Breast Cancer: An Examination of the Potential of ACKR3 to Modify the Response of CXCR4 to CXCL12

 , and
, and
Abstract
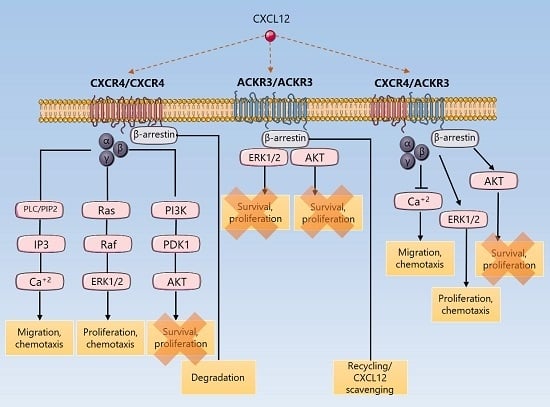
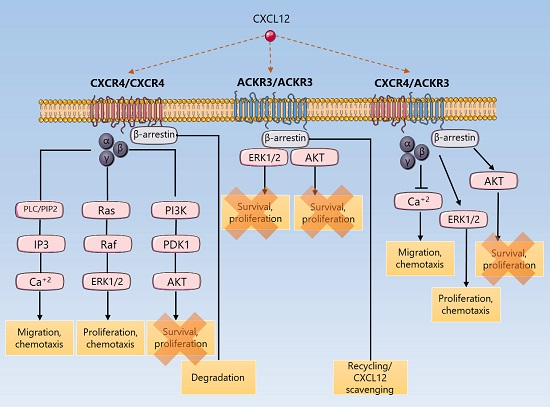{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
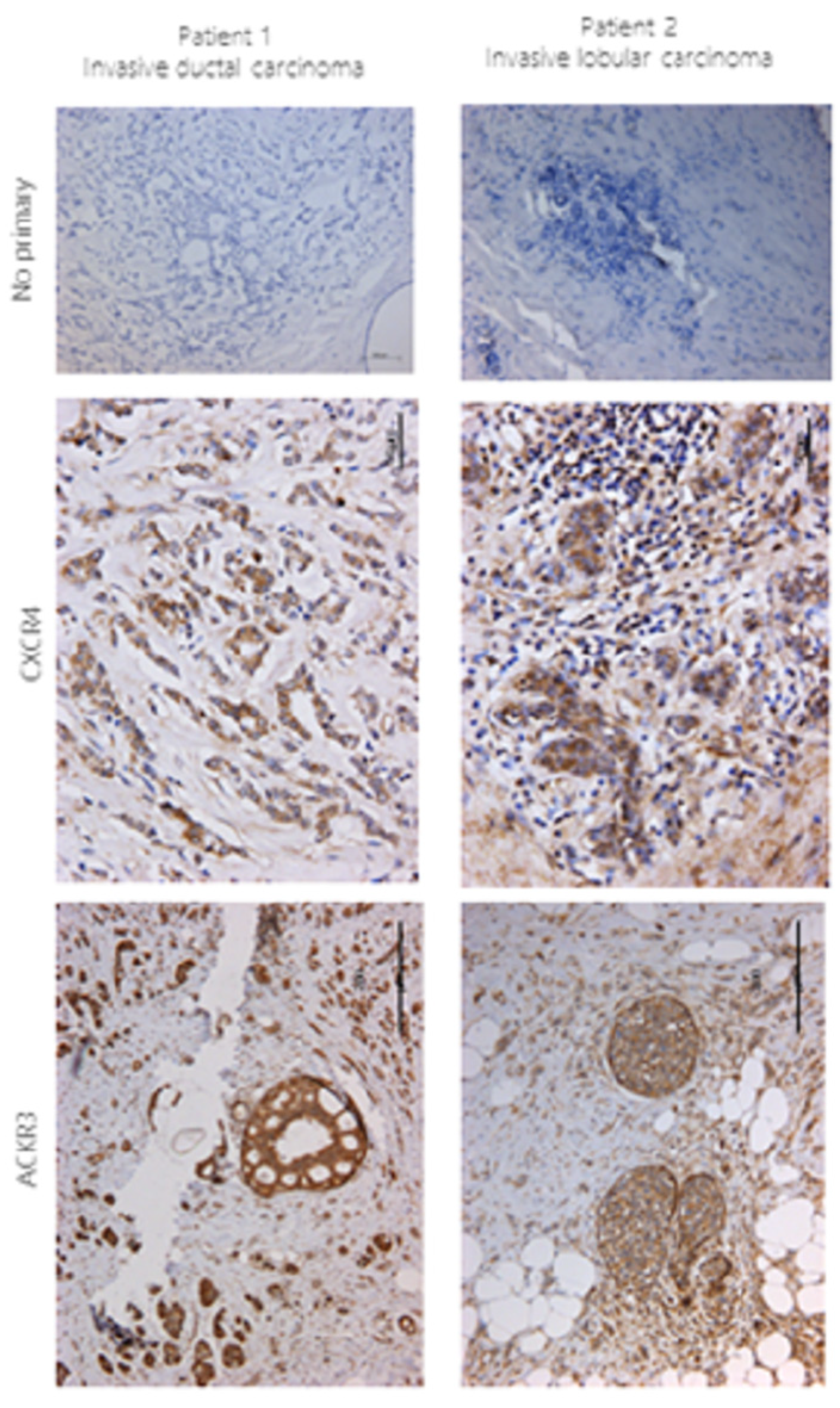
2.1. In Vivo CXCR4 and ACKR3 Expression in Breast Cancer Tissue
2.2. Expression of CXCR4 and ACKR3 on Transfected and Breast Cancer Cell Lines
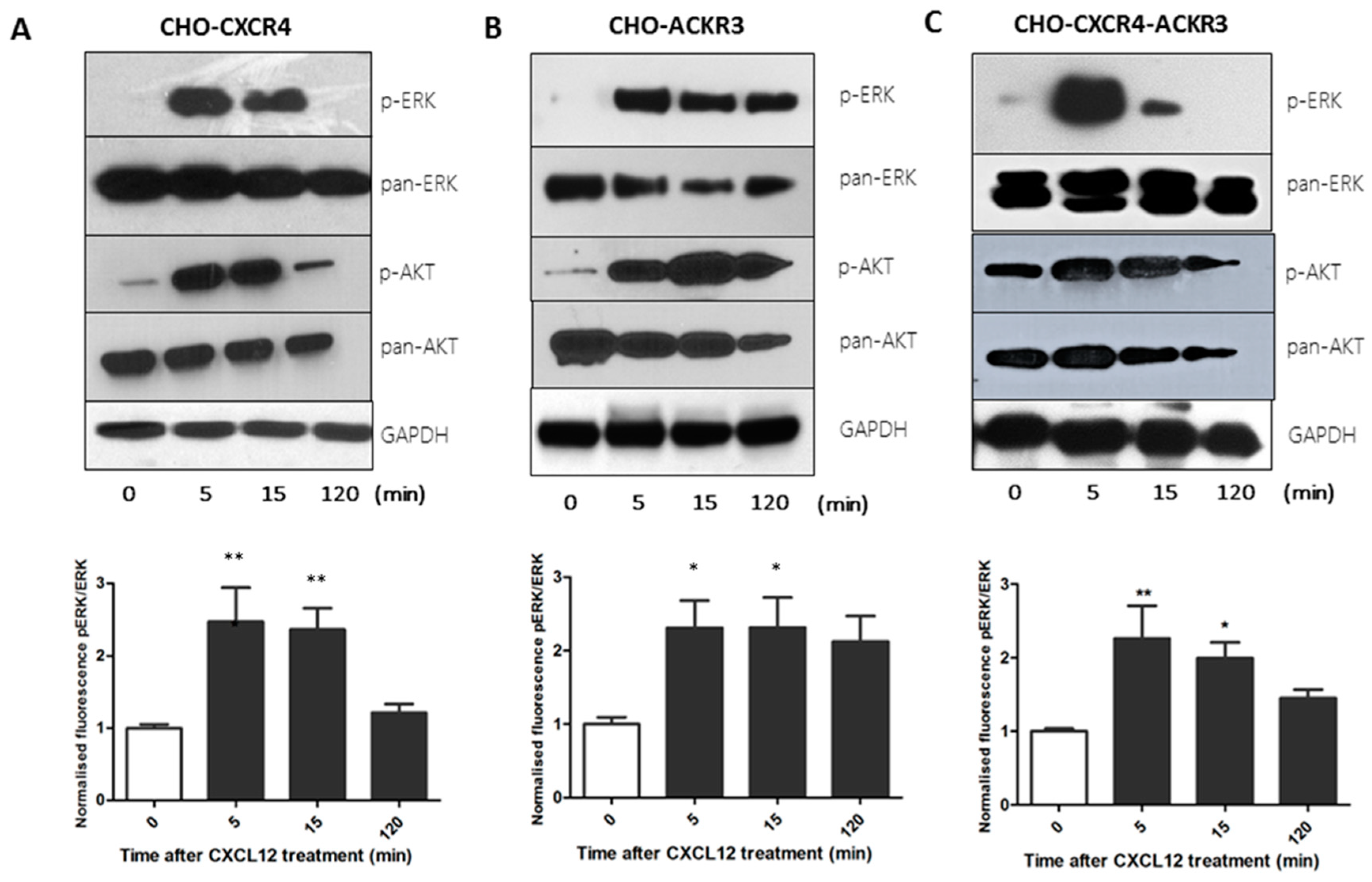
2.3. CXCR4 and ACKR3 Differentially Activate the Akt and ERK Oncogenic Pathways
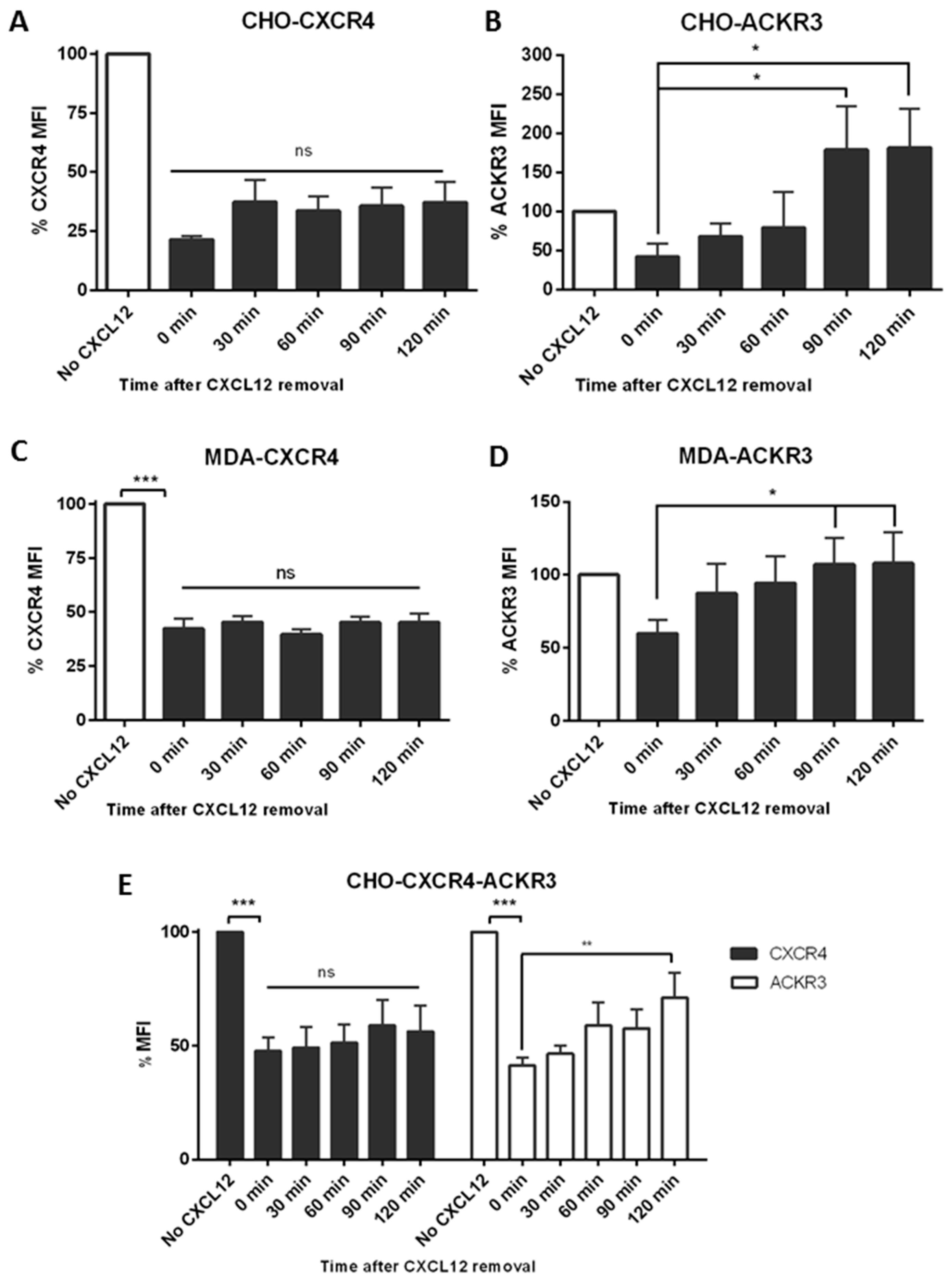
2.4. ACKR3 Expression Can Alter CXCR4’s Internalization Pathway
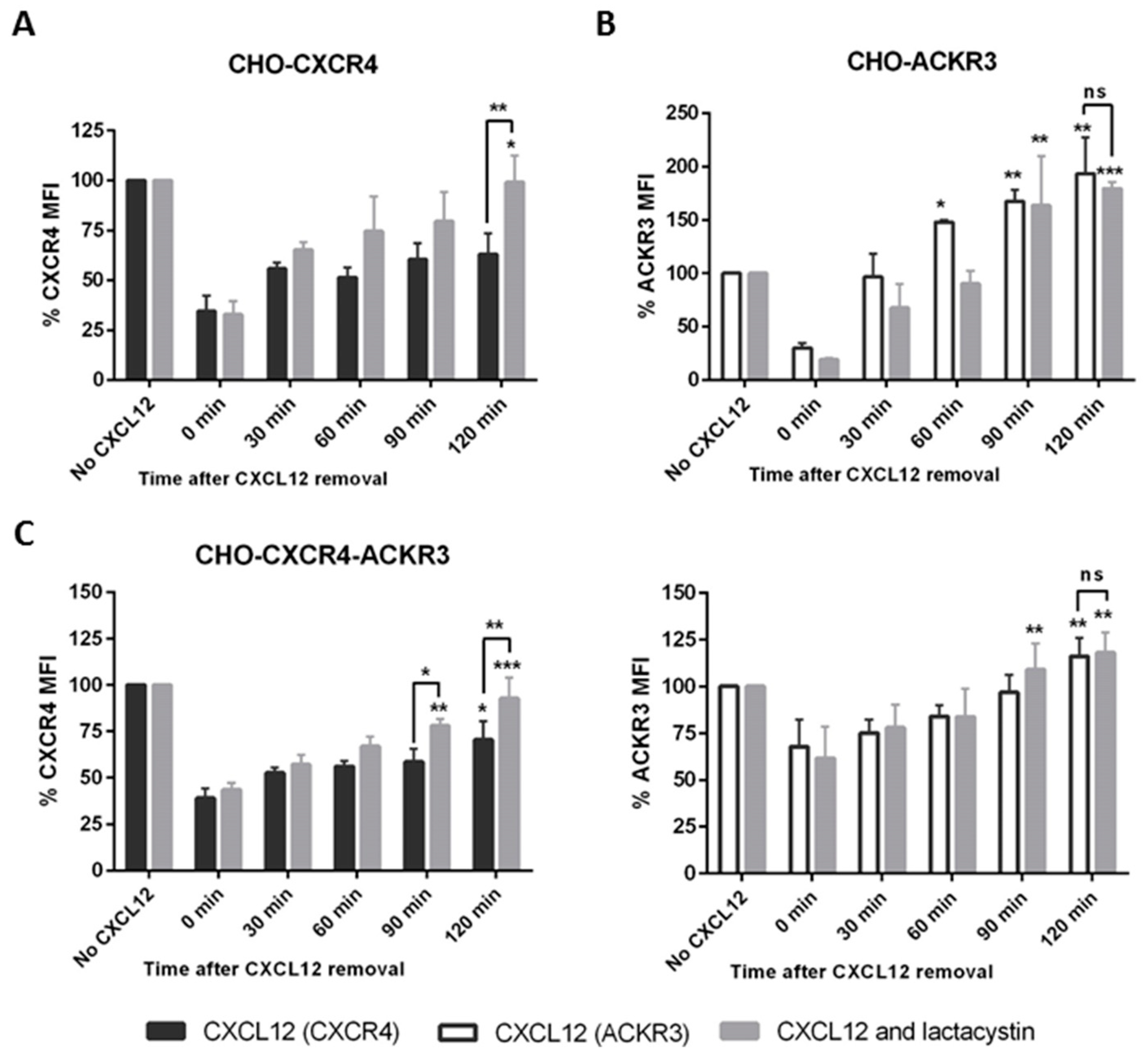
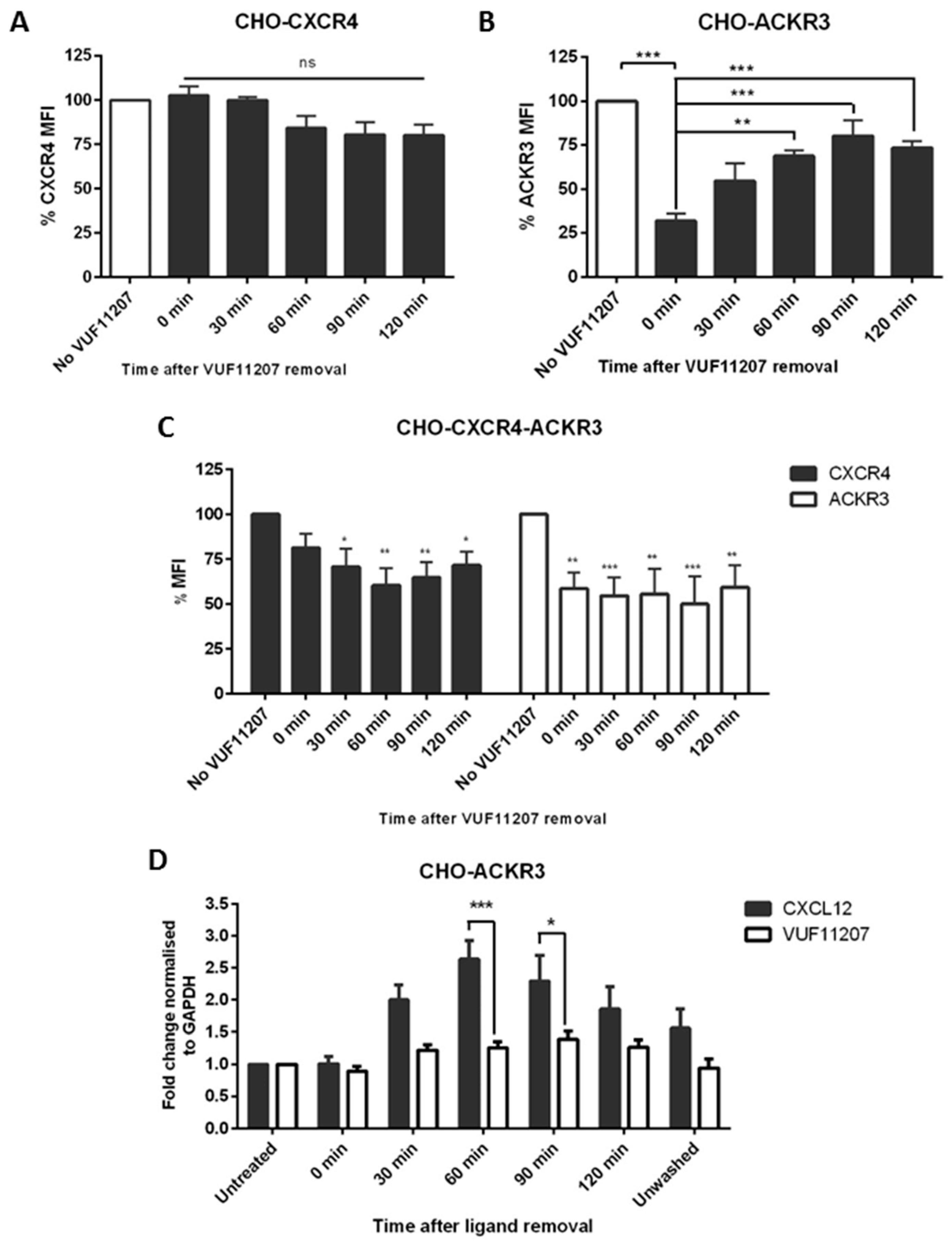
2.5. ACKR3 Agonist VUF11207 Functions Differently to CXCL12 and Can Cross-Desensitize CXCR4
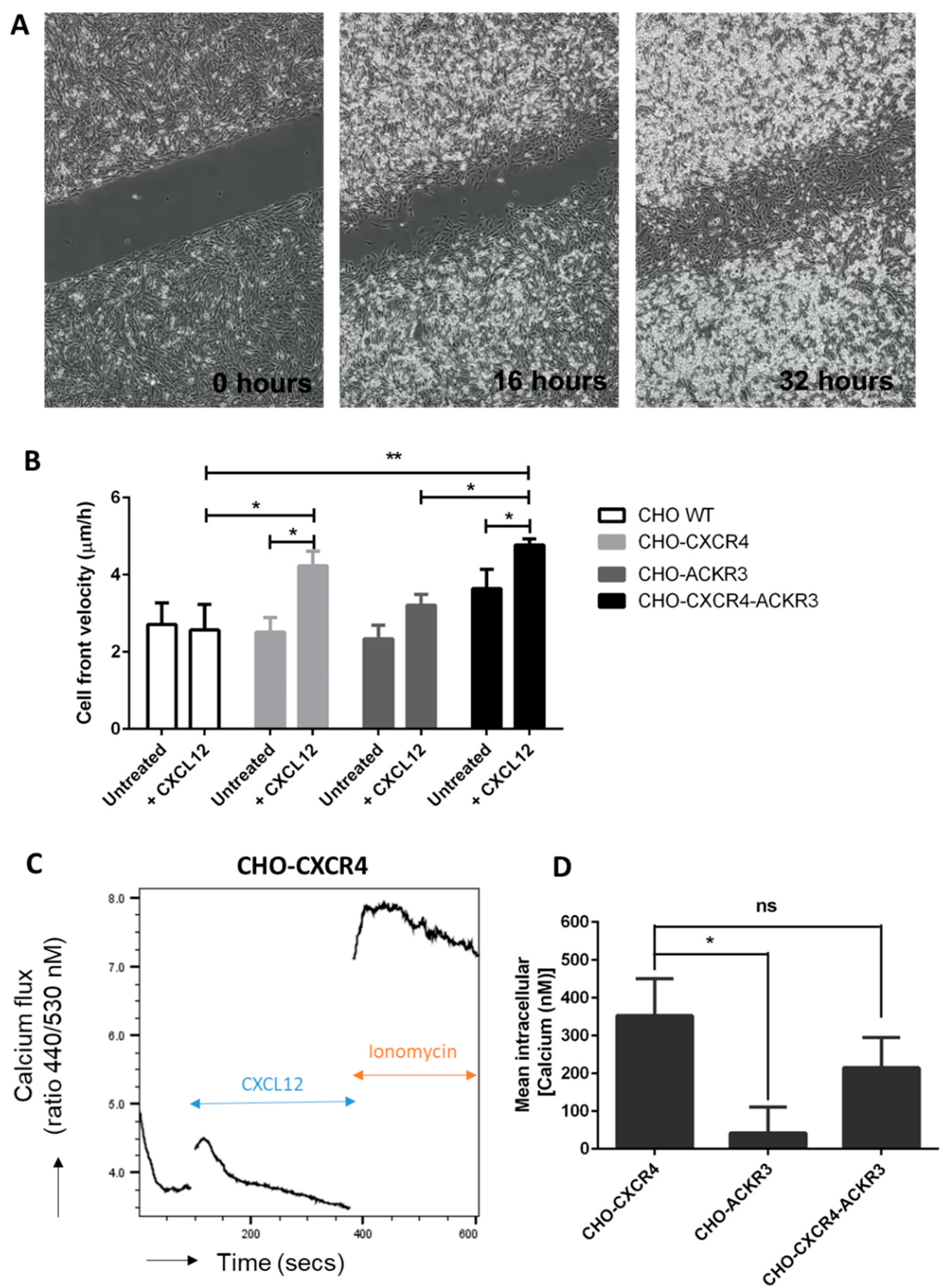
2.6. ACKR3 Increases In Vitro Migration to CXCL12 in the Presence of CXCR4
2.7. Diminished Calcium Flux Induction in ACKR3 Expressing Cell Line
3. Discussion
4. Materials and Methods
4.1. Immunohistochemistry
4.2. Cell Culture
4.3. Construction of Stable and Transient Transfectants
4.4. Cell Surface Expression of Chemokine Receptors
4.5. FRET
4.6. Western Blot
4.7. Cell-Based ELISA
4.8. qPCR
4.9. Wound Healing Assay
4.10. Calcium Flux
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Cronin, K.A.; Plevritis, S.K.; Fryback, D.G.; Clarke, L.; Zelen, M.; Mandelblatt, J.S.; Yakovlev, A.Y.; Habbema, J.D.F.; Feuer, E.J. Effect of screening and adjuvant therapy on mortality from breast cancer. N. Engl. J. Med. 2005, 353, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.; Gøtzsche, P.C. Cochrane review on screening for breast cancer with mammography. Lancet 2001, 358, 1340–1342. [Google Scholar] [CrossRef]
- Greene, F.L.; Page, D.L.; Fleming, I.D.; Fritz, A.G.; Balch, C.M.; Haller, D.G.; Morrow, M. Breast. In AJCC Cancer Staging Manual, 6th ed.; Springer: New York, NY, USA, 2002; pp. 223–240. [Google Scholar]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Yoon, Y.; Votaw, J.; Goodman, M.M.; Williams, L.; Shim, H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005, 65, 967–971. [Google Scholar] [PubMed]
- Liang, Z.; Wu, T.; Lou, H.; Yu, X.; Taichman, R.S.; Lau, S.K.; Nie, S.; Umbreit, J.; Shim, H. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004, 64, 4302–4308. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.P.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D. CXCR7 (RDC1) promotes breast and lung tumour growth in vivo and is expressed on tumour-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Qian, L.; Chen, X.; Ding, B. Prognostic significance of CXCL12, CXCR4 and CXCR7 in patients with breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13217–13224. [Google Scholar] [PubMed]
- Thelen, M.; Thelen, S. CXCR7, CXCR4 and CXCL12: An eccentric trio? J. Neuroimmunol. 2008, 198, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Steele, J.M.; Mihalko, L.A.; Ray, P.; Luker, G.D. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 2010, 29, 4599–4610. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Tohgo, A.; Pierce, K.L.; Choy, E.W.; Lefkowitz, R.J.; Luttrell, L.M. β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 2002, 277, 9429–9436. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shiozawa, Y.; Wang, J.; Wang, Y.; Jung, Y.; Pienta, K.J.; Mehra, R.; Loberg, R.; Taichman, R.S. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 2008, 283, 4283–4294. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Gupta, M.; Luker, G.D. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009, 23, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Sierro, F.; Biben, C.; Martínez-Muñoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signalling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Rodríguez-Frade, J.M.; Vila-Coro, A.J.; Fernández, S.; de Ana, A.M.; Jones, D.R.; Torán, J.L.; Martínez-A, C. Chemokine receptor homo-or heterodimerization activates distinct signalling pathways. EMBO J. 2001, 20, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Décaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 heterodimer constitutively recruits β-arrestin to enhance cell migration. J. Biol. Chem. 2011, 286, 32188–32197. [Google Scholar] [CrossRef] [PubMed]
- Floridi, F.; Trettel, F.; Di Bartolomeo, S.; Ciotti, M.T.; Limatola, C. Signalling pathways involved in the chemotactic activity of CXCL12 in cultured rat cerebellar neurons and CHP100 neuroepithelioma cells. J. Neuroimmunol. 2003, 135, 38–46. [Google Scholar] [CrossRef]
- Lazarini, F.; Casanova, P.; Tham, T.N.; De Clercq, E.; Arenzana-Seisdedos, F.; Baleux, F.; Dubois-Dalcq, M. Differential signalling of the chemokine receptor CXCR4 by stromal cell-derived factor 1 and the HIV glycoprotein in rat neurons and astrocytes. Eur. J. Neurosci. 2000, 12, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Peng, H.; Cui, M.; Whitney, N.P.; Huang, Y.; Zheng, J.C. CXCL12 increases human neural progenitor cell proliferation through Akt-1/FOXO3a signaling pathway. J. Neurochem. 2009, 109, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Boucek, M.M.; Snyderman, R. Calcium influx requirement for human neutrophil chemotaxis: Inhibition by lanthanum chloride. Science 1976, 193, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.F. Calcium and Chemotaxis. Sci. Signal. 2007, 2007, tw293. [Google Scholar] [CrossRef]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E. Role of high expression levels of CXCR4 in tumor growth, vascularization and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Tripathi, V.; Ahmad, M.; Nath, N.; Mir, R.A.; Chauhan, S.S.; Luthra, K. CXCR7 mediated Giα independent activation of ERK and Akt promotes cell survival and chemotaxis in T cells. Cell. Immunol. 2012, 272, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.N.; Grabovsky, V.; Pasvolsky, R.; Shulman, Z.; Buss, E.C.; Spiegel, A.; Nagler, A.; Lapidot, T.; Thelen, M.; Alon, R. A crosstalk between intracellular CXCR7 and CXCR4 involved in rapid CXCL12-triggered integrin activation but not in chemokine-triggered motility of human T lymphocytes and CD34+ cells. J. Leukoc. Biol. 2008, 84, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alcañiz, J.A.; Haege, S.; Mueller, W.; Pla, R.; Mackay, F.; Schulz, S.; López-Bendito, G.; Stumm, R.; Marín, O. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron 2011, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Uto-Konomi, A.; McKibben, B.; Wirtz, J.; Sato, Y.; Takano, A.; Nanki, T.; Suzuki, S. CXCR7 agonists inhibit the function of CXCL12 by down-regulation of CXCR4. Biochem. Biophys. Res. Commun. 2013, 431, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Lewin, S.A.; Mihalko, L.A.; Schmidt, B.T.; Winkler, J.S.; Coggins, N.L.; Thomas, D.G.; Luker, G.D. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene 2012, 31, 4750–4758. [Google Scholar] [CrossRef] [PubMed]
- Cabioglu, N.; Yazici, M.S.; Arun, B.; Broglio, K.R.; Hortobagyi, G.N.; Price, J.E.; Sahin, A. CCR7 and CXCR4 as novel biomarkers predicting axillary lymph node metastasis in T1 breast cancer. Clin. Cancer Res. 2005, 11, 5686–5693. [Google Scholar] [CrossRef] [PubMed]
- Lagane, B.; Chow, K.Y.; Balabanian, K.; Levoye, A.; Harriague, J.; Planchenault, T.; Baleux, F.; Gunera-Saad, N.; Arenzana-Seisdedos, F.; Bachelerie, F. CXCR4 dimerization and β-arrestin–mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood 2008, 112, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Shenoy, S.K.; Wei, H.; Lefkowitz, R.J. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 2004, 279, 35518–35525. [Google Scholar] [CrossRef] [PubMed]
- Ebisuya, M.; Kondoh, K.; Nishida, E. The duration, magnitude and compartmentalization of ERK MAP kinase activity: Mechanisms for providing signaling specificity. J. Cell Sci. 2005, 118, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Ödemis, V.; Lipfert, J.; Kraft, R.; Hajek, P.; Abraham, G.; Hattermann, K.; Mentlein, R.; Engele, J. The presumed atypical chemokine receptor CXCR7 signals through Gi/o proteins in primary rodent astrocytes and human glioma cells. Glia 2012, 60, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Zheng, J.; Hou, K.; Wang, J.; Chen, X.; Lu, X.; Bo, J.; Xu, C.; Shen, K.; Wang, J. Role of chemokine receptor CXCR7 in bladder cancer progression. Biochem. Pharmacol. 2012, 84, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Han, M.; Wang, F.; Xu, L.; Yu, H.; Yang, P. CXCR7 stimulates MAPK signaling to regulate hepatocellular carcinoma progression. Cell Death Dis. 2014, 5, e1488. [Google Scholar] [CrossRef] [PubMed]
- Liberman, J.; Sartelet, H.; Flahaut, M.; Mühlethaler-Mottet, A.; Coulon, A.; Nyalendo, C.; Vassal, G.; Joseph, J.-M.; Gross, N. Involvement of the CXCR7/CXCR4/CXCL12 axis in the malignant progression of human neuroblastoma. PLoS ONE 2012, 7, e43665. [Google Scholar] [CrossRef] [PubMed]
- Hattermann, K.; Holzenburg, E.; Hans, F.; Lucius, R.; Held-Feindt, J.; Mentlein, R. Effects of the chemokine CXCL12 and combined internalization of its receptors CXCR4 and CXCR7 in human MCF-7 breast cancer cells. Cell Tissue Res. 2014, 357, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Wurth, R.; Tarn, K.; Jernigan, D.; Fernandez, S.V.; Cristofanilli, M.; Fatatis, A.; Meucci, O. A preclinical model of inflammatory breast cancer to study the involvement of CXCR4 and ACKR3 in the metastatic process. Transl. Oncol. 2015, 8, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, Y.; Zuo, K.; Wang, Y.; Zhang, S.; Kuang, D.; Duan, Y.; Zhao, X.; Wang, G. Crosstalk between SDF-1/CXCR4 and SDF-1/CXCR7 in cardiac stem cell migration. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Riku, M.; Ito, H.; Tsunoda, T.; Ikeda, H.; Kasai, K. GLI1 orchestrates CXCR4/CXCR7 signaling to enhance migration and metastasis of breast cancer cells. Oncotarget 2015, 6, 33648. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, E.L.; Lee, W.; Lu, J.; Lowy, A.M.; Kim, J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med. 2012, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Boldajipour, B.; Mahabaleshwar, H.; Kardash, E.; Reichman-Fried, M.; Blaser, H.; Minina, S.; Wilson, D.; Xu, Q.; Raz, E. Control of chemokine-guided cell migration by ligand sequestration. Cell 2008, 132, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Grymula, K.; Tarnowski, M.; Wysoczynski, M.; Drukala, J.; Barr, F.G.; Ratajczak, J.; Kucia, M.; Ratajczak, M.Z. Overlapping and distinct role of CXCR7-SDF-1/ITAC and CXCR4-SDF-1 axes in regulating metastatic behavior of human rhabdomyosarcomas. Int. J. Cancer 2010, 127, 2554–2568. [Google Scholar] [CrossRef] [PubMed]
- Minina, S.; Reichman-Fried, M.; Raz, E. Control of receptor internalization, signaling level and precise arrival at the target in guided cell migration. Curr. Biol. 2007, 17, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Fernandis, A.Z.; Cherla, R.P.; Chernock, R.D.; Ganju, R.K. CXCR4/CCR5 down-modulation and chemotaxis are regulated by the proteasome pathway. J. Biol. Chem. 2002, 277, 18111–18117. [Google Scholar] [CrossRef] [PubMed]
- Lapham, C.K.; Romantseva, T.; Petricoin, E.; King, L.R.; Manischewitz, J.; Zaitseva, M.B.; Golding, H. CXCR4 heterogeneity in primary cells: Possible role of ubiquitination. J. Leukoc. Biol. 2002, 72, 1206–1214. [Google Scholar] [PubMed]
- Tamamura, H.; Hori, A.; Kanzaki, N.; Hiramatsu, K.; Mizumoto, M.; Nakashima, H.; Yamamoto, N.; Otaka, A.; Fujii, N. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett. 2003, 550, 79–83. [Google Scholar] [CrossRef]
- Burger, J.A.; Peled, A. CXCR4 antagonists: Targeting the microenvironment in leukemia and other cancers. Leukemia 2009, 23, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1α)-CXCR4/CXCR7 pathway inhibition: An emerging sensitizer for anticancer therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Wang, Y.; Lewén, S.; Berahovich, R.D.; Penfold, M.E.T.; Zhang, P.; Powers, J.; Summers, B.C.; Miao, Z.; Zhao, B. Elucidation of CXCR7-mediated signalling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 2009, 183, 3204–3211. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; O’Boyle, G.; Mellor, P.; Kirby, J.A. An apparent paradox: Chemokine receptor agonists can be used for anti-inflammatory therapy. Mol. Immunol. 2007, 44, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Seizer, P.; Borst, O.; Schönberger, T.; Mack, A.; Geisler, T.; Langer, H.F.; May, A.E.; Vogel, S.; Lang, F. SDF-1α induces differential trafficking of CXCR4-CXCR7 involving cyclophilin A, CXCR7 ubiquitination and promotes platelet survival. FASEB J. 2014, 28, 2864–2878. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Oishi, S.; Kubo, T.; Tanahara, N.; Fujii, N.; Furuya, T. Optimized method of G-protein-coupled receptor homology modeling: Its application to the discovery of novel CXCR7 ligands. J. Med. Chem. 2013, 56, 4236–4251. [Google Scholar] [CrossRef] [PubMed]
- Lodhish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Receptor-mediated endocytosis and the sorting of internalized proteins. In Molecular Cell Biology; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Hatse, S.; Princen, K.; Bridger, G.; De Clercq, E.; Schols, D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002, 527, 255–262. [Google Scholar] [CrossRef]
- Mazzinghi, B.; Ronconi, E.; Lazzeri, E.; Sagrinati, C.; Ballerini, L.; Angelotti, M.L.; Parente, E.; Mancina, R.; Netti, G.S.; Becherucci, F. Essential but differential role for CXCR4 and CXCR7 in the therapeutic homingof human renal progenitor cells. J. Exp. Med. 2008, 205, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Hattermann, K.; Held-Feindt, J.; Lucius, R.; Müerköster, S.S.; Penfold, M.E.; Schall, T.J.; Mentlein, R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010, 70, 3299–3308. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xiao, D.; Liu, H.; Zheng, X.; Liu, L.; Liu, S. Interfering with CXCR4 expression inhibits proliferation, adhesion and migration of breast cancer MDA-MB-231 cells. Oncol. Lett. 2014, 8, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, H.; Reddy, S.; Zhu, A.; Wang, S.; Blevins, D.; Yoon, Y.; Zhang, Y.; Shim, H. Blockade of invasion and metastasis of breast cancer cells via targeting CXCR4 with an artificial microRNA. Biochem. Biophys. Res. Commun. 2007, 363, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R. Regulation of leukocyte locomotion by Ca 2+. Trends Cell Biol. 1993, 3, 386–391. [Google Scholar] [CrossRef]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.; Chen, J.H.; Wang, B.; Zhang, R. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Haraldsen, G.; Rot, A. Coy decoy with a new ploy: Interceptor controls the levels of homeostatic chemokines. Eur. J. Immunol. 2006, 36, 1659–1661. [Google Scholar] [CrossRef] [PubMed]
- Long, P.; Sun, F.; Ma, Y.; Huang, Y. Inhibition of CXCR4 and CXCR7 for reduction of cell proliferation and invasion in human endometrial cancer. Tumor Biol. 2016, 37, 7473–7480. [Google Scholar] [CrossRef] [PubMed]
- Begley, L.A.; MacDonald, J.W.; Day, M.L.; Macoska, J.A. CXCL12 activates a robust transcriptional response in human prostate epithelial cells. J. Biol. Chem. 2007, 282, 26767–26774. [Google Scholar] [CrossRef] [PubMed]
- Worster, D.T.; Schmelzle, T.; Solimini, N.L.; Lightcap, E.S.; Millard, B.; Mills, G.B.; Brugge, J.S.; Albeck, J.G. Akt and ERK control the proliferative response of mammary epithelial cells to the growth factors IGF-1 and EGF through the cell cycle inhibitor p57Kip2. Sci. Signal. 2012, 5, ra19. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.R.; Mellor, P.; Eldaly, H.; Lennard, T.W.J.; Kirby, J.A.; Ali, S. Inhibition of CXCR4-mediated breast cancer metastasis: A potential role for heparinoids? Clin. Cancer Res. 2007, 13, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Batard, P.; Szollosi, J.; Luescher, I.; Cerottini, J.C.; MacDonald, R.; Romero, P. Use of phycoerythrin and allophycocyanin for fluorescence resonance energy transfer analyzed by flow cytometry: Advantages and limitations. Cytometry 2002, 48, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.T. Fluorescent and Luminescent Probes for Biological Activity: A Practical Guide to Technology for Quantitative Real-Time Analysis; Academic Press: Cambridge, MA, USA, 1999. [Google Scholar]
- Bassani, J.; Bassani, R.A.; Bers, D.M. Calibration of indo-1 and resting intracellular [Ca] i in intact rabbit cardiac myocytes. Biophys. J. 1995, 68, 1453–1460. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Molino del Barrio, I.; Wilkins, G.C.; Meeson, A.; Ali, S.; Kirby, J.A. Breast Cancer: An Examination of the Potential of ACKR3 to Modify the Response of CXCR4 to CXCL12. Int. J. Mol. Sci. 2018, 19, 3592. https://doi.org/10.3390/ijms19113592
del Molino del Barrio I, Wilkins GC, Meeson A, Ali S, Kirby JA. Breast Cancer: An Examination of the Potential of ACKR3 to Modify the Response of CXCR4 to CXCL12. International Journal of Molecular Sciences. 2018; 19(11):3592. https://doi.org/10.3390/ijms19113592
Chicago/Turabian Styledel Molino del Barrio, Irene, Georgina C. Wilkins, Annette Meeson, Simi Ali, and John A. Kirby. 2018. "Breast Cancer: An Examination of the Potential of ACKR3 to Modify the Response of CXCR4 to CXCL12" International Journal of Molecular Sciences 19, no. 11: 3592. https://doi.org/10.3390/ijms19113592
APA Styledel Molino del Barrio, I., Wilkins, G. C., Meeson, A., Ali, S., & Kirby, J. A. (2018). Breast Cancer: An Examination of the Potential of ACKR3 to Modify the Response of CXCR4 to CXCL12. International Journal of Molecular Sciences, 19(11), 3592. https://doi.org/10.3390/ijms19113592