Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
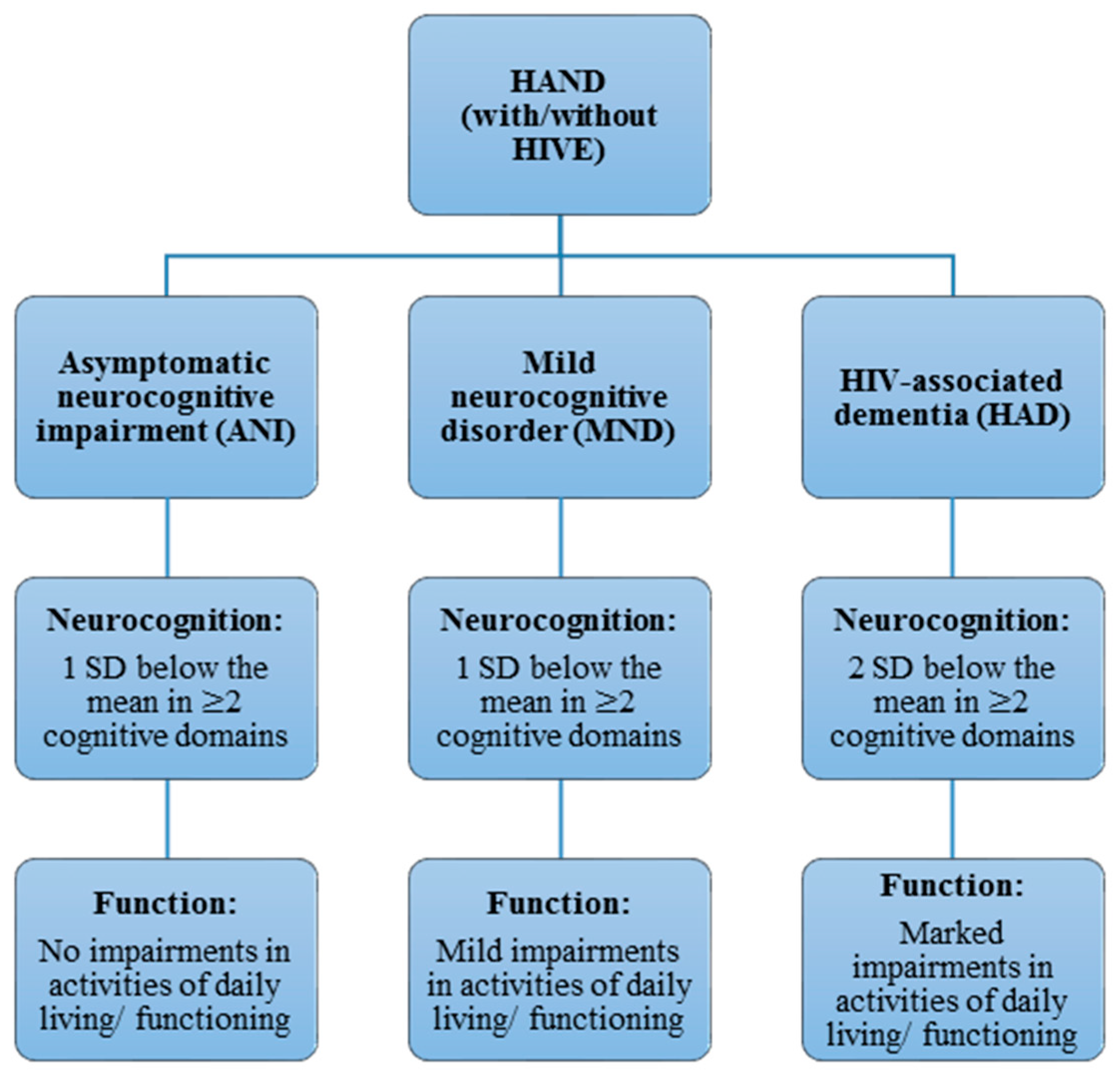
2. Classification and Diagnosis of Human Immunodeficiency Virus-Associated Neurocognitive Disorder (HAND) Phenotypes
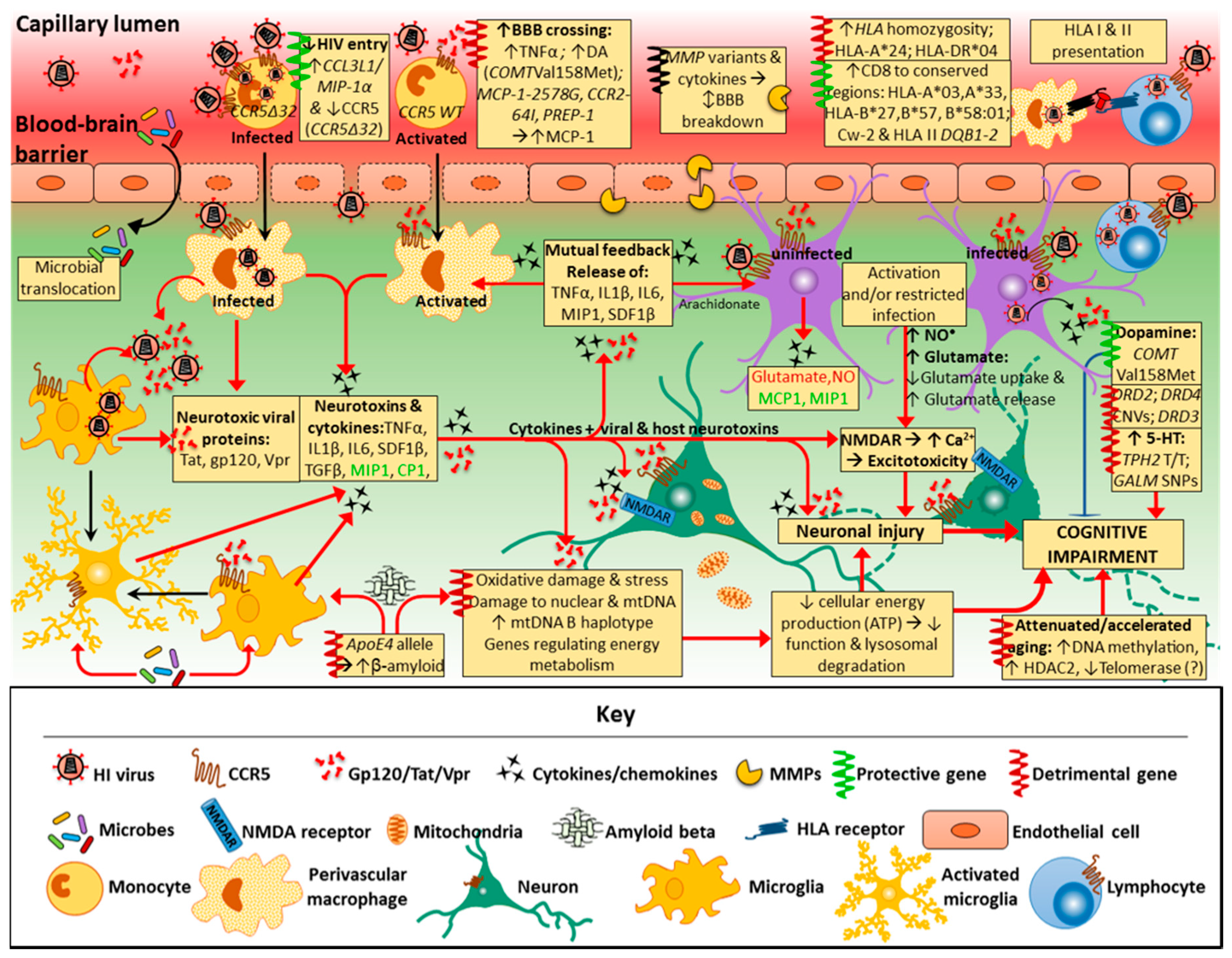
3. Pathogenesis of HAND
4. Risk Factors for HAND
5. Genes Involved in Neurotransmitter Systems
5.1. Genes Related to Serotonin Neurotransmission
5.2. Genes Related to Dopamine Neurotransmission
6. Genes Affecting Integrity of Mitochondrial and Nuclear DNA in HAND
7. Telomere Length and Cell Age
8. Genes Related to Cytokines and Associated Receptors of the Immune System
8.1. Gene Variants of CCR2 and Associated Ligand CCL2 (MCP-1)
8.2. Gene Variants of CCR5 and Associated Ligand CCL3 (MIP-1α)
8.3. Gene Variants of TNF-α
8.4. Gene Variants of the Chemokine Ligand CXCL12 (SDF-1)
8.5. Genetic Variation of Interleukins in HAND
9. Mannose Binding Lectin (MBL-2)
10. Human Leukocyte Antigen Class I and II (HLA) Genes
11. Matrix Metalloproteinase (MMP) Genes
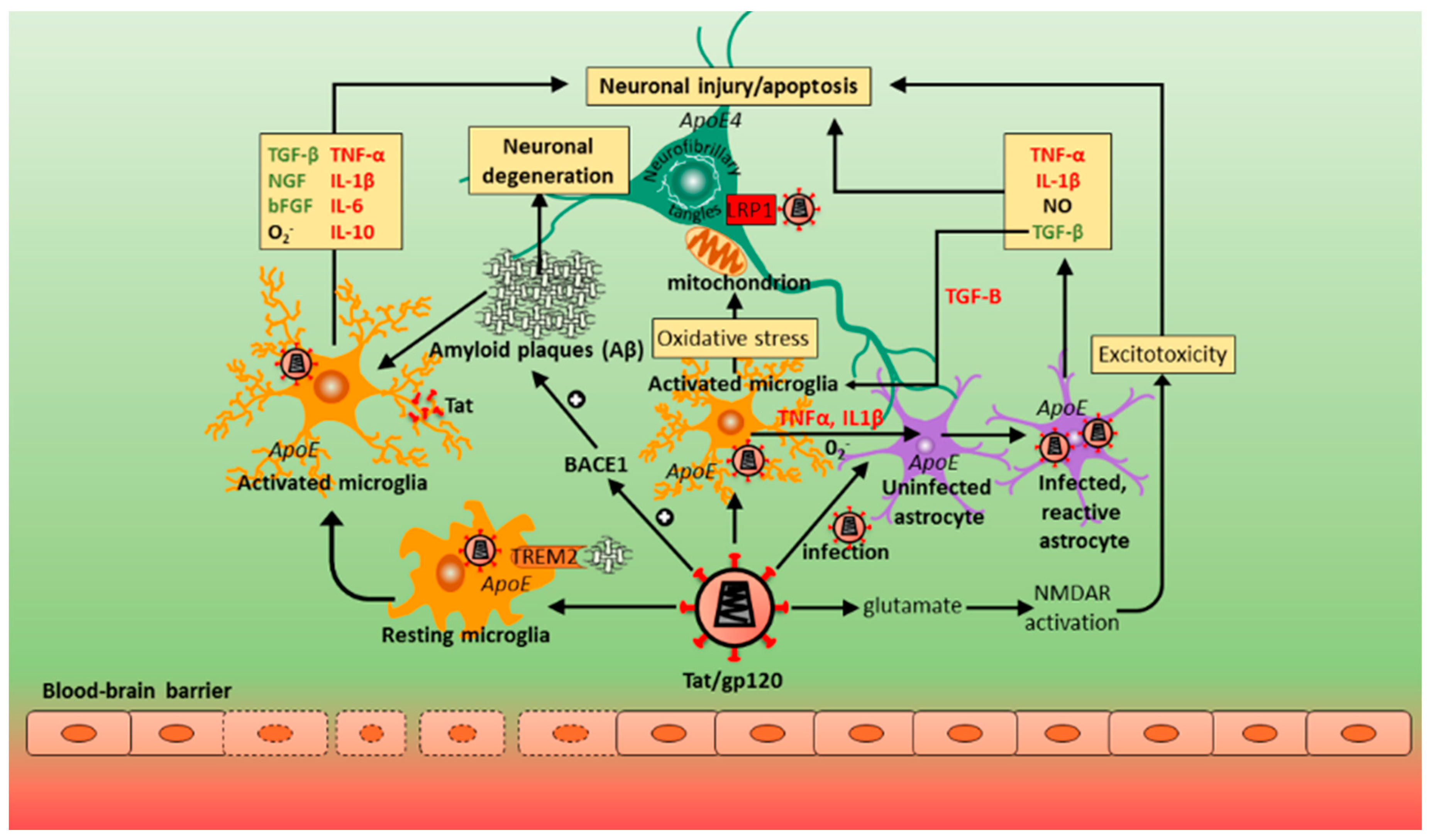
12. Contribution of Host Genes in Alzheimer’s Disease (AD) and HAND
13. Drug Metabolism/Transporter Genes
14. Summary
Funding
Conflicts of Interest
References
- Danforth, K.; Granich, R.; Wiedeman, D.; Baxi, S.; Padian, N. Global mortality and morbidity of HIV/AIDS. In Major Infectious Diseases; Holmes, K.K., Bertozzi, S., Bloom, B.R., Jha, P., Eds.; World Health Organization: Washington, DC, USA, 2017. [Google Scholar]
- Clifford, D.B. HIV-associated neurocognitive disorder. Curr. Opin. Infect. Dis. 2017, 30, 117–122. [Google Scholar] [PubMed]
- Arendt, G.; Grauer, O.; Hahn, K.; Maschke, M.; Obermann, M.; Husstedt, I. Neues bei HIV und neuro-AIDS. Aktuelle Neurol. 2015, 42, 445–455. [Google Scholar] [CrossRef]
- Joska, J.A.; Fincham, D.S.; Stein, D.J.; Paul, R.H.; Seedat, S. Clinical correlates of HIV-associated neurocognitive disorders in south Africa. AIDS Behav. 2010, 14, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Mogambery, J.C.; Dawood, H.; Wilson, D.; Moodley, A. HIV-associated neurocognitive disorder in a kwazulu-natal HIV clinic: A prospective study. S. Afr. J. HIV Med. 2017, 18, 732. [Google Scholar] [CrossRef] [PubMed]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E.; et al. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Wu, Y.T.; Fratiglioni, L.; Matthews, F.E.; Lobo, A.; Breteler, M.M.; Skoog, I.; Brayne, C. Dementia in western europe: Epidemiological evidence and implications for policy making. Lancet Neurol. 2016, 15, 116–124. [Google Scholar] [CrossRef]
- Langa, K.M.; Larson, E.B.; Crimmins, E.M.; Faul, J.D.; Levine, D.A.; Kabeto, M.U.; Weir, D.R. A comparison of the prevalence of dementia in the united states in 2000 and 2012. JAMA Intern. Med. 2017, 177, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report, Improving Healthcare for People Living with Dementia; Alzheimer’s Disease International: London, UK, 2016. [Google Scholar]
- Wilmshurst, J.M.; Badoe, E.; Wammanda, R.D.; Mallewa, M.; Kakooza-Mwesige, A.; Venter, A.; Newton, C.R. Child neurology services in Africa. J. Child. Neurol. 2011, 26, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Kalula, S.; Petros, G. Responses to dementia in less developed countries with a focus on south Africa. Glob. Aging 2011, 7, 31–40. [Google Scholar]
- Kalula, S.Z.; Ferreira, M.; Thomas, K.G.; de Villiers, L.; Joska, J.A.; Geffen, L.N. Profile and management of patients at a memory clinic. S. Afr. Med. J. 2010, 100, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Wilmshurst, J.M.; Kakooza-Mwesige, A.; Newton, C.R. The challenges of managing children with epilepsy in Africa. Semin. Pediatr. Neurol. 2014, 21, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder–pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Gisslen, M.; Price, R.W.; Nilsson, S. The definition of HIV-associated neurocognitive disorders: Are we overestimating the real prevalence? BMC Infect. Dis 2011, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Grant, I.; Franklin, D.R., Jr.; Deutsch, R.; Woods, S.P.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; Collier, A.C.; et al. Asymptomatic HIV-associated neurocognitive impairment increases risk for symptomatic decline. Neurology 2014, 82, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Soontornniyomkij, V.; Achim, C.L.; Masliah, E.; Gelman, B.B.; Sinsheimer, J.S.; Singer, E.J.; Moore, D.J. Multilevel analysis of neuropathogenesis of neurocognitive impairment in HIV. J. Neurovirol. 2016, 22, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Milanini, B.; Paul, R.; Bahemana, E.; Adamu, Y.; Kiweewa, F.; Langat, R.; Owuoth, J.; Allen, E.; Polyak, C.; Ake, J.; et al. Limitations of the international HIV dementia scale in the current era. AIDS (Lond. Engl.) 2018, 32, 2477–2483. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Joska, J.A.; Goodkin, K.; Lopez, E.; Myer, L.; Paul, R.H.; John, S.; Sunpath, H. Normative scores for a brief neuropsychological battery for the detection of HIV-associated neurocognitive disorder (hand) among South Africans. BMC Res. Notes 2010, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, K.; Hardy, D.J.; Singh, D.; Lopez, E. Diagnostic utility of the international HIV dementia scale for HIV-associated neurocognitive impairment and disorder in south Africa. J. Neuropsychiatry Clin. Neurosci. 2014, 26, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Joska, J.A.; Westgarth-Taylor, J.; Hoare, J.; Thomas, K.G.; Paul, R.; Myer, L.; Stein, D.J. Validity of the international HIV dementia scale in south Africa. AIDS Patient Care STDs 2011, 25, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Cross, H.M.; Combrinck, M.I.; Joska, J.A. HIV-associated neurocognitive disorders: Antiretroviral regimen, central nervous system penetration effectiveness, and cognitive outcomes. S. Afr. Med. J. 2013, 103, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Schouten, J.; Su, T.; Wit, F.W.; Kootstra, N.A.; Caan, M.W.; Geurtsen, G.J.; Schmand, B.A.; Stolte, I.G.; Prins, M.; Majoie, C.B.; et al. Determinants of reduced cognitive performance in HIV-1-infected middle-aged men on combination antiretroviral therapy. AIDS 2016, 30, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Persidsky, Y.; Ghorpade, A.; Limoges, J.; Stins, M.; Fiala, M.; Morrisett, R. The neuropathogenesis of the AIDS dementia complex. AIDS (Lond. Engl.) 1997, 11 (Suppl. A), S35–S45. [Google Scholar]
- Salgado, M.; Kwon, M.; Galvez, C.; Badiola, J.; Nijhuis, M.; Bandera, A.; Balsalobre, P.; Miralles, P.; Buno, I.; Martinez-Laperche, C.; et al. Mechanisms that contribute to a profound reduction of the HIV-1 reservoir after allogeneic stem cell transplant. Ann. Intern. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Satishchandra, P.; Nalini, A.; Gourie-Devi, M.; Khanna, N.; Santosh, V.; Ravi, V.; Desai, A.; Chandramuki, A.; Jayakumar, P.N.; Shankar, S.K. Profile of neurologic disorders associated with HIV/AIDS from Bangalore, South India (1989–1996). Indian J. Med. Res. 2000, 111, 14–23. [Google Scholar] [PubMed]
- Sacktor, N.; Lyles, R.H.; Skolasky, R.; Kleeberger, C.; Selnes, O.A.; Miller, E.N.; Becker, J.T.; Cohen, B.; McArthur, J.C.; Multicenter, A.C.S. HIV-associated neurologic disease incidence changes: Multicenter AIDS cohort study, 1990–1998. Neurology 2001, 56, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.J.; Kulkarni, H.; Camargo, J.F.; He, W.; Smith, A.; Anaya, J.M.; Miura, T.; Hecht, F.M.; Mamtani, M.; Pereyra, F.; et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat. Immunol. 2007, 8, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Hammerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Thylin, M.R.; Ghorpade, A.; Xiong, H.; Persidsky, Y.; Cotter, R.; Niemann, D.; Che, M.; Zeng, Y.C.; Gelbard, H.A.; et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J. Neuroimmunol. 1999, 98, 185–200. [Google Scholar] [CrossRef]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 protein tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Nath, A.; Mattson, M.P.; Slevin, J.T.; Geiger, J.D. HIV-1 tat through phosphorylation of nmda receptors potentiates glutamate excitotoxicity. J. Neurochem. 2001, 78, 457–467. [Google Scholar] [CrossRef] [PubMed]
- King, J.E.; Eugenin, E.A.; Buckner, C.M.; Berman, J.W. HIV tat and neurotoxicity. Microbes Infect. 2006, 8, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Conant, K.; Garzino-Demo, A.; Nath, A.; McArthur, J.C.; Halliday, W.; Power, C.; Gallo, R.C.; Major, E.O. Induction of monocyte chemoattractant protein-1 in HIV-1 tat-stimulated astrocytes and elevation in AIDS dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 3117–3121. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Banks, W.A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 2015, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.H.; Kim, H.J.; Thayer, S.A. Subtype selective nmda receptor antagonists induce recovery of synapses lost following exposure to HIV-1 tat. Br. J. Pharmacol. 2012, 166, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, Y.; Reid, R.; Steiner, J.; Malpica-Llanos, T.; Darden, T.A.; Shankar, S.K.; Mahadevan, A.; Satishchandra, P.; Nath, A. Nmda receptor activation by HIV-tat protein is clade dependent. J. Neurosci. 2008, 28, 12190–12198. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Sas, A.R.; Eugenin, E.A.; Siddappa, N.B.; Bimonte-Nelson, H.; Berman, J.W.; Ranga, U.; Tyor, W.R.; Prasad, V.R. HIV-1 clade-specific differences in the induction of neuropathogenesis. J. Neurosci. 2008, 28, 10010–10016. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Ruiz, A.P.; Prasad, V.R. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (hand). AIDS Res. Ther. 2014, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Vetrivel, S.; Siddappa, N.B.; Ranga, U.; Seth, P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 b and c tat of human neurons: Significance of dicysteine C30C31 motif. Ann. Neurol. 2008, 63, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Maurya, V.K.; Dandu, H.R.; Bhatt, M.L.; Saxena, S.K. Global perspective of novel therapeutic strategies for the management of neuroAIDS. Biomol. Concepts 2018, 9, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Saksena, N.K. HIV associated neurocognitive disorders. Infect. Dis. Rep. 2013, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Brabers, N.A.; Nottet, H.S. Role of the pro-inflammatory cytokines TNF-alpha and il-1beta in HIV-associated dementia. Eur. J. Clin. Investig. 2006, 36, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Lipton, S.A. Experimental and potential future therapeutic approaches for HIV-1 associated dementia targeting receptors for chemokines, glutamate and erythropoietin. Neurotox. Res. 2005, 8, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Letendre, S.L.; Zheng, J.C.; Kaul, M.; Yiannoutsos, C.T.; Ellis, R.J.; Taylor, M.J.; Marquie-Beck, J.; Navia, B. Chemokines in cerebrospinal fluid correlate with cerebral metabolite patterns in HIV-infected individuals. J. Neurovirol. 2011, 17, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Lee, M.H.; Song, B.J. Neuronal cell death and degeneration through increased nitroxidative stress and tau phosphorylation in HIV-1 transgenic rats. PLoS ONE 2017, 12, e0169945. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Neogi, U.; Talboom, J.S.; Padilla, L.; Rahman, M.; Fritz-French, C.; Gonzalez-Ramirez, S.; Verma, A.; Wood, C.; Ruprecht, R.M.; et al. Clade C HIV-1 isolates circulating in southern Africa exhibit a greater frequency of dicysteine motif-containing tat variants than those in southeast asia and cause increased neurovirulence. Retrovirology 2013, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Saiyed, Z.; Thangavel, S.; Rodriguez, J.; Rao, K.V.; Nair, M.P. Differential effects of HIV type 1 clade b and clade c tat protein on expression of proinflammatory and antiinflammatory cytokines by primary monocytes. AIDS Res. Hum. Retrovir. 2009, 25, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Zhao, Z.; Hulgan, T.; Bush, W.S.; Samuels, D.C.; Bloss, C.S.; Heaton, R.K.; Ellis, R.J.; Schork, N.; Marra, C.M.; et al. Genome-wide association study of HIV-associated neurocognitive disorder (hand): A charter group study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Service, S.; Miller, E.N.; Reynolds, S.M.; Singer, E.J.; Shapshak, P.; Martin, E.M.; Sacktor, N.; Becker, J.T.; Jacobson, L.P.; et al. Genome-wide association study of neurocognitive impairment and dementia in HIV-infected adults. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Kallianpur, A.R.; Levine, A.J. Host genetic factors predisposing to HIV-associated neurocognitive disorder. Curr. HIV/AIDS Rep. 2014, 11, 336–352. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Panos, S.E.; Horvath, S. Genetic, transcriptomic, and epigenetic studies of HIV-associated neurocognitive disorder. J. Acquir. Immune Defic. Syndr. 2014, 65, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Kestler, L.J.; Mazzanti, C.; Bates, J.A.; Goldberg, T.; Goldman, D. A functional polymorphism in the comt gene and performance on a test of prefrontal cognition. Am. J. Psychiatry 2002, 159, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Reynolds, S.; Cox, C.; Miller, E.N.; Sinsheimer, J.S.; Becker, J.T.; Martin, E.; Sacktor, N. Neuropsychology Working Group of the Multicenter, A.C.S. The longitudinal and interactive effects of HIV status, stimulant use, and host genotype upon neurocognitive functioning. J. Neurovirol. 2014, 20, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Villalba, K.; Devieux, J.G.; Rosenberg, R.; Cadet, J.L. Serotonin-related gene polymorphisms and asymptomatic neurocognitive impairment in HIV-infected alcohol abusers. Genet. Res. Int. 2016, 2016, 7169172. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Scheller, C.; du Plessis, S.; Burger, R.; Arendt, G.; Joska, J.; Sopper, S.; Maschke, C.M.; Obermann, M.; Husstedt, I.W.; et al. The dopamine-related polymorphisms BDNF, COMT, DRd2, DRD3, and DRD4 are not linked with changes in CSF dopamine levels and frequency of HIV infection. J. Neural. Transm. (Vienna) 2017, 124, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Enge, S.; Fleischhauer, M.; Lesch, K.P.; Reif, A.; Strobel, A. Serotonergic modulation in executive functioning: Linking genetic variations to working memory performance. Neuropsychologia 2011, 49, 3776–3785. [Google Scholar] [CrossRef] [PubMed]
- Villalba, K.; Devieux, J.G.; Rosenberg, R.; Cadet, J.L. DRD2 and DRD4 genes related to cognitive deficits in HIV-infected adults who abuse alcohol. Behav. Brain Funct. 2015, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; Knapp, P.E. Interactions of HIV and drugs of abuse: The importance of glia, neural progenitors, and host genetic factors. Int. Rev. Neurobiol. 2014, 118, 231–313. [Google Scholar] [PubMed]
- Bousman, C.A.; Cherner, M.; Glatt, S.J.; Atkinson, J.H.; Grant, I.; Tsuang, M.T.; Everall, I.P. Impact of comt val158met on executive functioning in the context of HIV and methamphetamine. Neurobehav. HIV Med. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Bousman, C.A.; Chana, G.; Cherner, M.; Heaton, R.K.; Deutsch, R.; Ellis, R.J.; Grant, I.; Everall, I.P. Dopamine receptor D3 genetic polymorphism (RS6280TC) is associated with rates of cognitive impairment in methamphetamine-dependent men with HIV: Preliminary findings. J. Neurovirol. 2011, 17, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.M.; Murray, J.; Byrd, D.A.; Hurd, Y.L.; Morgello, S. HIV-related cognitive impairment shows bi-directional association with dopamine receptor DRD1 and DRD2 polymorphisms in substance-dependent and substance-independent populations. J. Neurovirol. 2013, 19, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Coley, J.S.; Calderon, T.M.; Gaskill, P.J.; Eugenin, E.A.; Berman, J.W. Dopamine increases CD14+CD16+ monocyte migration and adhesion in the context of substance abuse and HIV neuropathogenesis. PLoS ONE 2015, 10, e0117450. [Google Scholar] [CrossRef] [PubMed]
- Var, S.R.; Day, T.R.; Vitomirov, A.; Smith, D.M.; Soontornniyomkij, V.; Moore, D.J.; Achim, C.L.; Mehta, S.R.; Perez-Santiago, J. Mitochondrial injury and cognitive function in HIV infection and methamphetamine use. AIDS 2016, 30, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Rozzi, S.J.; Avdoshina, V.; Fields, J.A.; Trejo, M.; Ton, H.T.; Ahern, G.P.; Mocchetti, I. Human immunodeficiency virus promotes mitochondrial toxicity. Neurotox. Res. 2017, 32, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.B.; Samuels, D.C.; Hulgan, T. The other genome: A systematic review of studies of mitochondrial DNA haplogroups and outcomes of HIV infection and antiretroviral therapy. AIDS Rev. 2013, 15, 213–220. [Google Scholar] [PubMed]
- Louboutin, J.P.; Strayer, D. Role of oxidative stress in HIV-1-associated neurocognitive disorder and protection by gene delivery of antioxidant enzymes. Antioxidants (Basel) 2014, 3, 770–797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, M.; Li, H.; Zhang, H.; Shi, Y.; Wei, F.; Liu, D.; Liu, K.; Chen, D. Accumulation of nuclear and mitochondrial DNA damage in the frontal cortex cells of patients with HIV-associated neurocognitive disorders. Brain Res. 2012, 1458, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perez-Santiago, J.; Schrier, R.D.; de Oliveira, M.F.; Gianella, S.; Var, S.R.; Day, T.R.; Ramirez-Gaona, M.; Suben, J.D.; Murrell, B.; Massanella, M.; et al. Cell-free mitochondrial DNA in csf is associated with early viral rebound, inflammation, and severity of neurocognitive deficits in HIV infection. J. Neurovirol. 2016, 22, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Hulgan, T.; Samuels, D.C.; Bush, W.; Ellis, R.J.; Letendre, S.L.; Heaton, R.K.; Franklin, D.R.; Straub, P.; Murdock, D.G.; Clifford, D.B.; et al. Mitochondrial DNA haplogroups and neurocognitive impairment during HIV infection. Clin. Infect. Dis. 2015, 61, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Malan-Muller, S.; Hemmings, S.M.; Spies, G.; Kidd, M.; Fennema-Notestine, C.; Seedat, S. Shorter telomere length—A potential susceptibility factor for HIV-associated neurocognitive impairments in south African women [corrected]. PLoS ONE 2013, 8, e58351. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Sauce, D. Assessing immune aging in HIV-infected patients. Virulence 2017, 8, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Giesbrecht, C.J.; Thornton, A.E.; Hall-Patch, C.; Maan, E.J.; Cote, H.C.; Money, D.M.; Murray, M.; Pick, N. Select neurocognitive impairment in HIV-infected women: Associations with HIV viral load, hepatitis c virus, and depression, but not leukocyte telomere length. PLoS ONE 2014, 9, e89556. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Quach, A.; Moore, D.J.; Achim, C.L.; Soontornniyomkij, V.; Masliah, E.; Singer, E.J.; Gelman, B.; Nemanim, N.; Horvath, S. Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J. Neurovirol. 2016, 22, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Saiyed, Z.M.; Gandhi, N.; Agudelo, M.; Napuri, J.; Samikkannu, T.; Reddy, P.V.; Khatavkar, P.; Yndart, A.; Saxena, S.K.; Nair, M.P. HIV-1 tat upregulates expression of histone deacetylase-2 (HDAC2) in human neurons: Implication for HIV-associated neurocognitive disorder (hand). Neurochem. Int. 2011, 58, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Thames, A.D.; Briones, M.S.; Magpantay, L.I.; Martinez-Maza, O.; Singer, E.J.; Hinkin, C.H.; Morgello, S.; Gelman, B.B.; Moore, D.J.; Heizerling, K.; et al. The role of chemokine C–C motif ligand 2 genotype and cerebrospinal fluid chemokine c-c motif ligand 2 in neurocognition among HIV-infected patients. AIDS 2015, 29, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: A potential mechanism of HIV-CNS invasion and neuroAIDS. J. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Rovin, B.H.; Sen, L.; Cooke, G.; Dhanda, R.; Mummidi, S.; Kulkarni, H.; Bamshad, M.J.; Telles, V.; Anderson, S.A.; et al. HIV-1 infection and AIDS dementia are influenced by a mutant mcp-1 allele linked to increased monocyte infiltration of tissues and mcp-1 levels. Proc. Natl. Acad. Sci. USA 2002, 99, 13795–13800. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Barroga, C.F.; Hughes, M.D.; Chen, J.; Raskino, C.; McKinney, R.E.; Spector, S.A. Genetic influence of CCR5, CCR2, and SDF1 variants on human immunodeficiency virus 1 (HIV-1)-related disease progression and neurological impairment, in children with symptomatic HIV-1 infection. J. Infect. Dis. 2003, 188, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Letendre, S.; Marquie-Beck, J.; Singh, K.K.; de Almeida, S.; Zimmerman, J.; Spector, S.A.; Grant, I.; Ellis, R.; Group, H. The monocyte chemotactic protein-1 -2578 g allele is associated with elevated mcp-1 concentrations in cerebrospinal fluid. J. Neuroimmunol. 2004, 157, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Ellis, R.J.; Marquie-Beck, J.; Letendre, S.; Heaton, R.K.; Grant, I.; Spector, S.A. CCR2 polymorphisms affect neuropsychological impairment in HIV-1-infected adults. J. Neuroimmunol. 2004, 157, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Singer, E.J.; Sinsheimer, J.S.; Hinkin, C.H.; Papp, J.; Dandekar, S.; Giovanelli, A.; Shapshak, P. Ccl3 genotype and current depression increase risk of HIV-associated dementia. Neurobehav. HIV Med. 2009, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Singh, K.K.; Gupta, S.; Cystique, L.A.; Jin, H.; Letendre, S.; Schrier, R.; Wu, Z.; Hong, K.X.; Yu, X.; et al. Apoe epsilon4 and MBL-2 O/O genotypes are associated with neurocognitive impairment in HIV-infected plasma donors. AIDS 2010, 24, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Bol, S.M.; Booiman, T.; van Manen, D.; Bunnik, E.M.; van Sighem, A.I.; Sieberer, M.; Boeser-Nunnink, B.; de Wolf, F.; Schuitemaker, H.; Portegies, P.; et al. Single nucleotide polymorphism in gene encoding transcription factor prep1 is associated with HIV-1-associated dementia. PLoS ONE 2012, 7, e30990. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Sacktor, N.; Marder, K.; Cohen, B.; Schifitto, G.; Skolasky, R.L.; Creighton, J.; Guo, L.; McArthur, J.C. CCL3L1 gene copy number in individuals with and without HIV-associated neurocognitive disorder. Curr. Biomark. Find. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed]
- van Rij, R.P.; Portegies, P.; Hallaby, T.; Lange, J.M.; Visser, J.; de Roda Husman, A.M.; van’t Wout, A.B.; Schuitemaker, H. Reduced prevalence of the CCR5 delta32 heterozygous genotype in human immunodeficiency virus-infected individuals with AIDS dementia complex. J. Infect. Dis. 1999, 180, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Kulkarni, H.; Bolivar, H.; Mangano, A.; Sanchez, R.; Catano, G.; Nibbs, R.J.; Freedman, B.I.; Quinones, M.P.; Bamshad, M.J.; et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005, 307, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chao, D.; Nakayama, E.E.; Taguchi, H.; Goto, M.; Xin, X.; Takamatsu, J.K.; Saito, H.; Ishikawa, Y.; Akaza, T.; et al. Polymorphism in rantes chemokine promoter affects HIV-1 disease progression. Proc. Natl. Acad. Sci. USA 1999, 96, 4581–4585. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Beecroft, M.J.; Kleeberger, C.A.; Al-Sharif, F.M.; Ollier, W.E.; Zimmerman, P.A.; Boatin, B.A.; Leitman, S.F.; Detels, R.; Hajeer, A.H.; et al. Chemokine rantes promoter polymorphism affects risk of both HIV infection and disease progression in the multicenter AIDS cohort study. AIDS 2000, 14, 2671–2678. [Google Scholar] [CrossRef] [PubMed]
- McLaren, P.J.; Coulonges, C.; Bartha, I.; Lenz, T.L.; Deutsch, A.J.; Bashirova, A.; Buchbinder, S.; Carrington, M.N.; Cossarizza, A.; Dalmau, J.; et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. USA 2015, 112, 14658–14663. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the ccr-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.F.; Salgado, M.; Sanchez-Ortega, I.; Arnan, M.; Canals, C.; Domingo-Domenech, E.; Fernandez-de-Sevilla, A.; Gonzalez-Barca, E.; Moron-Lopez, S.; Nogues, N.; et al. CCR5 delta32 homozygous cord blood allogeneic transplantation in a patient with HIV: A case report. Lancet HIV 2015, 2, e236–e242. [Google Scholar] [CrossRef]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 delta32/delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Boven, L.A.; van der Bruggen, T.; van Asbeck, B.S.; Marx, J.J.; Nottet, H.S. Potential role of CCR5 polymorphism in the development of AIDS dementia complex. FEMS Immunol. Med. Microbiol. 1999, 26, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Umaki, T.; Chew, G.M.; Chow, D.C.; Agsalda, M.; Kallianpur, K.J.; Paul, R.; Zhang, G.; Ho, E.; Hanks, N.; et al. Treatment intensification with maraviroc (CCR5 antagonist) leads to declines in CD16-expressing monocytes in cart-suppressed chronic HIV-infected subjects and is associated with improvements in neurocognitive test performance: Implications for HIV-associated neurocognitive disease (hand). J. Neurovirol. 2014, 20, 571–582. [Google Scholar] [PubMed]
- Quasney, M.W.; Zhang, Q.; Sargent, S.; Mynatt, M.; Glass, J.; McArthur, J. Increased frequency of the tumor necrosis factor-alpha-308 a allele in adults with human immunodeficiency virus dementia. Ann. Neurol. 2001, 50, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, L.A.; Stone, E.; Price, P.; van Bockxmeer, F.; Brew, B.J. The relationship between apoe, tnfa, il1a, il1b and il12b genes and HIV-1-associated dementia. HIV Med. 2008, 9, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Sato-Matsumura, K.C.; Berger, J.; Hainfellner, J.A.; Mazal, P.; Budka, H. Development of HIV encephalitis in AIDS and tnf-alpha regulatory elements. J. Neuroimmunol. 1998, 91, 89–92. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Gong, Y.; Kelly, C.J.; Gelman, B.B. Host genetic polymorphisms in human immunodeficiency virus-related neurologic disease. J. Neurovirol. 2004, 10 (Suppl. 1), 67–73. [Google Scholar] [CrossRef]
- Sanna, P.P.; Repunte-Canonigo, V.; Masliah, E.; Lefebvre, C. Gene expression patterns associated with neurological disease in human HIV infection. PLoS ONE 2017, 12, e0175316. [Google Scholar] [CrossRef] [PubMed]
- Borjabad, A.; Brooks, A.I.; Volsky, D.J. Gene expression profiles of HIV-1-infected glia and brain: Toward better understanding of the role of astrocytes in HIV-1-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 2010, 5, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Siangphoe, U.; Archer, K.J. Gene expression in HIV-associated neurocognitive disorders: A meta-analysis. J. Acquir. Immune Defic. Syndr. 2015, 70, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Quach, A.; Horvath, S.; Nemanim, N.; Vatakis, D.; Witt, M.D.; Miller, E.N.; Detels, R.; Langfelder, P.; Shapshak, P.; Singer, E.J.; et al. No reliable gene expression biomarkers of current or impending neurocognitive impairment in peripheral blood monocytes of persons living with HIV. J. Neurovirol. 2018, 24, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Agace, W.W.; Amara, A.; Roberts, A.I.; Pablos, J.L.; Thelen, S.; Uguccioni, M.; Li, X.Y.; Marsal, J.; Arenzana-Seisdedos, F.; Delaunay, T.; et al. Constitutive expression of stromal derived factor-1 by mucosal epithelia and its role in HIV transmission and propagation. Curr. Biol. 2000, 10, 325–328. [Google Scholar] [CrossRef]
- Winkler, C.; Modi, W.; Smith, M.W.; Nelson, G.W.; Wu, X.; Carrington, M.; Dean, M.; Honjo, T.; Tashiro, K.; Yabe, D.; et al. Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. Alive study, hemophilia growth and development study (HGDS), multicenter AIDS cohort study (Macs), multicenter hemophilia cohort study (MHCS), san francisco city cohort (SFCC). Science 1998, 279, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.; Magierowska, M.; Hubert, J.B.; Theodorou, I.; van Rij, R.; Prins, M.; de Roda Husman, A.M.; Coutinho, R.; Schuitemaker, H. CC-chemokine receptor variants, SDF-1 polymorphism, and disease progression in 720 HIV-infected patients. Seroco cohort. Amsterdam cohort studies on AIDS. AIDS (Lond. Engl.) 1999, 13, 624–626. [Google Scholar] [CrossRef]
- Ioannidis, J.P.; Rosenberg, P.S.; Goedert, J.J.; Ashton, L.J.; Benfield, T.L.; Buchbinder, S.P.; Coutinho, R.A.; Eugen-Olsen, J.; Gallart, T.; Katzenstein, T.L.; et al. Effects of CCR5-delta32, CCR2-64I, and SDF-1 3′a alleles on HIV-1 disease progression: An international meta-analysis of individual-patient data. Ann. Intern. Med. 2001, 135, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Tresoldi, E.; Romiti, M.L.; Boniotto, M.; Crovella, S.; Salvatori, F.; Palomba, E.; Pastore, A.; Cancrini, C.; de Martino, M.; Plebani, A.; et al. Prognostic value of the stromal cell-derived factor 1 3′a mutation in pediatric human immunodeficiency virus type 1 infection. J. Infect. Dis. 2002, 185, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Sinsheimer, J.S.; Bilder, R.; Shapshak, P.; Singer, E.J. Functional polymorphisms in dopamine-related genes: Effect on neurocognitive functioning in HIV+ adults. J. Clin. Exp. Neuropsychol. 2012, 34, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Strathdee, S.A.; Galai, N.; Oleksyk, T.; Fallin, M.D.; Mehta, S.; Schaid, D.; Vlahov, D.; O’Brien, S.J.; Smith, M.W. Behavioral risk exposure and host genetics of susceptibility to HIV-1 infection. J. Infect. Dis. 2006, 193, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Vasilescu, A.; Diop, G.; Hirtzig, T.; Coulonges, C.; Labib, T.; Heath, S.C.; Spadoni, J.L.; Therwath, A.; Lathrop, M.; et al. Associations of the IL2Ralpha, IL4Ralpha, IL10Ralpha, and IFN (gamma) r1 cytokine receptor genes with AIDS progression in a french AIDS cohort. Immunogenetics 2006, 58, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Price, P.; James, I.; Fernandez, S.; French, M.A. Alleles of the gene encoding IL-1alpha may predict control of plasma viraemia in HIV-1 patients on highly active antiretroviral therapy. AIDS 2004, 18, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Garred, P.; Madsen, H.O.; Balslev, U.; Hofmann, B.; Pedersen, C.; Gerstoft, J.; Svejgaard, A. Susceptibility to HIV infection and progression of AIDS in relation to variant alleles of mannose-binding lectin. Lancet (Lond. Engl.) 1997, 349, 236–240. [Google Scholar] [CrossRef]
- Singh, K.K.; Lieser, A.; Ruan, P.K.; Fenton, T.; Spector, S.A. An age-dependent association of mannose-binding lectin-2 genetic variants on HIV-1-related disease in children. J. Allergy Clin. Immunol. 2008, 122, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Nathamu, S.; Adame, A.; Alire, T.U.; Dumaop, W.; Gouaux, B.; Moore, D.J.; Masliah, E. Expression of mannose binding lectin in HIV-1-infected brain: Implications for HIV-related neuronal damage and neuroAIDS. Neurobehav. HIV Med. 2011, 3, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Naranbhai, V.; Carrington, M. Host genetic variation and HIV disease: From mapping to mechanism. Immunogenetics 2017, 69, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.D.; Gupta, S.; Riggs, P.; Cysique, L.A.; Letendre, S.; Jin, H.; Spector, S.A.; Singh, K.K.; Wolfson, T.; Wu, Z.; et al. The influence of HLA on HIV-associated neurocognitive impairment in Anhui, China. PLoS ONE 2012, 7, e32303. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Gray, P.K.; Wang, Y.; Fenton, T.; Trout, R.N.; Spector, S.A. Hla alleles are associated with altered risk for disease progression and central nervous system impairment of HIV-infected children. J. Acquir. Immune Defic. Syndr. 2011, 57, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sagar, V.; Pilakka-Kanthikeel, S.; Martinez, P.C.; Atluri, V.S.R.; Nair, M. Common gene-network signature of different neurological disorders and their potential implications to neuroAIDS. PLoS ONE 2017, 12, e0181642. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Shepherd, N.; Lan, J.; Li, W.; Rane, S.; Gupta, S.K.; Zhang, S.; Dong, J.; Yu, Q. MMPS/timps imbalances in the peripheral blood and cerebrospinal fluid are associated with the pathogenesis of HIV-1-associated neurocognitive disorders. Brain Behav. Immun. 2017, 65, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Samani, D.; Nambiar, N.; Ghate, M.V.; Gangakhedkar, R.R. Effect of matrix metalloproteinase-21 (572C/T) polymorphism on HIV-associated neurocognitive disorder. APMIS 2018, 126, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.O.; Marathe, S.D.; Nain, S.; Samani, D.; Nema, V.; Ghate, M.V.; Gangakhedkar, R.R. Promoter polymorphism MMP-1 (-1607 2G/1G) and MMP-3 (-1612 5A/6A) in development of hand and modulation of pathogenesis of hand. J. Biosci. 2017, 42, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Marathe, S.D.; Nema, V.; Ghate, M.V.; Gangakhedkar, R.R. Genetic variation of MMP-2(-735 c>t) and MMP-9(-1562 c>t) gene in risk of development of hand and severity of hand. J. Gene Med. 2016, 18, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Marathe, S.D.; Nain, S.; Nema, V.; Angadi, M.; Bapat, S.S.; Ghate, M.V.; Gangakhedkar, R.R. Impact of variants of mmp-7(-181a>g) gene in susceptibility to the development of HIV-associated neurocognitive disorder and its severity. APMIS 2016, 124, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Samani, D.; Nambiar, N.; Ghate, M.V.; Gangakhedkar, R.R. Prevalence of mmp-8 gene polymorphisms in HIV-infected individuals and its association with HIV-associated neurocognitive disorder. Gene 2018, 646, 83–90. [Google Scholar] [CrossRef] [PubMed]
- De Jager, C.A.; Msemburi, W.; Pepper, K.; Combrinck, M.I. Dementia prevalence in a rural region of south Africa: A cross-sectional community study. J. Alzheimer’s Dis. JAD 2017, 60, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What happens with the circuit in Alzheimer’s disease in mice and humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenomics of Alzheimer’s and parkinson’s diseases. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and apoe receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Herz, J. Apoe, apoe receptors, and the synapse in Alzheimer’s disease. Trends Endocrinol. Metab. 2017, 28, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein e and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Bretsky, P.; Guralnik, J.M.; Launer, L.; Albert, M.; Seeman, T.E. The role of apoe-epsilon4 in longitudinal cognitive decline: Macarthur studies of successful aging. Neurology 2003, 60, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Lipnicki, D.M.; Crawford, J.; Kochan, N.A.; Trollor, J.N.; Draper, B.; Reppermund, S.; Maston, K.; Mather, K.A.; Brodaty, H.; Sachdev, P.S. Risk factors for mild cognitive impairment, dementia and mortality: The sydney memory and ageing study. J. Am. Med. Directors Assoc. 2017, 18, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. Apoe2, apoe3, and apoe4 differentially stimulate app transcription and abeta secretion. Cell 2017, 168, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Geffin, R.; McCarthy, M. Aging and apolipoprotein e in HIV infection. J. Neurovirol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Valcour, V.; Paul, R.; Neuhaus, J.; Shikuma, C. The effects of age and HIV on neuropsychological performance. J. Int. Neuropsychol. Soc. JINS 2011, 17, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Valcour, V.; Shikuma, C.; Shiramizu, B.; Watters, M.; Poff, P.; Selnes, O.A.; Grove, J.; Liu, Y.; Abdul-Majid, K.B.; Gartner, S.; et al. Age, apolipoprotein E4, and the risk of HIV dementia: The Hawaii aging with HIV cohort. J. Neuroimmunol. 2004, 157, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Biddolph, S.C.; Morris, C.S. Prevalence of alzheimer plaques in AIDS. J. Neurol. Neurosurg. Psychiatry 1998, 65, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Soontornniyomkij, V.; Moore, D.J.; Gouaux, B.; Soontornniyomkij, B.; Tatro, E.T.; Umlauf, A.; Masliah, E.; Levine, A.J.; Singer, E.J.; Vinters, H.V.; et al. Cerebral beta-amyloid deposition predicts HIV-associated neurocognitive disorders in apoe epsilon4 carriers. AIDS (Lond. Engl.) 2012, 26, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Bien-Ly, N.; Gillespie, A.K.; Walker, D.; Yoon, S.Y.; Huang, Y. Reducing human apolipoprotein e levels attenuates age-dependent abeta accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 2012, 32, 4803–4811. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.D.; Agan, B.K.; Marconi, V.C.; He, W.; Kulkarni, H.; Mold, J.E.; Cavrois, M.; Huang, Y.; Mahley, R.W.; Dolan, M.J.; et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the apoe epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc. Natl. Acad. Sci. USA 2008, 105, 8718–8723. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Robertson, K.; Lannfelt, L.; Bogdanovic, N.; Eggertsen, G.; Wilkins, J.; Hall, C. HIV-infected subjects with the E4 allele for apoe have excess dementia and peripheral neuropathy. Nat. Med. 1998, 4, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Green, D.A.; Masliah, E.; Vinters, H.V.; Beizai, P.; Moore, D.J.; Achim, C.L. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS (Lond. Engl.) 2005, 19, 407–411. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Mahley, R.W.; Huang, Y. Increased tau phosphorylation in apolipoprotein e4 transgenic mice is associated with activation of extracellular signal-regulated kinase: Modulation by zinc. J. Biol. Chem. 2004, 279, 44795–44801. [Google Scholar] [CrossRef] [PubMed]
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.Q.; Xu, Q.; Dee Fish, J.; Wyss-Coray, T.; Buttini, M.; Mucke, L.; et al. Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004, 24, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Digicaylioglu, M.; Russo, R.; Kaul, M.; Achim, C.L.; Fletcher, L.; Masliah, E.; Lipton, S.A. Erythropoietin plus insulin-like growth factor-i protects against neuronal damage in a murine model of human immunodeficiency virus-associated neurocognitive disorders. Ann. Neurol. 2010, 68, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Brew, B.J.; Pemberton, L.; Blennow, K.; Wallin, A.; Hagberg, L. CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology 2005, 65, 1490–1492. [Google Scholar] [CrossRef] [PubMed]
- Anthony, I.C.; Ramage, S.N.; Carnie, F.W.; Simmonds, P.; Bell, J.E. Accelerated tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006, 111, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Soontornniyomkij, V.; Umlauf, A.; Soontornniyomkij, B.; Gouaux, B.; Ellis, R.J.; Levine, A.J.; Moore, D.J.; Letendre, S.L. Association of antiretroviral therapy with brain aging changes among HIV-infected adults. AIDS (Lond. Engl.) 2018, 32, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, K.; Lowe, V.; Przybelski, S.A.; Weigand, S.D.; Senjem, M.L.; Ivnik, R.J.; Preboske, G.M.; Roberts, R.; Geda, Y.E.; Boeve, B.F.; et al. Apoe modifies the association between abeta load and cognition in cognitively normal older adults. Neurology 2012, 78, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Nir, T.M.; Jahanshad, N.; Busovaca, E.; Wendelken, L.; Nicolas, K.; Thompson, P.M.; Valcour, V.G. Mapping white matter integrity in elderly people with HIV. Hum. Brain Mapp. 2014, 35, 975–992. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A multifunctional protein with implications in various pathologies as a result of its structural features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein e: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Martin, I.; Dubois, M.C.; Saermark, T.; Ruysschaert, J.M. Apolipoprotein a-1 interacts with the n-terminal fusogenic domains of SIV (simian immunodeficiency virus) GP32 and HIV (human immunodeficiency virus) gp41: Implications in viral entry. Biochem. Biophys. Res. Commun. 1992, 186, 95–101. [Google Scholar] [CrossRef]
- Briz, V.; Poveda, E.; Soriano, V. HIV entry inhibitors: Mechanisms of action and resistance pathways. J. Antimicrob. Chemother. 2006, 57, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Ran Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein e4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Jiang, C.; Cunningham, E.; Buchthal, S.; Douet, V.; Andres, M.; Ernst, T. Effects of apoe epsilon4, age, and HIV on glial metabolites and cognitive deficits. Neurology 2014, 82, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.; Chang, L. Effect of aging on brain metabolism in antiretroviral-naive HIV patients. AIDS (Lond. Engl.) 2004, 18 (Suppl. 1), S61–S67. [Google Scholar] [CrossRef]
- French, M.A.; King, M.S.; Tschampa, J.M.; da Silva, B.A.; Landay, A.L. Serum immune activation markers are persistently increased in patients with HIV infection after 6 years of antiretroviral therapy despite suppression of viral replication and reconstitution of CD4+ T cells. J. Infect. Dis. 2009, 200, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Piconi, S.; Trabattoni, D.; Gori, A.; Parisotto, S.; Magni, C.; Meraviglia, P.; Bandera, A.; Capetti, A.; Rizzardini, G.; Clerici, M. Immune activation, apoptosis, and treg activity are associated with persistently reduced cd4+ t-cell counts during antiretroviral therapy. AIDS (Lond. Engl.) 2010, 24, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Buckner, C.M.; Luers, A.J.; Calderon, T.M.; Eugenin, E.A.; Berman, J.W. Neuroimmunity and the blood-brain barrier: Molecular regulation of leukocyte transmigration and viral entry into the nervous system with a focus on neuroAIDS. J. Neuroimmune Pharmacol. 2006, 1, 160–181. [Google Scholar] [CrossRef] [PubMed]
- Giunta, B.; Ehrhart, J.; Obregon, D.F.; Lam, L.; Le, L.; Jin, J.; Fernandez, F.; Tan, J.; Shytle, R.D. Antiretroviral medications disrupt microglial phagocytosis of beta-amyloid and increase its production by neurons: Implications for HIV-associated neurocognitive disorders. Mol. Brain 2011, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Kiyota, T.; Hanamsagar, R.; Huang, Y.; Andrews, S.; Peng, H.; Zheng, J.C.; Swindells, S.; Carlson, G.A.; Ikezu, T. The effect of HIV protease inhibitors on amyloid-beta peptide degradation and synthesis in human cells and Alzheimer’s disease animal model. J. Neuroimmune Pharmacol. 2012, 7, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Geffin, R.; Martinez, R.; de Las Pozas, A.; Issac, B.; McCarthy, M. Apolipoprotein e4 suppresses neuronal-specific gene expression in maturing neuronal progenitor cells exposed to HIV. J. Neuroimmune Pharmacol. 2017, 12, 462–483. [Google Scholar] [CrossRef] [PubMed]
- Theendakara, V.; Peters-Libeu, C.A.; Spilman, P.; Poksay, K.S.; Bredesen, D.E.; Rao, R.V. Direct transcriptional effects of apolipoprotein E. J. Neurosci. 2016, 36, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Tensaouti, Y.; Stephanz, E.P.; Yu, T.S.; Kernie, S.G. ApoE regulates the development of adult newborn hippocampal neurons. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of glutamate and nmda receptors in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hui, L.; Geiger, N.H.; Haughey, N.J.; Geiger, J.D. Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol. Aging 2013, 34, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Daily, A.; Nath, A.; Hersh, L.B. Tat peptides inhibit neprilysin. J. Neurovirol. 2006, 12, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yoon, J.H.; Kim, Y.S. HIV-1 tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS ONE 2013, 8, e77972. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.L.; Ghura, S.; Gannon, P.J.; Akay-Espinoza, C.; Phan, J.M.; Yee, A.C.; Vassar, R.; Gelman, B.B.; Kolson, D.L.; Jordan-Sciutto, K.L. Bace1 mediates HIV-associated and excitotoxic neuronal damage through an app-dependent mechanism. J. Neurosci. 2018, 38, 4288–4300. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, N.; Tanzi, R.E.; Moir, R.D.; Nath, A.; He, J.J. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 2000, 6, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Nivison, M.; Montine, K.S.; Maeda, N.; Montine, T.J. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein e gene and is mediated by microglial P38MAPK. FASEB J. 2006, 20, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Salat, D.H.; Tuch, D.S.; Greve, D.N.; van der Kouwe, A.J.; Hevelone, N.D.; Zaleta, A.K.; Rosen, B.R.; Fischl, B.; Corkin, S.; Rosas, H.D.; et al. Age-related alterations in white matter microstructure measured by diffusion tensor imaging. Neurobiol. Aging 2005, 26, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Salat, D.H.; Tuch, D.S.; van der Kouwe, A.J.; Greve, D.N.; Pappu, V.; Lee, S.Y.; Hevelone, N.D.; Zaleta, A.K.; Growdon, J.H.; Corkin, S.; et al. White matter pathology isolates the hippocampal formation in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Storey, P.; Cohen, B.A.; Epstein, L.G.; Edelman, R.R.; Ragin, A.B. Diffusion alterations in corpus callosum of patients with HIV. AJNR Am. J. Neuroradiol. 2006, 27, 656–660. [Google Scholar] [PubMed]
- Chang, L.; Wong, V.; Nakama, H.; Watters, M.; Ramones, D.; Miller, E.N.; Cloak, C.; Ernst, T. Greater than age-related changes in brain diffusion of HIV patients after 1 year. J. Neuroimmune Pharmacol. 2008, 3, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, M.M.; Castillo, M.; Stadler, A.; Rieger, A.; Schmid, B.; Sundgren, P.C. Diffusion-tensor MR imaging of the brain in human immunodeficiency virus-positive patients. AJNR Am. J. Neuroradiol. 2005, 26, 2275–2281. [Google Scholar] [PubMed]
- Taoka, T.; Iwasaki, S.; Sakamoto, M.; Nakagawa, H.; Fukusumi, A.; Myochin, K.; Hirohashi, S.; Hoshida, T.; Kichikawa, K. Diffusion anisotropy and diffusivity of white matter tracts within the temporal stem in alzheimer disease: Evaluation of the “tract of interest” by diffusion tensor tractography. AJNR Am. J. Neuroradiol. 2006, 27, 1040–1045. [Google Scholar] [PubMed]
- Persson, J.; Lind, J.; Larsson, A.; Ingvar, M.; Cruts, M.; Van Broeckhoven, C.; Adolfsson, R.; Nilsson, L.G.; Nyberg, L. Altered brain white matter integrity in healthy carriers of the apoE epsilon4 allele: A risk for AD? Neurology 2006, 66, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hoare, J.; Westgarth-Taylor, J.; Fouche, J.P.; Combrinck, M.; Spottiswoode, B.; Stein, D.J.; Joska, J.A. Relationship between apolipoprotein E4 genotype and white matter integrity in HIV-positive young adults in south Africa. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Underwood, J.; Cole, J.H.; Caan, M.; De Francesco, D.; Leech, R.; van Zoest, R.A.; Su, T.; Geurtsen, G.J.; Schmand, B.A.; Portegies, P.; et al. Gray and white matter abnormalities in treated human immunodeficiency virus disease and their relationship to cognitive function. Clin. Infect. Dis. 2017, 65, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Wendelken, L.A.; Jahanshad, N.; Rosen, H.J.; Busovaca, E.; Allen, I.; Coppola, G.; Adams, C.; Rankin, K.P.; Milanini, B.; Clifford, K.; et al. Apoe epsilon4 is associated with cognition, brain integrity, and atrophy in HIV over age 60. J. Acquir. Immune Defic. Syndr. 2016, 73, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.E.; Woods, S.P.; Letendre, S.L.; Franklin, D.R.; Bloss, C.; Goate, A.; Heaton, R.K.; Collier, A.C.; Marra, C.M.; Gelman, B.B.; et al. Apolipoprotein E4 genotype does not increase risk of HIV-associated neurocognitive disorders. J. Neurovirol. 2013, 19, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Londos, E.; Blennow, K.; Minthon, L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol. 2006, 5, 228–234. [Google Scholar] [CrossRef]
- Cysique, L.A.; Hewitt, T.; Croitoru-Lamoury, J.; Taddei, K.; Martins, R.N.; Chew, C.S.; Davies, N.N.; Price, P.; Brew, B.J. ApoE epsilon4 moderates abnormal CSF-abeta-42 levels, while neurocognitive impairment is associated with abnormal CSF tau levels in HIV+ individuals—A cross-sectional observational study. BMC Neurol. 2015, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.S.; Chadwick, M.; Horton, W.A.; Simon, G.L.; Jiang, X.; Esposito, G. An individual with human immunodeficiency virus, dementia, and central nervous system amyloid deposition. Alzheimers Dement. (Amst) 2016, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Andres, M.; Sadino, J.; Jiang, C.S.; Nakama, H.; Miller, E.; Ernst, T. Impact of apolipoprotein e epsilon4 and HIV on cognition and brain atrophy: Antagonistic pleiotropy and premature brain aging. Neuroimage 2011, 58, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Milanini, B.; Valcour, V. Differentiating HIV-associated neurocognitive disorders from Alzheimer’s disease: An emerging issue in geriatric neuroHIV. Curr. HIV/AIDS Rep. 2017, 14, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, O.; Goplen, A.K.; Liestol, K.; Myrvang, B.; Rootwelt, H.; Christophersen, B.; Kvittingen, E.A.; Maehlen, J. Hiv dementia and apolipoprotein e. Acta Neurol. Scand. 1997, 95, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Joska, J.A.; Combrinck, M.; Valcour, V.G.; Hoare, J.; Leisegang, F.; Mahne, A.C.; Myer, L.; Stein, D.J. Association between apolipoprotein e4 genotype and human immunodeficiency virus-associated dementia in younger adults starting antiretroviral therapy in South Africa. J. Neurovirol. 2010, 16, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Panos, S.E.; Hinkin, C.H.; Singer, E.J.; Thames, A.D.; Patel, S.M.; Sinsheimer, J.S.; Del Re, A.C.; Gelman, B.B.; Morgello, S.; Moore, D.J.; et al. Apolipoprotein-e genotype and human immunodeficiency virus-associated neurocognitive disorder: The modulating effects of older age and disease severity. Neurobehavioral HIV Med. 2013, 5, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.T.; Martinson, J.J.; Penugonda, S.; Kingsley, L.; Molsberry, S.; Reynolds, S.; Aronow, A.; Goodkin, K.; Levine, A.; Martin, E.; et al. No association between apoepsilon4 alleles, HIV infection, age, neuropsychological outcome, or death. J. Neurovirol. 2015, 21, 24–31. [Google Scholar] [CrossRef] [PubMed]
- WHO. Who guidelines approved by the guidelines review committee. In Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection: Recommendations for a Public Health Approach (Available at Pmid: 24716260); WHO: Geneva, Switzerland, 2013. [Google Scholar]
- WHO. Consolidated Guidelines on HIV Prevention, Diagnosis, Treatment and Care for Key Populations. 2016. Available online: https://www.Ncbi.Nlm.Nih.Gov/books/nbk379694/ (accessed on 14 November 2018).
- Gaida, R.; Truter, I.; Grobler, C.; Kotze, T.; Godman, B. A review of trials investigating efavirenz-induced neuropsychiatric side effects and the implications. Expert Rev. Anti-Infect. Ther. 2016, 14, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Vaida, F.; Wong, J.; Sanders, C.A.; Kao, Y.T.; Croteau, D.; Clifford, D.B.; Collier, A.C.; Gelman, B.B.; Marra, C.M.; et al. Long-term efavirenz use is associated with worse neurocognitive functioning in HIV-infected patients. J. Neurovirol. 2016, 22, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Funes, H.A.; Blas-Garcia, A.; Galindo, M.J.; Alvarez, A.; Esplugues, J.V. Efavirenz and the cns: What we already know and questions that need to be answered. J. Antimicrob. Chemother. 2015, 70, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Swart, M.; Skelton, M.; Ren, Y.; Smith, P.; Takuva, S.; Dandara, C. High predictive value of CYP2B6 SNPS for steady-state plasma efavirenz levels in south African HIV/AIDS patients. Pharmacogenet. Genom. 2013, 23, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.W.; Ribaudo, H.J.; Kim, R.B.; Tierney, C.; Wilkinson, G.R.; Gulick, R.M.; Clifford, D.B.; Hulgan, T.; Marzolini, C.; Acosta, E.P. Pharmacogenetics of efavirenz and central nervous system side effects: An adult AIDS clinical trials group study. AIDS (Lond. Engl.) 2004, 18, 2391–2400. [Google Scholar]
- EÁ, B.; Rodríguez, S.N. The pharmacogenetics of HIV treatment: A practical clinical approach. J. Pharmacogenom. Pharmacoproteomics 2013, 4, 116. [Google Scholar]
- Gallien, S.; Journot, V.; Loriot, M.A.; Sauvageon, H.; Morlat, P.; Reynes, J.; Reliquet, V.; Chene, G.; Molina, J.M. Cytochrome 2B6 polymorphism and efavirenz-induced central nervous system symptoms: A substudy of the ANRS Alize trial. HIV Med. 2017, 18, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Masimirembwa, C.; Dandara, C.; Leutscher, P.D. Rolling out efavirenz for HIV precision medicine in Africa: Are we ready for pharmacovigilance and tackling neuropsychiatric adverse effects? Omics 2016, 20, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Mukonzo, J.K.; Owen, J.S.; Ogwal-Okeng, J.; Kuteesa, R.B.; Nanzigu, S.; Sewankambo, N.; Thabane, L.; Gustafsson, L.L.; Ross, C.; Aklillu, E. Pharmacogenetic-based efavirenz dose modification: Suggestions for an African population and the different CYP2B6 genotypes. PLoS ONE 2014, 9, e86919. [Google Scholar] [CrossRef] [PubMed]
- Pinillos, F.; Dandara, C.; Swart, M.; Strehlau, R.; Kuhn, L.; Patel, F.; Coovadia, A.; Abrams, E. Case report: Severe central nervous system manifestations associated with aberrant efavirenz metabolism in children: The role of CYP2B6 genetic variation. BMC Infect. Dis. 2016, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Sinxadi, P.Z.; Leger, P.D.; McIlleron, H.M.; Smith, P.J.; Dave, J.A.; Levitt, N.S.; Maartens, G.; Haas, D.W. Pharmacogenetics of plasma efavirenz exposure in HIV-infected adults and children in south Africa. Br. J. Clin. Pharmacol. 2015, 80, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Gebretsadik, T.; Shintani, A.; Mayo, G.; Acosta, E.P.; Stein, C.M.; Haas, D.W. Neuropsychometric correlates of efavirenz pharmacokinetics and pharmacogenetics following a single oral dose. Br. J. Clin. Pharmacol. 2013, 75, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Sandkovsky, U.; Podany, A.T.; Fletcher, C.V.; Owen, A.; Felton-Coleman, A.; Winchester, L.C.; Robertson, K.; Swindells, S. Impact of efavirenz pharmacokinetics and pharmacogenomics on neuropsychological performance in older HIV-infected patients. J. Antimicrob. Chemother. 2017, 72, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Rippeth, J.D.; Heaton, R.K.; Carey, C.L.; Marcotte, T.D.; Moore, D.J.; Gonzalez, R.; Wolfson, T.; Grant, I. Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J. Int. Neuropsychol. Soc. JINS 2004, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Morgan, E.E.; Iudicello, J.E.; Blackstone, K.; Grant, I.; Ellis, R.J.; Letendre, S.L.; Little, S.; Morris, S.; Smith, D.M.; et al. Substance use is a risk factor for neurocognitive deficits and neuropsychiatric distress in acute and early HIV infection. J. Neurovirol. 2013, 19, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Passaro, R.C.; Pandhare, J.; Qian, H.Z.; Dash, C. The complex interaction between methamphetamine abuse and HIV-1 pathogenesis. J. Neuroimmune Pharmacol. 2015, 10, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Cherner, M.; Bousman, C.; Everall, I.; Barron, D.; Letendre, S.; Vaida, F.; Atkinson, J.H.; Heaton, R.; Grant, I. Cytochrome p450-2d6 extensive metabolizers are more vulnerable to methamphetamine-associated neurocognitive impairment: Preliminary findings. J. Int. Neuropsychol. Soc. JINS 2010, 16, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Fellay, J.; Marzolini, C.; Meaden, E.R.; Back, D.J.; Buclin, T.; Chave, J.P.; Decosterd, L.A.; Furrer, H.; Opravil, M.; Pantaleo, G.; et al. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: A pharmacogenetics study. Lancet 2002, 359, 30–36. [Google Scholar] [CrossRef]
- Vautier, S.; Fernandez, C. Abcb1: The role in parkinson’s disease and pharmacokinetics of antiparkinsonian drugs. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Sankatsing, S.U.; Beijnen, J.H.; Schinkel, A.H.; Lange, J.M.; Prins, J.M. P glycoprotein in human immunodeficiency virus type 1 infection and therapy. Antimicrob. Agents Chemother. 2004, 48, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Johnson, A.D.; Papp, A.C.; Kroetz, D.L.; Sadee, W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet. Genom. 2005, 15, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Dirson, G.; Fernandez, C.; Hindlet, P.; Roux, F.; German-Fattal, M.; Gimenez, F.; Farinotti, R. Efavirenz does not interact with the abcb1 transporter at the blood-brain barrier. Pharm. Res. 2006, 23, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Sutherland, D.; Acosta, E.P.; Erdem, H.; Richardson, D.; Haas, D.W. Genetic and non-genetic determinants of raltegravir penetration into cerebrospinal fluid: A single arm pharmacokinetic study. PLoS ONE 2013, 8, e82672. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 2018, 97, 1032–1048. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Spencer, B.; Swinton, M.; Qvale, E.M.; Marquine, M.J.; Alexeeva, A.; Gough, S.; Soontornniyomkij, B.; Valera, E.; Masliah, E.; et al. Alterations in brain TREM2 and amyloid-beta levels are associated with neurocognitive impairment in HIV-infected persons on antiretroviral therapy. J. Neurochem. 2018. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivier, I.S.; Cacabelos, R.; Naidoo, V. Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics. Int. J. Mol. Sci. 2018, 19, 3594. https://doi.org/10.3390/ijms19113594
Olivier IS, Cacabelos R, Naidoo V. Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics. International Journal of Molecular Sciences. 2018; 19(11):3594. https://doi.org/10.3390/ijms19113594
Chicago/Turabian StyleOlivier, Ian Simon, Ramón Cacabelos, and Vinogran Naidoo. 2018. "Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics" International Journal of Molecular Sciences 19, no. 11: 3594. https://doi.org/10.3390/ijms19113594
APA StyleOlivier, I. S., Cacabelos, R., & Naidoo, V. (2018). Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics. International Journal of Molecular Sciences, 19(11), 3594. https://doi.org/10.3390/ijms19113594