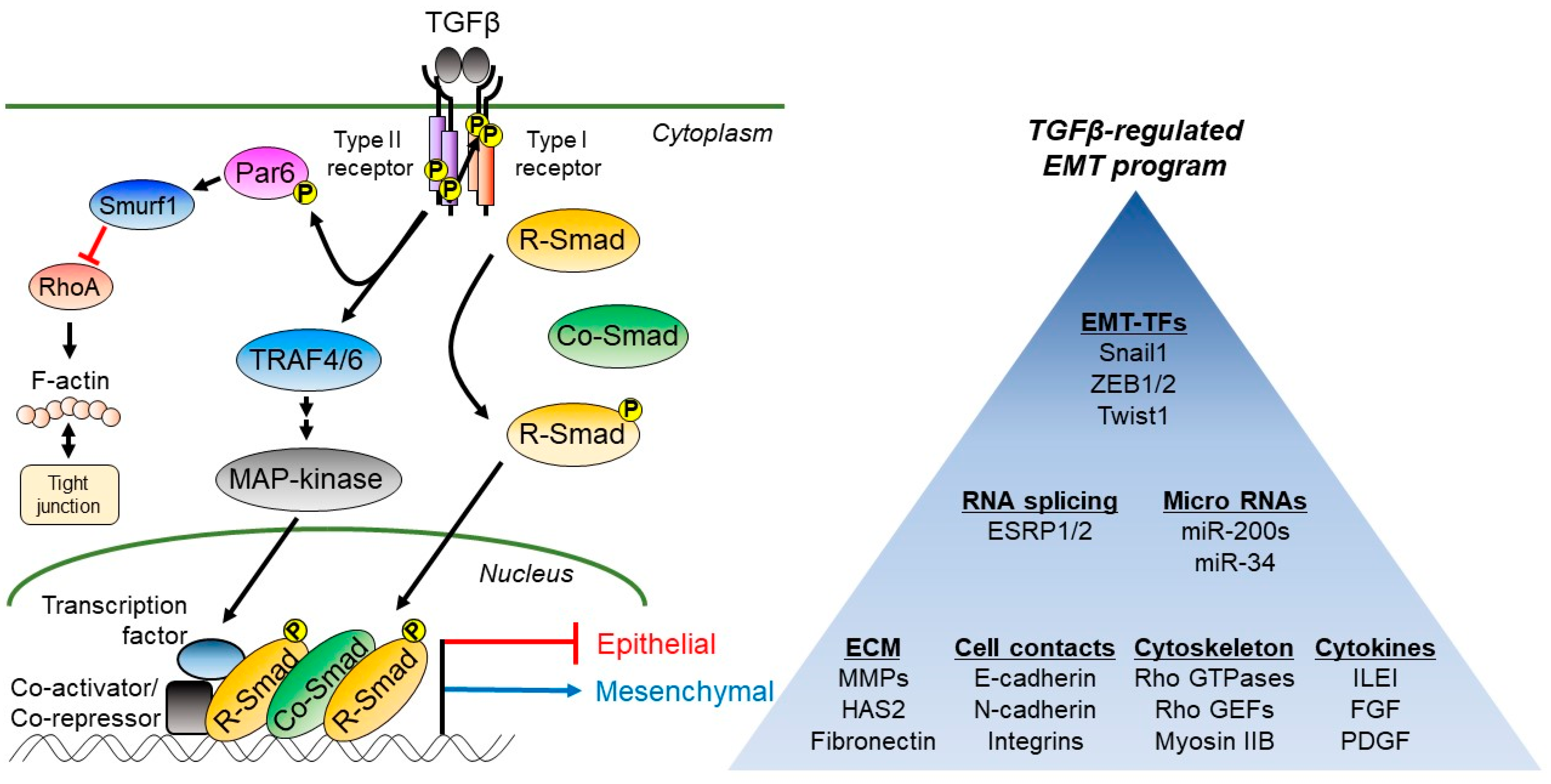{kind=link}
{kind=link}
1. Introduction
2. Regulation of EMT by TGF-β
2.1. Regulation of ECM Gene Expression by TGFβ
2.2. Regulation of Cell Contacts by TGFβ Signaling
2.3. Regulation of the Actin-Based Cytoskeleton during TGFβ-Induced EMT
2.4. TGFβ Controls the Expression of Many Other Growth Factors and Cytokines
3. Regulation of EMT-TF Expression and Activity by TGFβ
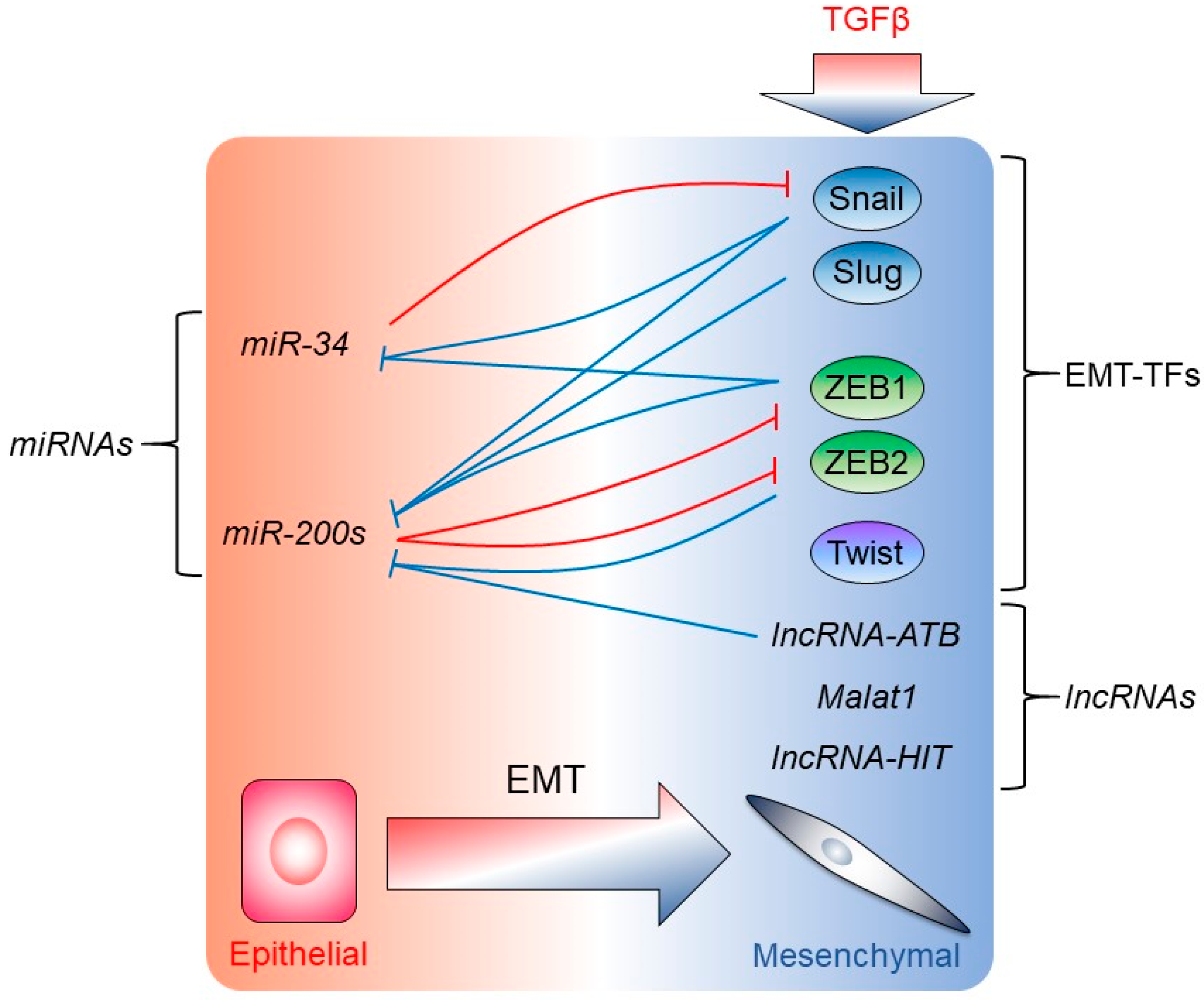
4. Regulation of EMT by miRNAs, lncRNAs, and mRNA Translational Mechanisms
5. The Importance of EMT in Tumor Metastasis
6. New Approaches Towards the Treatment of Metastasis
7. Concluding Remarks
Acknowledgments
Conflicts of interest
Abbreviations
| 4Ei-1 | eIF4E inhibitor 1 |
| ADCY9 | adenylyl cyclase 9 |
| AGO2 | argonaute 2 |
| Akt (1, 2, 3) | oncogene Akt serine/threonine kinase 1, 2, 3 (alias: protein kinase B, PKB) |
| AP-1 | activator protein 1 |
| ASPP2 | apoptosis-stimulating protein of p53-2 |
| ANGPTL4 | angiopoetin-like 4 |
| BMP | bone morphogenetic protein |
| CAF | cancer-associated fibroblast |
| CD51 | cell differentiation 51, alias: integrin αv |
| CD61 | cell differentiation 61, alias: integrin β3 |
| CD106 | cell differentiation 106, alias: vascular cell adhesion molecule-1, VCAM-1 |
| CdGAP | Cdc42 GTPase-activating protein |
| CDH1 | E-cadherin |
| CDH2 | N-cadherin |
| CDK2 | cyclin-dependent kinase 2 |
| ceRNA | competing endogenous RNA |
| CITED1 | Cbp/p300-interacting transactivator 1 |
| Cnt3 | concentrating nucleoside transporter 3 |
| CpG | cytosine-phosphate-guanine nukleotide |
| CRB3 | Crumbs3 |
| CRC | colorectal cancer |
| Cre | cyclization recombinase |
| CREB1 | cAMP-responsive element binding protein 1 |
| CRTC1 | CREB regulated transcription coactivator 1 |
| CSC | cancer stem cell |
| CtBP | c-terminal-binding protein |
| CTC | circulating tumor cell |
| CUBIC | clear, unobstructed brain/body imaging cocktails and computational analysis |
| CXCL | C-X-C motif chemokine ligand |
| Dab2 | disabled-2 |
| DUB | deubiquitinase |
| ECM | extracellular matrix |
| eEF1A1 | eukaryotic elongation factor 1A1 |
| EGF | epidermal growth factor |
| EMT | epithelial-mesenchymal transition |
| EMT-TFs | EMT transcription factors |
| EndMT | endothelial-mesenchymal transition |
| ENT1 | equilibrative nucleoside transporter 1 |
| EpCam | epithelial cell adhesion molecule |
| ERK2 | extracellular signal regulated kinase 2 |
| ESRP | epithelial splicing regulatory protein |
| ETS2 | ETS proto-oncogene 2 |
| Fbxo45 | F-box homologue 45 |
| FLT3 | Fms-related tyrosine kinase 3 |
| FN1 | fibronectin 1 |
| Fos | FBJ osteosarcoma oncogene |
| Fra-1 | Fos-related antigen 1 |
| Fsp1 | fibroblast-specific protein 1 |
| GEF-H1 | guanine exchange factor H1 |
| GFP | green fluorescent protein |
| GM-CSF | granulocyte-monocyte colony stimulating factor |
| GPNMB | glycoprotein nmb |
| GSK3β | glycogen synthase kinase 3β |
| H3K4 | histone 3, lysine 4 |
| H3K27me | histone 3, lysine 27, methylation |
| HAS2 | hyaluronan synthase 2 |
| HDAC1 | histone deacetylase 1 |
| Hic-5 | hydrogen peroxide-inducible clone 5 |
| HIF | hypoxia-inducible factor |
| HMGA2 | high mobility group A2 |
| HMLE | human mammary epithelial cells immortalized with the large T antigen |
| HNF4 | hepatocyte nuclear factor 4 |
| hnRNPE1 | heterogeneous ribonucleoprotein E1 |
| Id1/2 | inhibitor of differentiation 1/2 |
| IL-11 | interleukin 11 |
| ILEI | interleukin-like EMT-inducer |
| I-Smad | inhibitory Smad |
| ITGA1 | integrin α1 |
| JMJD3 | Jumonji domain-containing protein 3 |
| JNK | Jun N-terminal kinase |
| Jun | Jun proto-oncogene |
| Klf4 | Krüppel-like factor 4 |
| KPC | K-Ras and mutant p53 pancreatic carcinoma mouse model |
| LARG | leukemia-associated Rho guanine nucleotide exchange factor |
| Lgr4 | leucine-rich repeat-containing G protein-coupled receptor 4 |
| lncRNA | long non-coding RNA |
| lncRNA-ATB | lncRNA-induced by TGFβ |
| lncRNA-HIT | HOXa transcript induced by TGFβ |
| LOXL2 | lysyl oxidase-like 2 |
| LSD1 | lysine-specific histone demethylase 1, alias: KDM1A |
| MAF | musculoaponeurotic fibrosarcoma |
| MAFK | MAF oncogene family protein K |
| Malat1 | metastasis associated in lung adenocarcinoma transcript-1 |
| MAP-kinase | mitogen-activated protein kinase |
| MAPK1 | MAP-kinase 1 |
| MDM2 | mouse double minute 2 homolog |
| MEKK2 | mitogen-activated protein kinase kinase kinase 2 |
| MET | mesenchymal-epithelial transition |
| MIG6 | mitogen-inducible gene 6 |
| MiRNA | micro-RNA |
| MMP | matrix metalloprotease |
| MMTV-PyMT | mouse mammary tumor virus polyoma virus middle T antigen |
| MOF | males absent on the first (alias: KAT8, lysine acetyltransferase 8) |
| MT1-MMP | membrane type I-matrix metalloprotease |
| MYCBP | Myc binding protein |
| Net1 | neuroepithelial cell transforming 1 |
| NF-κB | nuclear factor-κB |
| Nkx2.8 | Nk2 homeobox 2.8 |
| PAI-1 | plasminogen activator inhibitor 1 |
| Par3/6 | partitioning-defective 3/6 |
| Pc2 | polycomb protein 2 |
| PDGF | platelet-derived growth factor |
| PIAS | protein inhibitor of activated Stat |
| PMEPAI | prostate transmembrane protein androgen induced-1 |
| PRC1/2 | polycomb-repressive complex 1 and 2 |
| PTBP3 | polypyrimidine tract binding protein 3 |
| R-Smad | receptor-activated Smad |
| Rad17 | radiation-induced 17 (alias: checkpoint clamp loader component) |
| RFP | red fluorescent protein |
| Rho | Ras homologue |
| RISC | RNA-induced silencing complex |
| RING | really interesting new gene |
| RNF8 | RING finger 8 |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| SAE1/2 | SUMO1 activating enzyme subunit 1 |
| Shh | sonic hedegehog |
| Siah | seven in absentia homolog |
| SIK1 | salt-inducible kinase 1 |
| Smurf | Smad ubiquitylation regulatory factor |
| SND1 | staphylococcal nuclease and tudor domain containing 1 |
| SNAG | Snail1/Gfi-1 |
| Sox5 | SRY-related high-mobility-group box 5 |
| SPRY | SP1a and the Ryanodine Receptor |
| Src | (Rous) sarcoma (virus oncogene) |
| STAT | signal transducer and activator of transcription |
| SUMO | small ubiquitin-related modifier |
| Suz12 | suppressor of zeste 12 |
| TAK1 | TGFβ-activated kinase 1 |
| TAZ | transcriptional coactivator with a PDZ-binding domain |
| TF | transcription factor |
| TGFβ | transforming growth factor β |
| TGIF | TGFβ-induced factor homeobox 1 |
| TIC | tumor initiating cell |
| TNBC | triple-negative breast cancer |
| TNFα | tumor necrosis factor-α |
| TOP1 | topoisomerase 1 |
| TRAF | TNFα receptor-associated factor |
| β-TrCP | β-transducin repeats-containing protein |
| Trx | thioredoxin |
| TXNIP | thioredoxin-interacting protein |
| UBC9 | ubiquitin-conjugating enzyme 9 |
| USP | ubiquitin-specific protease |
| UTR | untranslated region |
| Wnt | wingless and integration site |
| YAP | Yes-associated protein |
| ZEB1/2 | zinc finger E-box binding homeobox 1/2 |
| ZO | zonula occludens |
References
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.-P. EMT 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Kahata, K.; Dadras, M.S.; Moustakas, A. TGF-β family signaling in epithelial differentiation and epithelial-mesenchymal transition. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Sjöblom, T.; Fedorova, L.; Imreh, S.; Beug, H.; Moustakas, A. Sustained TGFβ exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008, 27, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; ten Dijke, P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Duan, J.; Pillai, I.C.; Rosa-Garrido, M.; Wu, Y.; Bargiacchi, F.; Lu, Y.; Stanbouly, S.; Huang, J.; Rojas, M.; et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 2014, 514, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhong, C.; Liu, D.; Yu, W.; Chen, W.; Wang, Y.; Shi, S.; Yuan, Y. Evidence for Kaposi sarcoma originating from mesenchymal stem cell through KSHV-induced mesenchymal-to-endothelial transition. Cancer Res. 2018, 78, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.-H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.-H. Induction of epithelial-mesenchymal transition by transforming growth factor β. Semin. Cancer Biol. 2012, 22, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.-H. The regulation of TGFβ signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed]
- Gudey, S.K.; Wallenius, A.; Landström, M. Regulated intramembrane proteolysis of the TGFβ type I receptor conveys oncogenic signals. Future Oncol. 2014, 10, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFβ type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.-H.; Landström, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z. Role for a3 integrin in EMT and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 7–10. [Google Scholar] [PubMed]
- Safina, A.; Ren, M.Q.; Vandette, E.; Bakin, A.V. TAK1 is required for TGF-β 1-mediated regulation of matrix metalloproteinase-9 and metastasis. Oncogene 2008, 27, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Zieba, A.; Vasilaki, E.; Herrera Hidalgo, C.; Soderberg, O.; Koinuma, D.; Miyazono, K.; Heldin, C.-H.; Landegren, U.; ten Dijke, P.; et al. Specific interactions between Smad proteins and AP-1 components determine TGFβ-induced breast cancer cell invasion. Oncogene 2013, 32, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Mauviel, A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-β: Implications for carcinogenesis. Oncogene 2005, 24, 5742–5750. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Lu, W.; Li, X.; Yang, G.; Guo, J.; Yu, H.; Li, Z.; Guan, F. Altered N-Glycan expression profile in epithelial-to-mesenchymal transition of NMuMG cells revealed by an integrated strategy using mass spectrometry and glycogene and lectin microarray analysis. J. Proteome Res. 2014, 13, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.; Takimoto, R.; Tamura, F.; Yoshida, M.; Ono, M.; Murase, K.; Sato, Y.; Osuga, T.; Sato, T.; Iyama, S.; et al. Fucosylated TGF-β receptors transduces a signal for epithelial-mesenchymal transition in colorectal cancer cells. Br. J. Cancer 2014, 110, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Kadam, P.; Cao, K.; Wu, S.; Samara, G.J.; Zhang, Q.; Zucker, S.; Cao, J. MT1-MMP Activation of TGF-β Signaling Enables Intercellular Activation of an Epithelial-mesenchymal Transition Program in Cancer. Curr. Cancer Drug Targets 2016, 16, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Mochizuki, N.; Kongtawelert, P.; Konno, K.; Itano, N. Excessive hyaluronan production promotes acquisition of cancer stem cell signatures through the coordinated regulation of Twist and the transforming growth factor β (TGF-β)-Snail signaling axis. J. Biol. Chem. 2014, 289, 26038–26056. [Google Scholar] [CrossRef] [PubMed]
- Porsch, H.; Bernert, B.; Mehic, M.; Theocharis, A.D.; Heldin, C.-H.; Heldin, P. Efficient TGFβ-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 2013, 32, 4355–4365. [Google Scholar] [CrossRef] [PubMed]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Wrana, J.L. Coordinating developmental signaling: Novel roles for the Hippo pathway. Trends Cell Biol. 2012, 22, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Taylor, M.A.; Schiemann, B.J.; Schiemann, W.P. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol. Biol. Cell 2011, 22, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J.; Wu, M.; Le, T.T.; Cho, S.H.; Brenner, M.B.; Blackburn, M.R.; Agarwal, S.K. Cadherin-11 contributes to pulmonary fibrosis: Potential role in TGF-β production and epithelial to mesenchymal transition. FASEB J. 2012, 26, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Sancisi, V.; Gandolfi, G.; Ragazzi, M.; Nicoli, D.; Tamagnini, I.; Piana, S.; Ciarrocchi, A. Cadherin 6 is a new RUNX2 target in TGF-β signalling pathway. PLoS ONE 2013, 8, e75489. [Google Scholar] [CrossRef] [PubMed]
- Viloria-Petit, A.M.; Wrana, J.L. The TGFβ-Par6 polarity pathway: Linking the Par complex to EMT and breast cancer progression. Cell Cycle 2010, 9, 623–624. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, E.L.; Liu, C.J.; Fearon, E.R.; Margolis, B. The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene 2008, 27, 3875–3879. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liu, X.; Cui, K.; Di, Y.; Xin, L.; Sun, X.; Zhang, W.; Yang, X.; Wei, M.; Yao, Z.; et al. SND1 Acts Downstream of TGFβ1 and Upstream of Smurf1 to Promote Breast Cancer Metastasis. Cancer Res. 2015, 75, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Fan, J.; Ding, X.; Peng, W.; Yu, X.; Chen, Y.; Nie, J. TGF-β-induced MiR-491-5p expression promotes Par-3 degradation in rat proximal tubular epithelial cells. J. Biol. Chem. 2010, 285, 40019–40027. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Lönn, P.; Vanlandewijck, M.; Kowanetz, K.; Heldin, C.-H.; Moustakas, A. TGFβ induces SIK to negatively regulate type I receptor kinase signaling. J. Cell Biol. 2008, 182, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; Vanlandewijck, M.; Raja, E.; Kowanetz, M.; Watanabe, Y.; Kowanetz, K.; Vasilaki, E.; Heldin, C.-H.; Moustakas, A. Transcriptional induction of salt-inducible kinase 1 by transforming growth factor β leads to negative regulation of type I receptor signaling in cooperation with the Smurf2 ubiquitin ligase. J. Biol. Chem. 2012, 287, 12867–12878. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; Dadras, M.S.; Lomnytska, M.; Mahzabin, T.; Lee Miller, M.; Busch, C.; Brunak, S.; Heldin, C.-H.; Moustakas, A. The protein kinase SIK downregulates the polarity protein Par3. Oncotarget 2018, 9, 5716–5735. [Google Scholar] [CrossRef] [PubMed]
- Parvani, J.G.; Galliher-Beckley, A.J.; Schiemann, B.J.; Schiemann, W.P. Targeted inactivation of b1 integrin induces b3 integrin switching, which drives breast cancer metastasis by TGF-β. Mol. Biol. Cell 2013, 24, 3449–3459. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, J.; Tumbarello, D.A.; Schmidt, R.P.; Turner, C.E. Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial-mesenchymal transition. J. Cell Biol. 2012, 197, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, E.; Vasilaki, E.; Vorvis, C.; Iliopoulos, D.; Moustakas, A.; Kardassis, D.; Stournaras, C. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-β and miR-24: Role in epithelial-to-mesenchymal transition. Oncogene 2012, 31, 2862–2875. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Aakre, M.; Brown, K.; Singh, V.; Moses, H.L. TGF-β-induced RhoA and p160ROCK activation is involved in the inhibition of Cdc25A with resultant cell-cycle arrest. Proc. Natl. Acad. Sci. USA 2003, 100, 15548–15553. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.G.; Ludwig, M.P.; Andrysik, Z.; Pandey, A.; Joshi, M.; Galbraith, M.D.; Sullivan, K.D.; Espinosa, J.M. DNp63a suppresses TGFB2 expression and RHOA activity to drive Ccll proliferation in squamous cell carcinomas. Cell Rep. 2018, 24, 3224–3236. [Google Scholar] [CrossRef] [PubMed]
- Osborne, L.D.; Li, G.Z.; How, T.; O’Brien, E.T.; Blobe, G.C.; Superfine, R.; Mythreye, K. TGF-β regulates LARG and GEF-H1 during EMT to affect stiffening response to force and cell invasion. Mol. Biol. Cell 2014, 25, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.; Northey, J.J.; Brown, C.M.; Ursini-Siegel, J.; Siegel, P.M. A complex containing LPP and α-actinin mediates TGFβ-induced migration and invasion of ErbB2-expressing breast cancer cells. J. Cell Sci. 2013, 126 Pt 9, 1981–1991. [Google Scholar] [CrossRef]
- Haynes, J.; Srivastava, J.; Madson, N.; Wittmann, T.; Barber, D.L. Dynamic actin remodeling during epithelial-mesenchymal transition depends on increased moesin expression. Mol. Biol. Cell 2011, 22, 4750–4764. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Nakagami, H.; Koibuchi, N.; Miura, K.; Takami, Y.; Koriyama, H.; Hayashi, H.; Sabe, H.; Mochizuki, N.; Morishita, R.; et al. Zyxin mediates actin fiber reorganization in epithelial-mesenchymal transition and contributes to endocardial morphogenesis. Mol. Biol. Cell 2009, 20, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Mise, N.; Savai, R.; Yu, H.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin is a transforming growth factor-β (TGF-β)/Smad3 target gene that regulates lung cancer cell motility via integrin α5β1. J. Biol. Chem. 2012, 287, 31393–31405. [Google Scholar] [CrossRef] [PubMed]
- Beach, J.R.; Hussey, G.S.; Miller, T.E.; Chaudhury, A.; Patel, P.; Monslow, J.; Zheng, Q.; Keri, R.A.; Reizes, O.; Bresnick, A.R.; et al. Myosin II isoform switching mediates invasiveness after TGF-β-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, 17991–17996. [Google Scholar] [CrossRef] [PubMed]
- Cantelli, G.; Orgaz, J.L.; Rodriguez-Hernandez, I.; Karagiannis, P.; Maiques, O.; Matias-Guiu, X.; Nestle, F.O.; Marti, R.M.; Karagiannis, S.N.; Sanz-Moreno, V. TGF-β-Induced Transcription Sustains Amoeboid Melanoma Migration and Dissemination. Curr. Biol. 2015, 25, 2899–2914. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and Autocrine Signals Induce and Maintain Mesenchymal and Stem Cell States in the Breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Chang, X.; Michailidi, C.; Kagohara, L.; Ravi, R.; Paz, K.; Brait, M.; Hoque, M.O.; Ling, S.; Bedi, A.; et al. The TGFβ-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res. 2014, 74, 3995–4005. [Google Scholar] [CrossRef] [PubMed]
- Maitah, M.Y.; Ali, S.; Ahmad, A.; Gadgeel, S.; Sarkar, F.H. Up-regulation of sonic hedgehog contributes to TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS ONE 2011, 6, e16068. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Zanudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P., Jr.; Albert, R. Network modeling of TGFβ signaling in hepatocellular carcinoma epithelial-to-mesenchymal transition reveals joint sonic hedgehog and Wnt pathway activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- Gotzmann, J.; Mikula, M.; Eger, A.; Schulte-Hermann, R.; Foisner, R.; Beug, H.; Mikulits, W. Molecular aspects of epithelial cell plasticity: Implications for local tumor invasion and metastasis. Mutat. Res. 2004, 566, 9–20. [Google Scholar] [CrossRef]
- Lahsnig, C.; Mikula, M.; Petz, M.; Zulehner, G.; Schneller, D.; van Zijl, F.; Huber, H.; Csiszar, A.; Beug, H.; Mikulits, W. ILEI requires oncogenic Ras for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene 2009, 28, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Van Zijl, F.; Mair, M.; Csiszar, A.; Schneller, D.; Zulehner, G.; Huber, H.; Eferl, R.; Beug, H.; Dolznig, H.; Mikulits, W. Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal transition at the tumor edge. Oncogene 2009, 28, 4022–4033. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.-H.; Moustakas, A. Transforming growth factor-β employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Tan, E.-J.; Peinado, H.; Cano, A.; Heldin, C.-H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Shen, H.; Jin, Y.; Lin, T.; Cai, Q.; Pinard, M.A.; Biswas, S.; Tran, Q.; Li, G.; Shenoy, A.K.; et al. The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition. J. Biol. Chem. 2013, 288, 27680–27691. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Shenoy, A.K.; Li, X.; Jin, Y.; Jin, L.; Cai, Q.; Tang, M.; Liu, Y.; Chen, H.; Reisman, D.; et al. MOF acetylates the histone demethylase LSD1 to suppress epithelial-to-mesenchymal transition. Cell Rep. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jung, S.M.; Yang, K.M.; Bae, E.; Ahn, S.G.; Park, J.S.; Seo, D.; Kim, M.; Ha, J.; Lee, J.; et al. A20 promotes metastasis of aggressive basal-like breast cancers through multi-monoubiquitylation of Snail1. Nat. Cell Biol. 2017, 19, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Estaràs, C.; Akizu, N.; Garcia, A.; Beltran, S.; de la Cruz, X.; Martinez-Balbas, M.A. Genome-wide analysis reveals that Smad3 and JMJD3 HDM co-activate the neural developmental program. Development 2012, 139, 2681–2691. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Qi, G.; Tang, F.; Yuan, S.; Wang, Z.; Liang, X.; Li, B.; Yu, S.; Liu, J.; Huang, Q.; et al. Aberrant JMJD3 expression upregulates Slug to promote migration, invasion, and stem cell-like behaviors in hepatocellular carcinoma. Cancer Res. 2016, 76, 6520–6532. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Huang, H.C.; Kao, S.H.; Hsu, Y.C.; Wang, Y.T.; Li, K.C.; Chen, Y.J.; Yu, S.L.; Wang, S.P.; Hsiao, T.H.; et al. Slug is temporally regulated by cyclin E in cell cycle and controls genome stability. Oncogene 2015, 34, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Lim, S.O.; Hsu, J.L.; Huo, L.; Wu, Y.; Li, L.Y.; Lai, C.C.; Chang, S.S.; Hsu, Y.H.; et al. AKT1 inhibits epithelial-to-mesenchymal transition in breast cancer through phosphorylation-dependent Twist1 degradation. Cancer Res. 2016, 76, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Liu, W.; Li, J. USP17 is upregulated in osteosarcoma and promotes cell proliferation, metastasis, and epithelial-mesenchymal transition through stabilizing SMAD4. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, Y.; Shi, Q.; Yu, Q.; Liu, C.; Feng, J.; Deng, J.; Evers, B.M.; Zhou, B.P.; Wu, Y. Stabilization of the transcription factors slug and twist by the deubiquitinase dub3 is a key requirement for tumor metastasis. Oncotarget 2017, 8, 75127–75140. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Li, C.F.; Ruan, D.; Powers, S.; Thompson, P.A.; Frohman, M.A.; Chan, C.H. The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through Twist activation. Mol. Cell 2016, 63, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, Z.; Chen, Q.; Li, Y.; Jiang, L.; Zhang, Z.; Zhou, F. Nkx2.8 inhibits epithelial-mesenchymal transition in bladder urothelial carcinoma via transcriptional repression of Twist1. Cancer Res. 2018, 78, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Tian, J.; Zhu, S.; Sun, L.; Yu, J.; Tian, H.; Dong, Q.; Luo, Q.; Jiang, N.; Niu, Y.; et al. Sox5 contributes to prostate cancer metastasis and is a master regulator of TGF-β-induced epithelial mesenchymal transition through controlling Twist1 expression. Br. J. Cancer 2018, 118, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guio-Carrion, A.; Hasenfuss, S.C.; Eger, A.; Muller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2015, 22, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, L.; Chen, F.; Chen, Y.; Liu, H.; Li, J.; Bai, J.; Zheng, J. PTBP3-mediated regulation of ZEB1 mRNA stability promotes epithelial-mesenchymal transition in breast cancer. Cancer Res. 2018, 78, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Wong, C.S.; Liu, M.C.; House, C.M.; Sceneay, J.; Bowtell, D.D.; Thompson, E.W.; Moller, A. The ubiquitin ligase Siah is a novel regulator of Zeb1 in breast cancer. Oncotarget 2015, 6, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Preca, B.T.; Bajdak, K.; Mock, K.; Lehmann, W.; Sundararajan, V.; Bronsert, P.; Matzge-Ogi, A.; Orian-Rousseau, V.; Brabletz, S.; Brabletz, T.; et al. A novel ZEB1/HAS2 positive feedback loop promotes EMT in breast cancer. Oncotarget 2017, 8, 11530–11543. [Google Scholar] [CrossRef] [PubMed]
- Bogachek, M.V.; De Andrade, J.P.; Weigel, R.J. Regulation of epithelial-mesenchymal transition through SUMOylation of transcription factors. Cancer Res. 2015, 75, 11–15. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Northey, J.J.; Pelletier, A.; Kos, Z.; Meunier, L.; Haibe-Kains, B.; Mes-Masson, A.M.; Cote, J.F.; Siegel, P.M.; Lamarche-Vane, N. The Cdc42/Rac1 regulator CdGAP is a novel E-cadherin transcriptional co-repressor with Zeb2 in breast cancer. Oncogene 2017, 36, 3490–3503. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Valcourt, U.; Bergström, R.; Heldin, C.-H.; Moustakas, A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor β and bone morphogenetic protein. Mol. Cell. Biol. 2004, 24, 4241–4254. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Z.; Zhang, X.; Yang, S.; Lin, X.; Yang, X.; Lin, X.; Shi, J.; Wang, S.; Zhao, W.; et al. Klf4 reduces stemness phenotype, triggers mesenchymal-epithelial transition (MET)-like molecular changes, and prevents tumor progression in nasopharygeal carcinoma. Oncotarget 2017, 8, 93924–93941. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Subramaniam, M.; Kari, V.; Pitel, K.S.; Baumgart, S.J.; Naylor, R.M.; Nagarajan, S.; Wegwitz, F.; Ellenrieder, V.; Hawse, J.R.; et al. Kruppel-like transcription factor KLF10 suppresses TGFβ-induced epithelial-to-mesenchymal transition via a negative feedback mechanism. Cancer Res. 2017, 77, 2387–2400. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; You, R.I.; Cheng, C.C.; Lee, M.C.; Lin, T.Y.; Hu, C.T. Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci. Rep. 2017, 7, 17753. [Google Scholar] [CrossRef] [PubMed]
- Díaz-López, A.; Diaz-Martin, J.; Moreno-Bueno, G.; Cuevas, E.P.; Santos, V.; Olmeda, D.; Portillo, F.; Palacios, J.; Cano, A. Zeb1 and Snail1 engage miR-200f transcriptional and epigenetic regulation during EMT. Int. J. Cancer 2015, 136, E62–E73. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, M.; Zhao, K.; He, M.; Ge, W.; Sun, Y.; Wang, Y.; Sun, H.; Hu, Y. Upregulation of MiR-205 under hypoxia promotes epithelial-mesenchymal transition by targeting ASPP2. Cell Death Dis. 2016, 7, e2517. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.Y.; Yoo, J.O.; An, H.J.; Bae, I.H.; Park, M.J.; Kim, J.; Han, Y.H. miR-5003-3p promotes epithelial-mesenchymal transition in breast cancer cells through Snail stabilization and direct targeting of E-cadherin. J. Mol. Cell Biol. 2016, 8, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Incarnato, D.; Savino, A.; Gai, M.; Marino, F.; Pensa, S.; Barbieri, I.; Stadler, M.B.; Provero, P.; Oliviero, S.; et al. MicroRNAs-143 and -145 induce epithelial to mesenchymal transition and modulate the expression of junction proteins. Cell Death Differ. 2017, 24, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, J.; Wang, X.; Meng, S.; Shen, J.; Wang, S.; Xu, X.; Xie, B.; Liu, B.; Xie, L. MiR-22 suppresses epithelial-mesenchymal transition in bladder cancer by inhibiting Snail and MAPK1/Slug/vimentin feedback loop. Cell Death Dis. 2018, 9, 209. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Bhagirath, D.; Sekhon, K.; Yang, T.; Fukuhara, S.; Majid, S.; Shahryari, V.; Tabatabai, Z.; Greene, K.L.; Hashimoto, Y.; et al. A novel microRNA regulator of prostate cancer epithelial-mesenchymal transition. Cell Death Differ. 2017, 24, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Dang, B.L.; Huang, J.Z.; Chen, M.; Wu, D.; Xu, M.L.; Li, R.; Yan, G.R. MiR-373 drives the epithelial-to-mesenchymal transition and metastasis via the miR-373-TXNIP-HIF1a-TWIST signaling axis in breast cancer. Oncotarget 2015, 6, 32701–32712. [Google Scholar] [PubMed]
- Xu, M.; Zhu, C.; Zhao, X.; Chen, C.; Zhang, H.; Yuan, H.; Deng, R.; Dou, J.; Wang, Y.; Huang, J.; et al. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget 2015, 6, 979–994. [Google Scholar] [CrossRef] [PubMed]
- Drasin, D.J.; Guarnieri, A.L.; Neelakantan, D.; Kim, J.; Cabrera, J.H.; Wang, C.A.; Zaberezhnyy, V.; Gasparini, P.; Cascione, L.; Huebner, K.; et al. TWIST1-induced miR-424 reversibly drives mesenchymal programming while inhibiting tumor initiation. Cancer Res. 2015, 75, 1908–1921. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J.; Zhang, G.; Li, Z.P.; Permuth-Wey, J.; Challa, S.; Li, Y.; Kong, W.; Dan, S.; Bui, M.M.; Coppola, D.; et al. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) β: LncRNA-hit-mediated TGFβ-induced epithelial to mesenchymal transition in mammary epithelia. J. Biol. Chem. 2015, 290, 6857–6867. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-β-Induced Upregulation of malat1 Promotes Bladder Cancer Metastasis by Associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Hussey, G.S.; Ray, P.S.; Jin, G.; Fox, P.L.; Howe, P.H. TGF-β-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat. Cell Biol. 2010, 12, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hussey, G.S.; Chaudhury, A.; Dawson, A.E.; Lindner, D.J.; Knudsen, C.R.; Wilce, M.C.; Merrick, W.C.; Howe, P.H. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol. Cell 2011, 41, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Howley, B.V.; Hussey, G.S.; Link, L.A.; Howe, P.H. Translational regulation of inhibin betaA by TGFβ via the RNA-binding protein hnRNP E1 enhances the invasiveness of epithelial-to-mesenchymal transitioned cells. Oncogene 2016, 35, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Hussey, G.S.; Link, L.A.; Brown, A.S.; Howley, B.V.; Chaudhury, A.; Howe, P.H. Establishment of a TGFβ-induced post-transcriptional EMT gene signature. PLoS ONE 2012, 7, e52624. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Tan, P.; Rodriguez, M.; He, L.; Tan, K.; Zeng, L.; Siwko, S.; Liu, M. Leucine-rich repeat-containing G protein-coupled receptor 4 (Lgr4) is necessary for prostate cancer metastasis via epithelial-mesenchymal transition. J. Biol. Chem. 2017, 292, 15525–15537. [Google Scholar] [CrossRef] [PubMed]
- Okita, Y.; Kimura, M.; Xie, R.; Chen, C.; Shen, L.T.; Kojima, Y.; Suzuki, H.; Muratani, M.; Saitoh, M.; Semba, K.; et al. The transcription factor MAFK induces EMT and malignant progression of triple-negative breast cancer cells through its target GPNMB. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Cuevas, E.P.; Santos, V.; Lopez-Menendez, C.; Moreno-Bueno, G.; Huang, Y.; Csiszar, K.; Portillo, F.; Peinado, H.; Lyden, D.; et al. Lysyl oxidase-like 2 (LOXL2) and E47 EMT factor: Novel partners in E-cadherin repression and early metastasis colonization. Oncogene 2015, 34, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Banerjee, P.; Liu, X.; Yu, J.; Gibbons, D.L.; Wu, P.; Scott, K.L.; Diao, L.; Zheng, X.; Wang, J.; et al. The epithelial-to-mesenchymal transition activator ZEB1 initiates a prometastatic competing endogenous RNA network. J. Clin. Investig. 2018, 128, 3198. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Banerjee, P.; Liu, X.; Yu, J.; Gibbons, D.L.; Wu, P.; Scott, K.L.; Diao, L.; Zheng, X.; Wang, J.; et al. The epithelial-to-mesenchymal transition activator ZEB1 initiates a prometastatic competing endogenous RNA network. J. Clin. Investig. 2018, 128, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Maturi, V.; Enroth, S.; Heldin, C.-H.; Moustakas, A. Genome-wide binding of transcription factor ZEB1 in triple-negative breast cancer cells. J. Cell. Physiol. 2018, 233, 7113–7127. [Google Scholar] [CrossRef] [PubMed]
- Maturi, V.; Moren, A.; Enroth, S.; Heldin, C.-H.; Moustakas, A. Genomewide binding of transcription factor Snail1 in triple-negative breast cancer cells. Mol. Oncol. 2018, 12, 1153–1174. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef] [PubMed]
- Zajac, O.; Raingeaud, J.; Libanje, F.; Lefebvre, C.; Sabino, D.; Martins, I.; Roy, P.; Benatar, C.; Canet-Jourdan, C.; Azorin, P.; et al. Tumour spheres with inverted polarity drive the formation of peritoneal metastases in patients with hypermethylated colorectal carcinomas. Nat. Cell Biol. 2018, 20, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lee, D.K.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massagué, J.; Benezra, R. TGF-β-Id1 Signaling Opposes Twist1 and Promotes Metastatic Colonization via a Mesenchymal-to-Epithelial Transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Fournier, P.G.; Juarez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-β signaling regulator PMEPA1 suppresses prostate cancer metastases to bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF β signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Alexander, S.; Schacht, V.; Coussens, L.M.; von Andrian, U.H.; van Rheenen, J.; Deryugina, E.; Friedl, P. Collagen-based cell migration models in vitro and in vivo. Semin. Cell Dev. Biol. 2009, 20, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Sahai, E.; Weiss, S.; Yamada, K.M. New dimensions in cell migration. Nat. Rev. Mol. Cell Biol. 2012, 13, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Ghajar, C.M.; Bissell, M.J. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Deliv. Rev. 2014, 69–70, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Furuta, S.; Ren, G.; Mao, J.H.; Bissell, M.J. Laminin signals initiate the reciprocal loop that informs breast-specific gene expression and homeostasis by activating NO, p53 and microRNAs. eLife 2018, 7, e26148. [Google Scholar] [CrossRef] [PubMed]
- Weigelin, B.; Bakker, G.J.; Friedl, P. Third harmonic generation microscopy of cells and tissue organization. J. Cell Sci. 2016, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Entenberg, D.; Pastoriza, J.M.; Oktay, M.H.; Voiculescu, S.; Wang, Y.; Sosa, M.S.; Aguirre-Ghiso, J.; Condeelis, J. Time-lapsed, large-volume, high-resolution intravital imaging for tissue-wide analysis of single cell dynamics. Methods 2017, 128, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Saitou, T.; Kawakami, R. In vivo optical imaging of cancer cell function and tumor microenvironment. Cancer Sci. 2018, 109, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Kubota, S.I.; Takahashi, K.; Nishida, J.; Morishita, Y.; Ehata, S.; Tainaka, K.; Miyazono, K.; Ueda, H.R. Whole-body profiling of cancer metastasis with single-cell resolution. Cell Rep. 2017, 20, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Ell, B.J.; Buffa, F.M.; Ibrahim, T.; Blanco, M.A.; Celia-Terrassa, T.; Mercatali, L.; Khan, Z.; Goodarzi, H.; Hua, Y.; et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 2011, 17, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Banno, A.; Garcia, D.A.; van Baarsel, E.D.; Metz, P.J.; Fisch, K.; Widjaja, C.E.; Kim, S.H.; Lopez, J.; Chang, A.N.; Geurink, P.P.; et al. Downregulation of 26S proteasome catalytic activity promotes epithelial-mesenchymal transition. Oncotarget 2016, 7, 21527–21541. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, M.J.; Park, S.A.; Kim, J.S.; Min, K.N.; Kim, D.K.; Lim, W.; Nam, J.S.; Sheen, Y.Y. Combinatorial TGF-β attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget 2015, 6, 37526–37543. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Zhou, B.; Avdulov, S.; Benyumov, A.; Peterson, M.; Liu, Y.; Okon, A.; Hergert, P.; Braziunas, J.; Wagner, C.R.; et al. Transforming Growth Factor-β1 Induced Epithelial Mesenchymal Transition is blocked by a chemical antagonist of translation factor eIF4E. Sci. Rep. 2015, 5, 18233. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fu, Y.; Zheng, L.; Lin, G.; Ma, J.; Lou, J.; Zhu, H.; He, Q.; Yang, B. Nutlin-3 inhibits epithelial-mesenchymal transition by interfering with canonical transforming growth factor-β1-Smad-Snail/Slug axis. Cancer Lett. 2014, 342, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Termén, S.; Tan, E.-J.; Heldin, C.-H.; Moustakas, A. p53 regulates epithelial-mesenchymal transition induced by transforming growth factor β. J. Cell. Physiol. 2013, 228, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wu, G.; Chang, C.; Zhu, F.; Xiao, Y.; Li, Q.; Zhang, T.; Zhang, L. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-kB/Snail pathway. Oncotarget 2015, 6, 40907–40919. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotoh, K.; Asaoka, T.; Wada, H.; et al. A Histone Deacetylase Inhibitor Suppresses Epithelial-Mesenchymal Transition and Attenuates Chemoresistance in Biliary Tract Cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, M.; Imamura, T.; Chiba, T.; Ebisawa, T.; Kawabata, M.; Tanaka, K.; Miyazono, K. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol. Biol. Cell 2001, 12, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Ke, X.; Tan, S.; Liu, T.; Wang, S.; Ma, J.; Lu, H. The natural compound codonolactone attenuates TGF-β1-mediated epithelial-to-mesenchymal transition and motility of breast cancer cells. Oncol. Rep. 2016, 35, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, X.; Gao, Y.; Zhang, C.; Bao, J.; Guan, H.; Yu, H.; Lu, R.; Xu, Q.; Sun, Y. Curcumin inhibits metastasis in human papillary thyroid carcinoma BCPAP cells via down-regulation of the TGF-β/Smad2/3 signaling pathway. Exp. Cell Res. 2016, 341, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Han, J.; Hou, B.; Deng, C.; Wu, H.; Shen, L. Sulforaphane inhibits TGF-β-induced epithelial-mesenchymal transition of hepatocellular carcinoma cells via the reactive oxygen species-dependent pathway. Oncol. Rep. 2016, 35, 2977–2983. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. https://doi.org/10.3390/ijms19113672
Tsubakihara Y, Moustakas A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. International Journal of Molecular Sciences. 2018; 19(11):3672. https://doi.org/10.3390/ijms19113672
Chicago/Turabian StyleTsubakihara, Yutaro, and Aristidis Moustakas. 2018. "Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β" International Journal of Molecular Sciences 19, no. 11: 3672. https://doi.org/10.3390/ijms19113672
APA StyleTsubakihara, Y., & Moustakas, A. (2018). Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. International Journal of Molecular Sciences, 19(11), 3672. https://doi.org/10.3390/ijms19113672