Causal Transcription Regulatory Network Inference Using Enhancer Activity as a Causal Anchor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
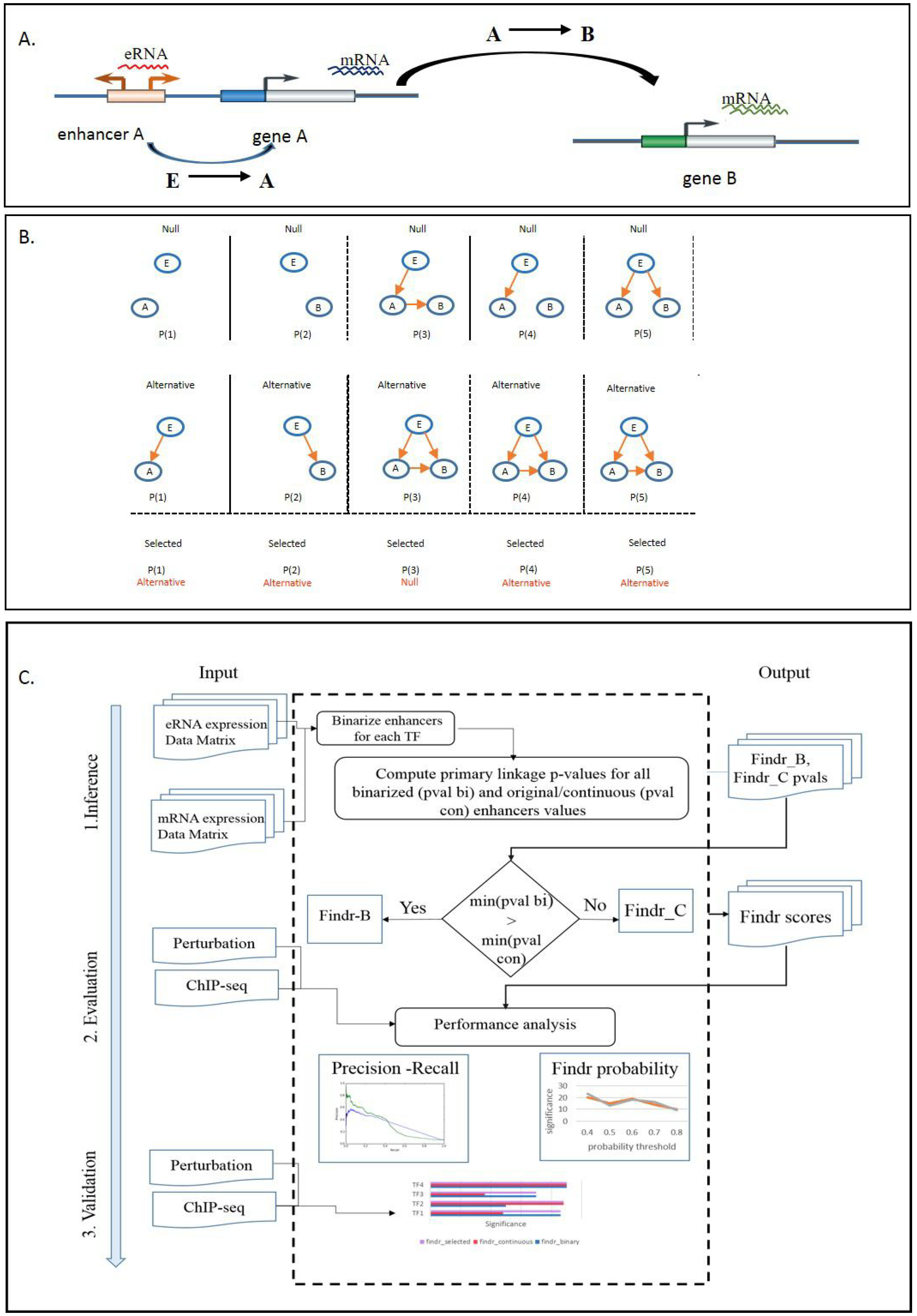
2.1. Development of a Causal Transcription Network Inference Framework
2.2. Causal Inference from Enhancer and Transcript/Gene Expression CAGE Data
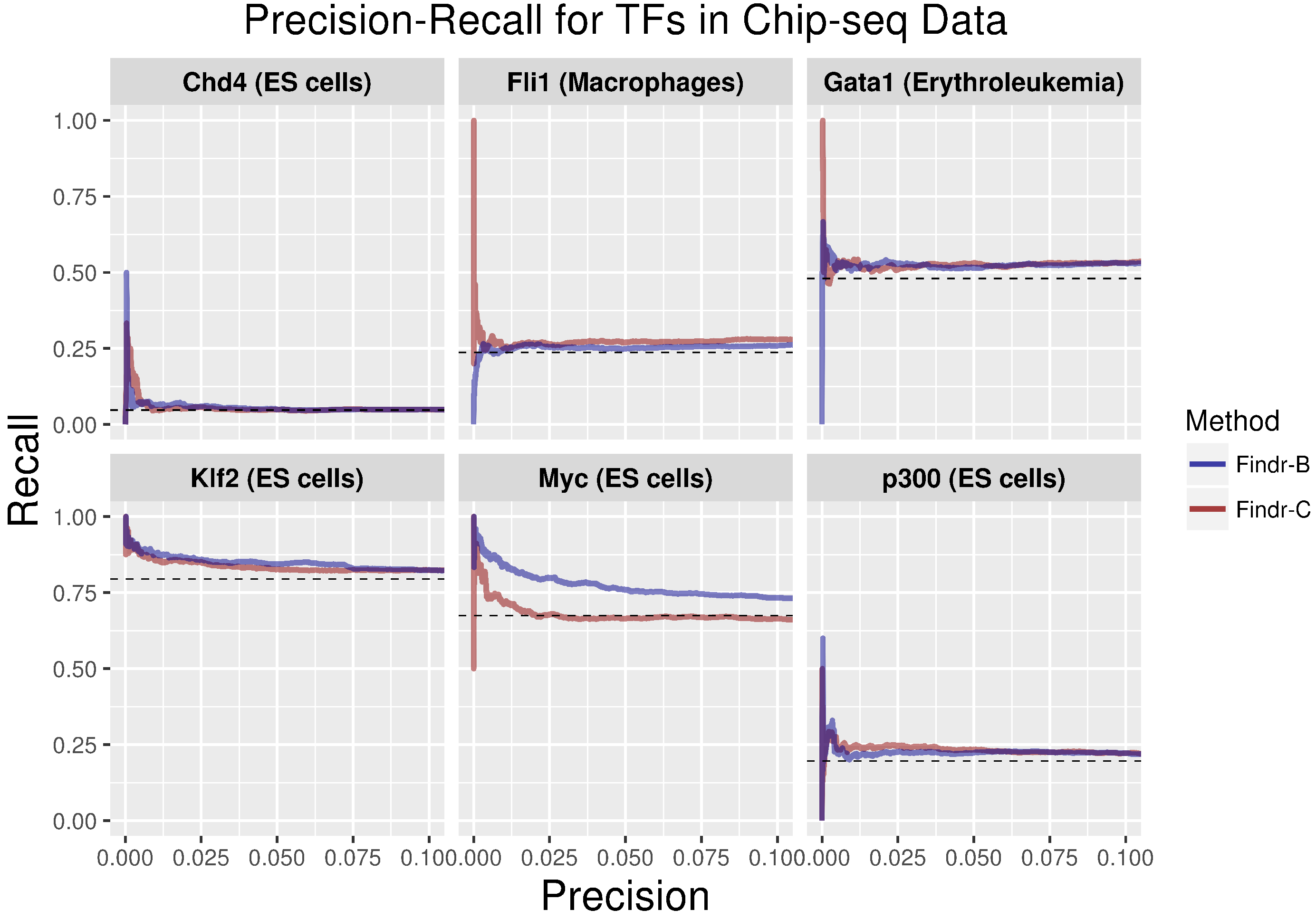
2.3. Development and Validation of an Adaptive Model-Selection Approach for Causal Inference Using Discretized or Continuous Data
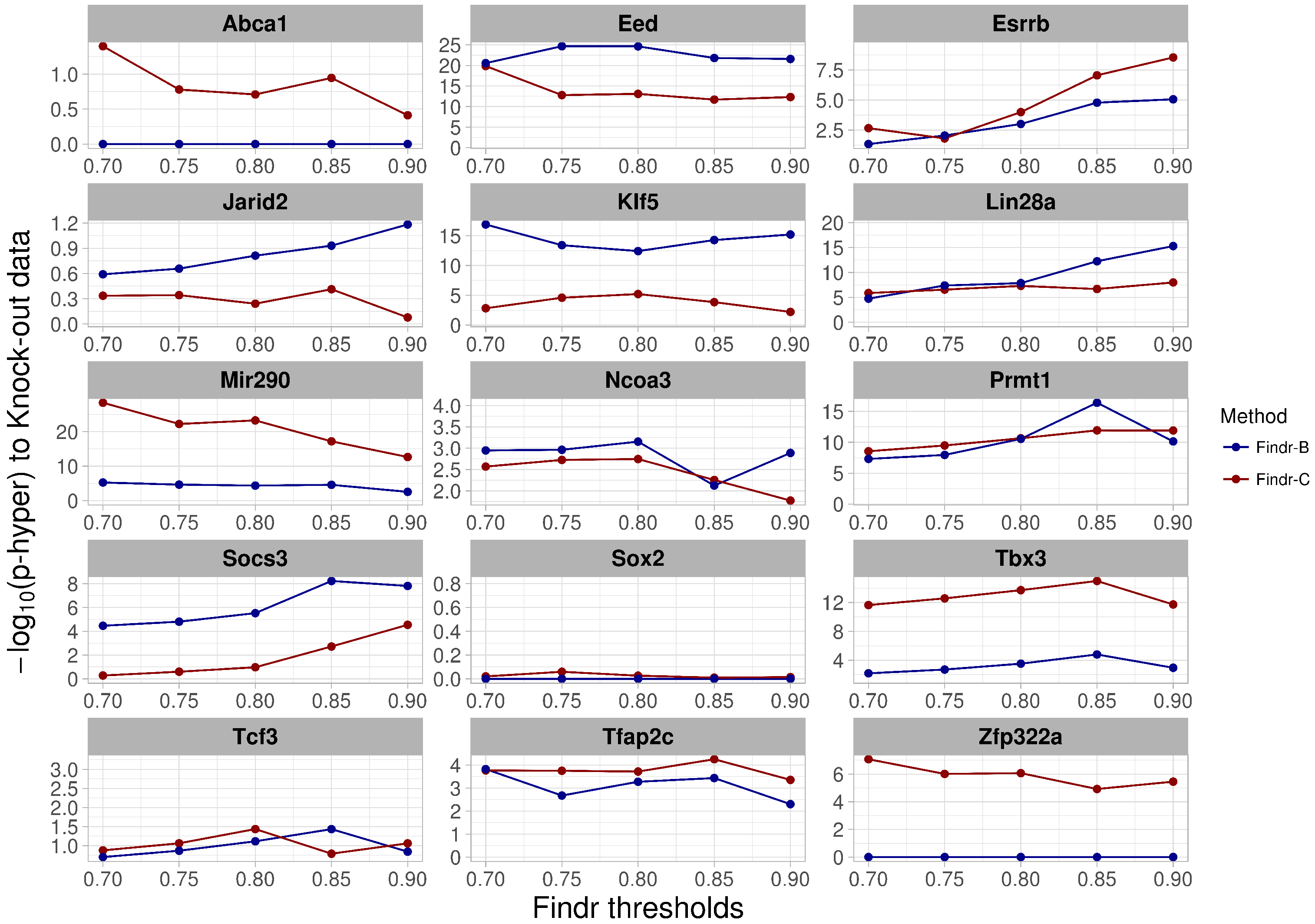
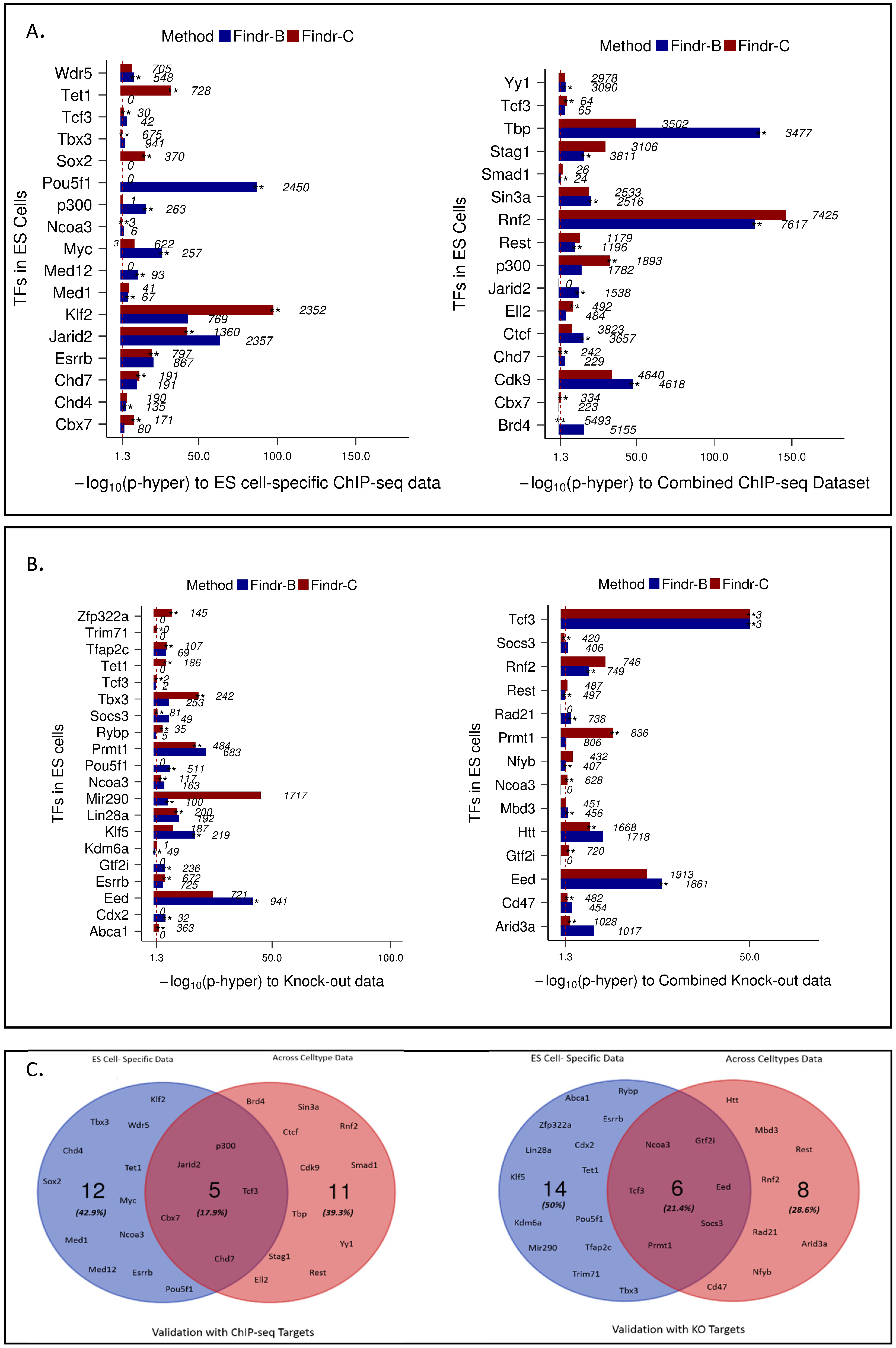
2.4. Perturbations within and across Cell Types Provide Causal Targets for a Distinct Set of Transcription Factors
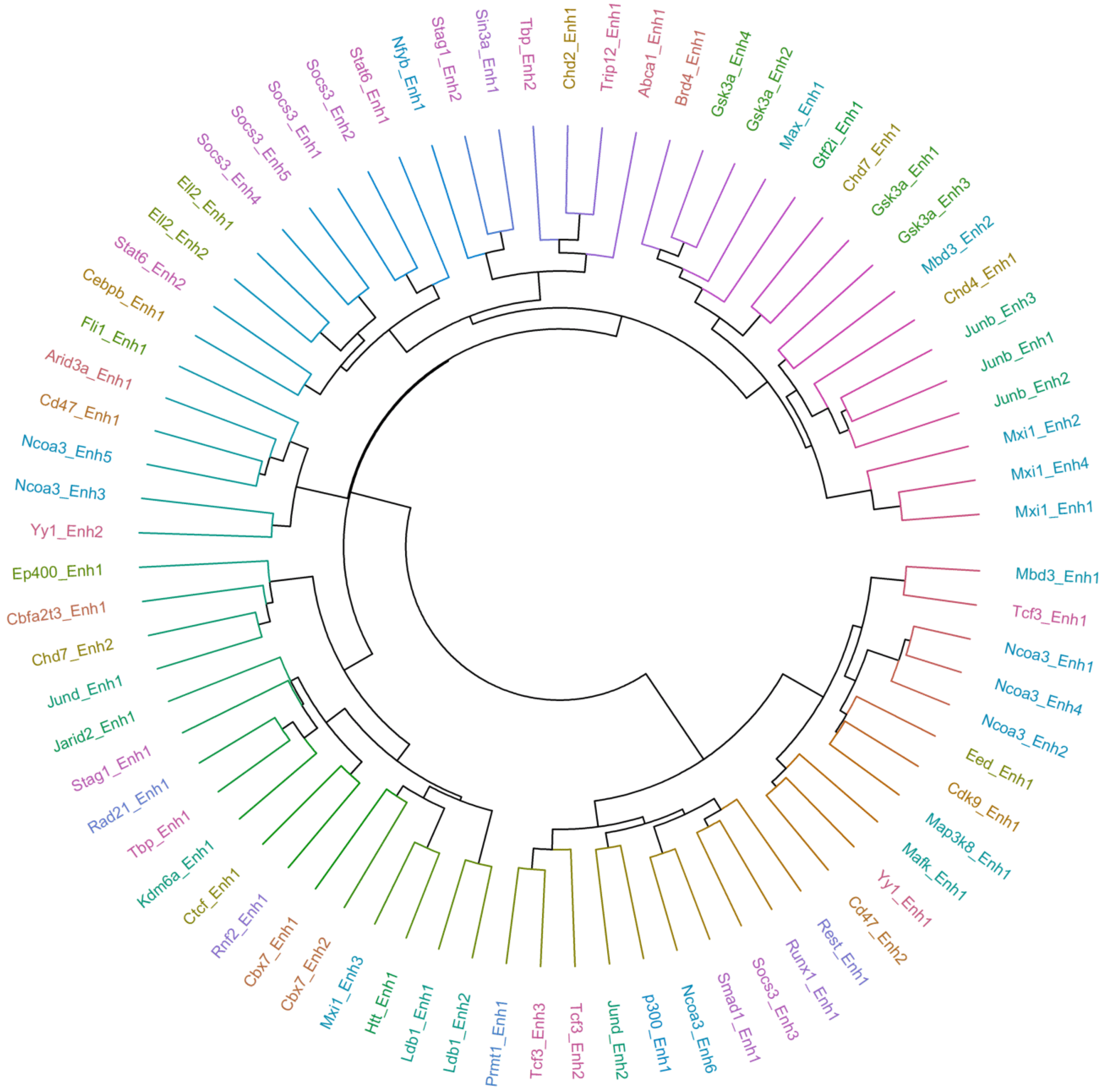
2.5. Multiple Enhancers of the Same Factor Have a Highly Correlated Expression
2.6. Causal Inference Using CAGE Expression Data across Human Cell Types
3. Discussion
4. Materials and Methods
4.1. Datasets
- We used Cap Analysis of Gene Expression (CAGE) data (TPM expression values) from the FANTOM5 Consortium for enhancer and transcription start sites (TSS) in mouse embryonic stem (ES) cells (36 experiments), macrophages (224 experiments) and erythroblastic leukaemia (52 experiments) [6,17]. We also selected 1036 samples from all cell types and tissues in mouse.
- We use ChIP-seq data from the Codex Consortium [20]. Data available for 78 TFs in mouse ES cells, 12 TFs in macrophages and 17 TFs in erythroleukaemia cells were used.
- For validation using knock-out data, we have collected differentially-expressed gene lists after perturbation of factors from published studies in mouse and gene lists after overexpression of factors in mouse ES cells from [27].
- We obtained CAGE data (TPM expression values) from the FANTOM5 Consortium for enhancer and TSSs for human cell types and tissues [6,17]. The enhancer regions identified using bi-directional expression were obtained from [1]. The FANTOM5 Consortium has provided data for 1826 samples from which we selected 360 samples from all cell types and tissues, one sample for each cell and tissue type with the highest sequencing depth (removing technical and biological replicates).
4.2. Data Processing
- CAGE data were processed to clear unannotated and non-expressed genes, and expression levels were log-transformed. Genes with a TPM value of 1 or more in at least one sample were considered expressed. Only enhancers expressed in more than one third of experiments were retained.
- For each TF, we selected the promoter with the highest median expression level as the promoter for that TF.
- For each TF, all enhancers within 50 kb of the TF promoter region were detected using the GenomicRanges package in Bioconductor [28] and considered as candidate causal anchor enhancers for that TF. The Findr framework (described below in detail) includes a “primary linkage” step such that targets are only predicted for TFs with significantly correlated eRNAs.
- Enhancer data were binarized by setting all experiments with zero read count to zero and all others to one.
- For the ChIP-seq data, genes with a TF binding site within 1 kb of their TSS were defined as targets for that TF.
- For the knock-out and over-expression data, genes with differential expression q-value were defined as targets for the TF.
4.3. Likelihood Ratio Tests with Continuous Causal Anchor Data
- Primary linkage test: The primary linkage test verifies that the enhancer E regulates the regulator gene A. Its null and alternative hypotheses are:The log likelihood ratio (LLR) and its null distribution are identical to the correlation test in [18]. Therefore, the LLR is:where:Its null distribution is:The probability density function (PDF) for is defined as: for ,and for , , where is the Beta function.
- Secondary linkage test: The secondary linkage test verifies that the enhancer E regulates the target gene B. The LLR and its null distribution are identical to those of the primary linkage test, except by replacing A with B.
- Conditional independence test: The conditional independence test verifies that E and B become independent after conditioning on A, with its null and alternative hypotheses as:Correlated genes are modelled as having a multi-variant normal distribution, whose mean linearly depends on their regulator gene. Therefore,Following the same definition of the null data, their null distribution is:
- Relevance test: The relevance test verifies that B is regulated by either E or A. Its hypotheses are:Similarly,
- Controlled test: The controlled test verifies that E regulates B through A, partially or fully, with the hypotheses as:Its LLR iswith the null distribution:
4.4. Findr-B and Findr-C
4.5. Adaptive Method Findr-A
- Compute the primary linkage test p-value for all candidate enhancers of A, both continuous and binarized.
- Find the enhancer E with the lowest p-value overall.
- If the lowest p-value occurred for a binarized enhancer, use Findr-B for TF A with E as its causal anchor, else use Findr-C.
4.6. Validation Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–E2199. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.; Kawaji, H.; Rehli, M.; Baillie, J.; de Hoon, M.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Pearl, J. Causality; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Schadt, E.E.; Lamb, J.; Yang, X.; Zhu, J.; Edwards, S.; GuhaThakurta, D.; Sieberts, S.K.; Monks, S.; Reitman, M.; Zhang, C.; et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 2005, 37, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Emmert-Streib, F.; Storey, J.D. Harnessing naturally randomized transcription to infer regulatory relationships among genes. Genome Biol. 2007, 8, R219. [Google Scholar] [CrossRef] [PubMed]
- Rockman, M.V. Reverse engineering the genotype–phenotype map with natural genetic variation. Nature 2008, 456, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tesson, B.M.; Churchill, G.A.; Jansen, R.C. Critical reasoning on causal inference in genome-wide linkage and association studies. Trends Genet. 2010, 26, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E. Molecular networks as sensors and drivers of common human diseases. Nature 2009, 461, 218. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Andrau, J.C. Noncoding transcription at enhancers: general principles and functional models. Annu. Rev. Genet. 2012, 46, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.T.; Cho, H.; Lesch, H.P.; Gosselin, D.; Heinz, S.; Tanaka-Oishi, Y.; Benner, C.; Kaikkonen, M.U.; Kim, A.S.; Kosaka, M.; et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013, 498, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Danko, C.G.; Hyland, S.L.; Core, L.J.; Martins, A.L.; Waters, C.T.; Lee, H.W.; Cheung, V.G.; Kraus, W.L.; Lis, J.T.; Siepel, A. Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods 2015, 12, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, J.G.; Allen, M.A.; Hendrix, J.R.; Read, T.; Rubin, J.D.; Dowell, R.D. Enhancer RNA profiling predicts transcription factor activity. Genome Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Daub, C.O.; Vitting-Seerup, K.; Andersson, R.; Lilje, B.; Drablos, F.; Lennartsson, A.; Ronnerblad, M.; Hrydziuszko, O.; Vitezic, M.; et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015, 347, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Michoel, T. Efficient and accurate causal inference with hidden confounders from genome-transcriptome variation data. PLoS Comput. Biol. 2017, 13, e1005703. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Castillo, M.; Ruau, D.; Wilkinson, A.C.; Ng, F.S.; Hannah, R.; Diamanti, E.; Lombard, P.; Wilson, N.K.; Gottgens, B. CODEX: A next-generation sequencing experiment database for the haematopoietic and embryonic stem cell communities. Nucleic Acids Res. 2015, 43, D1117–D1123. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Pavlovic, B.; Pritchard, J.K.; Gilad, Y. The functional consequences of variation in transcription factor binding. PLoS Genet. 2014, 10, e1004226. [Google Scholar] [CrossRef] [PubMed]
- Healy, S.; Khan, P.; Davie, J.R. Immediate early response genes and cell transformation. Pharmacol. Ther. 2013, 137, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Mantsoki, A.; Devailly, G.; Joshi, A. CpG island erosion, polycomb occupancy and sequence motif enrichment at bivalent promoters in mammalian embryonic stem cells. Sci. Rep. 2015, 5, 16791. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.J.; Martello, G.; Yordanov, B.; Emmott, S.; Smith, A.G. Defining an essential transcription factor program for naïve pluripotency. Science 2014, 344, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Shim, H.; Shin, D.; Shim, J.E.; Ko, Y.; Shin, J.; Kim, H.; Cho, A.; Kim, E.; Lee, T.; et al. TRRUST: A reference database of human transcriptional regulatory interactions. Sci. Rep. 2015, 5, 11432. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Baroukh, C.; Dannenfelser, R.; Chen, E.Y.; Tan, C.M.; Kou, Y.; Kim, Y.E.; Lemischka, I.R.; Ma’ayan, A. ESCAPE: database for integrating high-content published data collected from human and mouse embryonic stem cells. Database (Oxford) 2013, 2013, bat045. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vipin, D.; Wang, L.; Devailly, G.; Michoel, T.; Joshi, A. Causal Transcription Regulatory Network Inference Using Enhancer Activity as a Causal Anchor. Int. J. Mol. Sci. 2018, 19, 3609. https://doi.org/10.3390/ijms19113609
Vipin D, Wang L, Devailly G, Michoel T, Joshi A. Causal Transcription Regulatory Network Inference Using Enhancer Activity as a Causal Anchor. International Journal of Molecular Sciences. 2018; 19(11):3609. https://doi.org/10.3390/ijms19113609
Chicago/Turabian StyleVipin, Deepti, Lingfei Wang, Guillaume Devailly, Tom Michoel, and Anagha Joshi. 2018. "Causal Transcription Regulatory Network Inference Using Enhancer Activity as a Causal Anchor" International Journal of Molecular Sciences 19, no. 11: 3609. https://doi.org/10.3390/ijms19113609
APA StyleVipin, D., Wang, L., Devailly, G., Michoel, T., & Joshi, A. (2018). Causal Transcription Regulatory Network Inference Using Enhancer Activity as a Causal Anchor. International Journal of Molecular Sciences, 19(11), 3609. https://doi.org/10.3390/ijms19113609