Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue

 ,
,  and
and
Abstract
1. Introduction
2. Peroxisome Proliferator-Activated Receptor γ (PPARγ)/Cyclooxygenase-2 (COX-2) Expression in Tumors
3. PPARγ Expression in Tumor Stroma
4. Induction of Anakoinosis with Master Modulators
5. Keys for Uncovering the Therapeutic Potential of PPARγ Agonists: Selecting the Appropriate, Histology-Independent Combination of Master Modulators
5.1. Poor Monoactivity of PPARγ Agonists Across Different Tumor Histologies
5.2. PPARγ Agonists in Pro-Anakoinotic Combination Therapy with Master Modulators
5.2.1. PPARγ Agonists Combined with Metronomic Low-Dose Chemotherapy/Demethylating Agents
5.2.2. PPARγ Agonists Plus Dexamethasone
5.2.3. PPARγ Agonists Plus All-Trans Retinoic Acid
5.2.4. PPARγ Agonists Plus Interferon-α
5.2.5. PPARγ Agonists Plus COX-2 Inhibitor
5.2.6. PPARγ Agonists and IMiDs
5.3. PPARγ Agonists in Pro-Anakoinotic Combination Therapy Combined with Targeted Therapy
5.3.1. Pioglitazone and Imatinib
5.3.2. PPARγ and Mechanistic Target of Rapamycin (mTOR) Inhibitor
6. Specific Methodological Aspects of Anakoinosis Inducing Therapies
6.1. Communication Tools
6.2. What Is the Appropriate Model System: From Histology to ‘Evolution-Adjusted’ Tumor Pathophysiology?
6.3. What Is the Appropriate Dosage of Pro-Anakoinotic Therapy?
6.4. Pro-Anakoinotic Therapy Schedules: Indications and Diagnostics
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.A.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefèbvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Walter, I.; Schulz, U.; Vogelhuber, M.; Wiedmann, K.; Endlicher, E.; Klebl, F.; Andreesen, R.; Herr, W.; Ghibelli, L.; Hackl, C.; et al. Communicative reprogramming non-curative hepatocellular carcinoma with low-dose metronomic chemotherapy, COX-2 inhibitor and PPAR-γ agonist: A phase II trial. Med. Oncol. 2017, 34, 192. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.; Schrettenbrunner, I.; Vogelhuber, M.; Grassinger, J.; Bross, K.; Wilke, J.; Suedhoff, T.; Berand, A.; Wieland, W.F.; Rogenhofer, S.; et al. Pioglitazone, etoricoxib, interferon-α, and metronomic capecitabine for metastatic renal cell carcinoma: Final results of a prospective phase II trial. Med. Oncol. 2012, 29, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, V.K.; Bolt, M.J.; White, K.P. Nuclear receptors in cancer—Uncovering new and evolving roles through genomic analysis. Nat. Rev. Genet. 2018, 19, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Capper, C.P.; Rae, J.M.; Auchus, R.J. The metabolism, analysis, and targeting of steroid hormones in breast and prostate cancer. Horm. Cancer 2016, 7, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Kfir-Erenfeld, S.; Yefenof, E. Non-genomic events determining the sensitivity of hemopoietic malignancies to glucocorticoid-induced apoptosis. Cancer Immunol. Immunother. 2014, 63, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Spokoini, R.; Kfir, R.S.; Cohen, O.; Yefenof, E. Mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoid-induced apoptosis. Adv. Cancer Res. 2008, 101, 127–248. [Google Scholar] [PubMed]
- Photiou, L.; van der Weyden, C.; McCormack, C.; Miles Prince, H. Systemic treatment options for advanced-stage mycosis fungoides and sézary syndrome. Curr. Oncol. Rep. 2018, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Avvisati, G.; Cicconi, L.; Thiede, C.; Paoloni, F.; Vignetti, M.; Ferrara, F.; Divona, M.; Albano, F.; Efficace, F.; et al. Improved Outcomes With Retinoic Acid and Arsenic Trioxide Compared With Retinoic Acid and Chemotherapy in Non-High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J. Clin. Oncol. 2017, 35, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.; Vogelhuber, M.; Wolff, D.; Klobuch, S.; Ghibelli, L.; Foell, J.; Corbacioglu, S.; Rehe, K.; Haegeman, G.; Thomas, S.; et al. Anakoinosis: Communicative Reprogramming of Tumor Systems—for Rescuing from Chemorefractory Neoplasia. Cancer Microenviron. 2015, 8, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Hildebrandt, G.C. Principles of modular tumor therapy. Cancer Microenviron. 2009, 2, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor γ and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activated receptors and cancers: Complex stories. Nat. Rev. Cancer 2004, 4, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A. (Ed.) From Molecular to Modular Tumor Therapy; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural Peroxisome Proliferator-Activated Receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Exp. Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Bosscher, K. Selective Glucocorticoid Receptor modulators. J. Steroid Biochem. Mol. Biol. 2010, 120, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Lonard, D.M.; O’Malley, B.W. Nuclear receptor coactivators: Master regulators of human health and disease. Ann. Rev. Med. 2014, 65, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Akbiyik, F.; Ray, D.M.; Gettings, K.F.; Blumberg, N.; Francis, C.W.; Phipps, R.P. Human bone marrow megakaryocytes and platelets express PPARγ, and PPARγ agonists blunt platelet release of CD40 ligand and thromboxanes. Blood 2004, 104, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.-g.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.-D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Koeffler, H.P. Peroxisome proliferator-activated receptor γ and cancers. Clin. Cancer Res. 2003, 9, 1–9. [Google Scholar] [PubMed]
- Danielian, P.S.; White, R.; Lees, J.A.; Parker, M.G. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992, 11, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Zannini, A.; Ingallina, E.; Bertolio, R.; Marotta, C.; Neri, C.; Cappuzzello, E.; Forcato, M.; Rosato, A.; et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat. Commun. 2017, 8, 14073. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.S.; Badr, M.Z.; Badawi, A.F. Inhibition of cyclooxygenase-2 and activation of peroxisome proliferator-activated receptor-γ synergistically induces apoptosis and inhibits growth of human breast cancer cells. Int. J. Mol. Med. 2003, 11, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Lenhard, J.M.; Oliver, B.B.; Ringold, G.M.; Kliewer, S.A. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997, 272, 3406–3410. [Google Scholar] [CrossRef] [PubMed]
- Gelman, L.; Fruchart, J.C.; Auwerx, J. An update on the mechanisms of action of the peroxisome proliferator-activated receptors (PPARs) and their roles in inflammation and cancer. Cell. Mol. life Sci. 1999, 55, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Lin, D.T.; Hart, J.C.; Dannenberg, A.J. Peroxisome proliferator-activated receptor γ ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J. Biol. Chem. 2001, 276, 12440–12448. [Google Scholar] [CrossRef] [PubMed]
- Badawi, A.F.; Badr, M.Z. Expression of cyclooxygenase-2 and peroxisome proliferator-activated receptor-γ and levels of prostaglandin E2 and 15-deoxy-delta12,14-prostaglandin J2 in human breast cancer and metastasis. Int. J. Cancer 2003, 103, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Han, J.S.; Seo, C.Y.; Park, T.H.; Kwon, H.C.; Jeong, J.S.; Kim, I.H.; Yun, J.; Bae, Y.S.; Kwak, J.Y.; et al. Pioglitazone, a synthetic ligand for PPARγ, induces apoptosis in RB-deficient human colorectal cancer cells. Apoptosis 2006, 11, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Clay, C.E.; Namen, A.M.; Atsumi, G.; Willingham, M.C.; High, K.P.; Kute, T.E.; Trimboli, A.J.; Fonteh, A.N.; Dawson, P.A.; Chilton, F.H. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis 1999, 20, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Okano, J.; Murawaki, Y. Mechanisms of anti-proliferative effect of JTE-522, a selective cyclooxygenase-2 inhibitor, on human liver cancer cells. Oncol. Rep. 2007, 18, 1281–1290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meyer, S.; Vogt, T.; Landthaler, M.; Berand, A.; Reichle, A.; Bataille, F.; Marx, A.H.; Menz, A.; Hartmann, A.; Kunz-Schughart, L.A.; et al. Cyclooxygenase 2 (COX2) and peroxisome proliferator-activated receptor γ (PPARG) are stage-dependent prognostic markers of malignant melanoma. PPAR Res. 2009, 2009, 848645. [Google Scholar] [PubMed]
- Bundscherer, A.; Reichle, A.; Hafner, C.; Meyer, S.; Vogt, T. Targeting the tumor stroma with peroxisome proliferator activated receptor (PPAR) agonists. ACAMC 2009, 9, 816–821. [Google Scholar] [CrossRef]
- Knower, K.C.; Chand, A.L.; Eriksson, N.; Takagi, K.; Miki, Y.; Sasano, H.; Visvader, J.E.; Lindeman, G.J.; Funder, J.W.; Fuller, P.J.; et al. Distinct nuclear receptor expression in stroma adjacent to breast tumors. Breast Cancer Res. Treat. 2013, 142, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Basu-Roy, U.; Han, E.; Rattanakorn, K.; Gadi, A.; Verma, N.; Maurizi, G.; Gunaratne, P.H.; Coarfa, C.; Kennedy, O.D.; Garabedian, M.J.; et al. PPARγ agonists promote differentiation of cancer stem cells by restraining YAP transcriptional activity. Oncotarget 2016, 7, 60954–60970. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of nuclear receptors with the Wnt/β-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Opposite Interplay Between the Canonical WNT/β-Catenin Pathway and PPARγ: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018, 34, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Hong, O.-Y.; Youn, H.J.; Jang, H.-Y.; Jung, S.H.; Noh, E.-M.; Chae, H.S.; Jeong, Y.-J.; Kim, W.; Kim, C.-H.; Kim, J.-S. Troglitazone inhibits matrix metalloproteinase-9 expression and invasion of breast cancer cell through a peroxisome proliferator-activated receptor γ-dependent mechanism. J. Breast Cancer 2018, 21, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Domschke, C.; Stoiber, N.; Schott, S.; Heil, J.; Rom, J.; Blumenstein, M.; Thum, J.; Sohn, C.; Schneeweiss, A.; et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: Immunological effects and clinical outcome. Cancer Immunol. Immunother. 2012, 61, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Singer, S.; Forman, B.M.; Sarraf, P.; Fletcher, J.A.; Fletcher, C.D.; Brun, R.P.; Mueller, E.; Altiok, S.; Oppenheim, H.; et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Fletcher, C.D.M.; Mueller, E.; Sarraf, P.; Naujoks, R.; Campbell, N.; Spiegelman, B.M.; Singer, S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-γ ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. USA 1999, 96, 3951–3956. [Google Scholar] [CrossRef] [PubMed]
- Debrock, G.; Vanhentenrijk, V.; Sciot, R.; Debiec-Rychter, M.; Oyen, R.; van Oosterom, A. A phase II trial with rosiglitazone in liposarcoma patients. Br. J. Cancer 2003, 89, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Coras, B.; Hafner, C.; Reichle, A.; Hohenleutner, U.; Szeimies, R.-M.; Landthaler, M.; Vogt, T. Antiangiogenic therapy with pioglitazone, rofecoxib, and trofosfamide in a patient with endemic kaposi sarcoma. Arch. Dermatol. 2004, 140, 1504–1507. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T.; Hafner, C.; Bross, K.; Bataille, F.; Jauch, K.-W.; Berand, A.; Landthaler, M.; Andreesen, R.; Reichle, A. Antiangiogenetic therapy with pioglitazone, rofecoxib, and metronomic trofosfamide in patients with advanced malignant vascular tumors. Cancer 2003, 98, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Demetri, G.D.; Mueller, E.; Sarraf, P.; Spiegelman, B.M.; Winer, E.P. Use of the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Res. Treat. 2003, 79, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.; Vogelhuber, M.; Hafner, C.; Landthaler, M.; Berneburg, M.; Haferkamp, S.; Herr, W.; Reichle, A. Biomodulatory metronomic therapy in stage IV melanoma is well-tolerated and may induce prolonged progression-free survival, a phase I trial. J. Eur. Acad. Dermatol. Venereol. 2016, 30, e119–e121. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Vogt, T.; Coras, B.; Terheyden, P.; Neuber, K.; Trefzer, U.; Schultz, E.; Berand, A.; Bröcker, E.B.; Landthaler, M.; et al. Targeted combined anti-inflammatory and angiostatic therapy in advanced melanoma: A randomized phase II trial. Melanoma Res. 2007, 17, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Demetri, G.D.; Sharpless, N.E.; Ryan, D.P.; Shivdasani, R.; Clark, J.S.; Spiegelman, B.M.; Kim, H.; Mayer, R.J.; Fuchs, C.S. A phase II study of troglitazone, an activator of the PPARγ receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002, 8, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Grassinger, J.; Bross, K.; Wilke, J.; Suedhoff, T.; Walter, B.; Wieland, W.-F.; Berand, A.; Andreesen, R. C-reactive protein in patients with metastatic clear cell renal carcinoma: An important biomarker for tumor-associated inflammation. Biomark. Insights 2007, 1, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.; Smith, M.; Sarraf, P.; Kroll, T.; Aiyer, A.; Kaufman, D.S.; Oh, W.; Demetri, G.; Figg, W.D.; Zhou, X.P.; et al. Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 10990–10995. [Google Scholar] [CrossRef] [PubMed]
- Vogelhuber, M.; Feyerabend, S.; Stenzl, A.; Suedhoff, T.; Schulze, M.; Huebner, J.; Oberneder, R.; Wieland, W.; Mueller, S.; Eichhorn, F.; et al. Biomodulatory treatment of patients with castration-resistant prostate cancer: A phase II study of imatinib with pioglitazone, etoricoxib, dexamethasone and low-dose treosulfan. Cancer Microenviron. 2015, 8, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.; Rogenhofer, S.; Vogelhuber, M.; Berand, A.; Wieland, W.F.; Andreesen, R.; Reichle, A. Modular therapy approach in metastatic castration-refractory prostate cancer. World J. Urol. 2010, 28, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T.; Coras, B.; Hafner, C.; Landthaler, M.; Reichle, A. Antiangiogenic therapy in metastatic prostate carcinoma complicated by cutaneous lupus erythematodes. Lancet Oncol. 2006, 7, 695–697. [Google Scholar] [CrossRef]
- Smith, M.R.; Manola, J.; Kaufman, D.S.; George, D.; Oh, W.K.; Mueller, E.; Slovin, S.; Spiegelman, B.; Small, E.; Kantoff, P.W. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer 2004, 101, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Lugner, A.; Ott, C.; Klebl, F.; Vogelhuber, M.; Berand, A.; Andreesen, R. Control of cancer-associated inflammation and survival: Results from a prospective randomized phase II trial in gastric cancer. J. Clin. Oncol. 2009, 27, e15584. [Google Scholar]
- Hau, P.; Kunz-Schughart, L.; Bogdahn, U.; Baumgart, U.; Hirschmann, B.; Weimann, E.; Muhleisen, H.; Ruemmele, P.; Steinbrecher, A.; Reichle, A. Low-dose chemotherapy in combination with COX-2 inhibitors and PPAR-γ agonists in recurrent high-grade gliomas—A phase II study. Oncology 2007, 73, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Hart, C.; Grube, M.; Andreesen, R. Anti-inflammatory, immuno-modulatory and angiostatic treatment as third-line therapy for multiple myeloma (MM)—A combined treatment setting of lenalidomide with pioglitazone, dexamethasone and low-dose treosulfan (phase I/II). Blood 2012, 120, 5029. [Google Scholar]
- Heudobler, D.; Rehe, K.; Foell, J.; Corbacioglu, S.; Hildebrandt, G.; Herr, W.; Reichle, A.; Vogelhuber, M. Biomodulatory metronomic therapy shows remarkable activity in chemorefractory multi-system langerhans cell histiocytosis. Blood 2016, 128, 4254. [Google Scholar]
- Reichle, A.; Vogt, T.; Kunz-Schughart, L.; Bretschneider, T.; Bachthaler, M.; Bross, K.; Freund, S.; Andreesen, R. Anti-inflammatory and angiostatic therapy in chemorefractory multisystem Langerhans’ cell histiocytosis of adults. Br. J. Haematol. 2005, 128, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Ugocsai, P.; Wolff, D.; Menhart, K.; Hellwig, D.; Holler, E.; Herr, W.; Reichle, A. Biomodulatory metronomic therapy induces PET-negative remission in chemo- and brentuximab-refractory Hodgkin lymphoma. Br. J. Haematol. 2016, 172, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.-P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Schelker, R.; Klobuch, S.; Zaiss, S.; Troppmann, M.; Rehli, M.; Haferlach, T.; Herr, W.; Reichle, A. Biomodulatory therapy induces complete molecular remission in chemorefractory acute myeloid leukemia. Haematologica 2015, 100, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Heudobler, D.; Klobuch, S.; Thomas, S.; Hahn, J.; Herr, W.; Reichle, A. Cutaneous leukemic infiltrates successfully treated with biomodulatory therapy in a rare case of therapy-related high risk MDS/AML. Front. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Simkens, L.H.J.; van Tinteren, H.; May, A.; Tije, A.J.; Creemers, G.-J.M.; Loosveld, O.J.L.; Jongh, F.E.; Erdkamp, F.L.G.; van der Torren, A.M.; Tol, J. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): A phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 2015, 385, 1843–1852. [Google Scholar] [CrossRef]
- Pramanik, R.; Agarwala, S.; Gupta, Y.K.; Thulkar, S.; Vishnubhatla, S.; Batra, A.; Dhawan, D.; Bakhshi, S. Metronomic Chemotherapy vs Best Supportive Care in Progressive Pediatric Solid Malignant Tumors: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Rochlitz, C.; Bigler, M.; Moos, R.; Bernhard, J.; Matter-Walstra, K.; Wicki, A.; Zaman, K.; Anchisi, S.; Küng, M.; Na, K.-J.; et al. SAKK 24/09: Safety and tolerability of bevacizumab plus paclitaxel vs. bevacizumab plus metronomic cyclophosphamide and capecitabine as first-line therapy in patients with HER2-negative advanced stage breast cancer—A multicenter, randomized phase III trial. BMC Cancer 2016, 16, 780. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Wade, J.L.; Oza, A.M.; Sullivan, D.; Chen, A.P.; Gandara, D.R.; Ji, J.; Kinders, R.J.; Wang, L.; Allen, D.; et al. Randomized phase II trial of cyclophosphamide and the oral poly (ADP-ribose) polymerase inhibitor veliparib in patients with recurrent, advanced triple-negative breast cancer. Investig. New Drugs 2016, 34, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Bottini, A.; Generali, D.; Brizzi, M.P.; Fox, S.B.; Bersiga, A.; Bonardi, S.; Allevi, G.; Aguggini, S.; Bodini, G.; Milani, M.; et al. Randomized phase II trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J. Clin. Oncol. 2006, 24, 3623–3628. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.L.; Iwamoto, F.M.; Sul, J.; Panageas, K.; Lassman, A.B.; DeAngelis, L.M.; Hormigo, A.; Nolan, C.P.; Gavrilovic, I.; Karimi, S.; et al. Randomized phase II trial of chemoradiotherapy followed by either dose-dense or metronomic temozolomide for newly diagnosed glioblastoma. J. Clin. Oncol. 2009, 27, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Senerchia, A.A.; Macedo, C.R.; Ferman, S.; Scopinaro, M.; Cacciavillano, W.; Boldrini, E.; Lins de Moraes, V.L.; Rey, G.; Oliveira, C.T.; Castillo, L.; et al. Results of a randomized, prospective clinical trial evaluating metronomic chemotherapy in nonmetastatic patients with high-grade, operable osteosarcomas of the extremities: A report from the Latin American Group of Osteosarcoma Treatment. Cancer 2017, 123, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Andreeff, M. Role of peroxisome proliferator-activated receptor-γ in hematologic malignancies. Curr. Opin. Hematol. 2002, 9, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Elrod, H.A.; Sun, S.-Y. PPARγ and Apoptosis in Cancer. PPAR Res. 2008, 2008, 704165. [Google Scholar] [CrossRef] [PubMed]
- Nemenoff, R.A.; Winn, R.A. Role of nuclear receptors in lung tumourigenesis. Eur. J. Cancer 2005, 41, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Rumi, M.A.K.; Ishihara, S.; Kazumori, H.; Kadowaki, Y.; Kinoshita, Y. Can PPARγ ligands be used in cancer therapy? Curr. Med. Chem. 2004, 4, 465–477. [Google Scholar]
- Schmidt, M.V.; Brüne, B.; Knethen, A. The nuclear hormone receptor PPARγ as a therapeutic target in major diseases. Sci. World J. 2010, 10, 2181–2197. [Google Scholar] [CrossRef] [PubMed]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Haematol. 2011, 164, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Skelhorne-Gross, G.; Nicol, C.J.B. The Key to Unlocking the Chemotherapeutic Potential of PPARγ Ligands: Having the Right Combination. PPAR Res. 2012, 2012, 946943. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Bencini, L.; Galli, A. Peroxisome proliferator activated receptors at the crossroad of obesity, diabetes, and pancreatic cancer. World J. Gastroenterol. 2016, 22, 2441–2459. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Nagelli, L.V.; Rosen, S.T.; Kuzel, T.M.; Guitart, J. Bexarotene in the treatment of cutaneous T-cell lymphoma. Exp. Opin. Pharmacother. 2006, 7, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Wahl, R. Chemotherapy and chemoprevention by thiazolidinediones. BioMed Res. Int. 2015, 2015, 845340. [Google Scholar] [CrossRef] [PubMed]
- Higurashi, T.; Hosono, K.; Takahashi, H.; Komiya, Y.; Umezawa, S.; Sakai, E.; Uchiyama, T.; Taniguchi, L.; Hata, Y.; Uchiyama, S.; et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: A multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016, 17, 475–483. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Di, H.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; Leboffe, L.; Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Aspects Med. 2015, 41, 1–115. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.D.; Richards, S.M.; Kinsey, S.E.; Lilleyman, J.; Vora, A.; Eden, T.O.B. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: Results of the UK Medical Research Council ALL97 randomized trial. Br. J. Haematol. 2005, 129, 734–745. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Regan, M.M.; Clark, J.I.; Flaherty, L.E.; Weiss, G.R.; Logan, T.F.; Kirkwood, J.M.; Gordon, M.S.; Sosman, J.A.; Ernstoff, M.S.; et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2005, 23, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Pineda, C.M.; Xin, T.; Boucher, J.; Suozzi, K.C.; Park, S.; Matte-Martone, C.; Gonzalez, D.G.; Rytlewski, J.; Beronja, S.; et al. Correction of aberrant growth preserves tissue homeostasis. Nature 2017, 548, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.C.; Man, S.; Xu, P.; Francia, G.; Hashimoto, K.; Emmenegger, U.; Kerbel, R.S. Development of a resistance-like phenotype to sorafenib by human hepatocellular carcinoma cells is reversible and can be delayed by metronomic UFT chemotherapy. Neoplasia 2010, 12, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, X.; Wang, Q.; Xue, P.; Jiao, X.; Peng, H.; Lu, H.; Zheng, Q.; Chen, X.; Huang, X.; et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARγ signaling pathway. Acta Pharmacol. Sin. 2009, 30, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-q.; Chen, X.-l.; Wang, Q.; Huang, X.-h.; Zhen, M.-C.; Zhang, L.-J.; Li, W.; Bi, J. Upregulation of PTEN involved in rosiglitazone-induced apoptosis in human hepatocellular carcinoma cells. Acta Pharmacol. Sin. 2007, 28, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Yap, R.; Veliceasa, D.; Emmenegger, U.; Kerbel, R.S.; McKay, L.M.; Henkin, J.; Volpert, O.V. Metronomic low-dose chemotherapy boosts CD95-dependent antiangiogenic effect of the thrombospondin peptide ABT-510: A complementation antiangiogenic strategy. Clin. Cancer Res. 2005, 11, 6678–6685. [Google Scholar] [CrossRef] [PubMed]
- Biziota, E.; Briasoulis, E.; Mavroeidis, L.; Marselos, M.; Harris, A.L.; Pappas, P. Cellular and molecular effects of metronomic vinorelbine and 4-O-deacetylvinorelbine on human umbilical vein endothelial cells. Anti-Cancer Drugs 2016, 27, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Lozach, J.; Benner, C.; Pascual, G.; Tangirala, R.K.; Westin, S.; Hoffmann, A.; Subramaniam, S.; David, M.; Rosenfeld, M.G.; et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell 2005, 122, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.; Richardson, P.G.; Treon, S.P.; Anderson, K.C. Novel biologically based therapies for Waldenstrom’s macroglobulinemia. Semin. Oncol. 2003, 30, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Hisatake, J.I.; Ikezoe, T.; Carey, M.; Holden, S.; Tomoyasu, S.; Koeffler, H.P. Down-Regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor γ in human prostate cancer. Cancer Res. 2000, 60, 5494–5498. [Google Scholar] [PubMed]
- Narayanan, S.; Srinivas, S.; Feldman, D. Androgen-glucocorticoid interactions in the era of novel prostate cancer therapy. Nat. Rev. Urol. 2016, 13, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.; Schwartz, D.; Judd, R.; Akingbemi, B.; Braden, T.; Morrison, E.; Dennis, J.; Bartol, F.; Hazi, A.; Napier, I.; et al. Thiazolidinediones/PPARγ agonists and fatty acid synthase inhibitors as an experimental combination therapy for prostate cancer. Int. J. Oncol. 2011, 38, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012, 19, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Rocchi, P.; Ferreri, A.M.; Orlandi, M. RXRγ and PPARγ ligands in combination to inhibit proliferation and invasiveness in colon cancer cells. Cancer Lett. 2010, 297, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Storci, G.; Guarnieri, T.; Carolis, S.; Bertoni, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; Santini, D.; Ceccarelli, C.; et al. Peroxisome proliferator activated receptor-α/hypoxia inducible factor-1α interplay sustains carbonic anhydrase IX and apoliprotein E expression in breast cancer stem cells. PLoS ONE 2013, 8, e54968. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Tatenhorst, L.; Terwel, D.; Hermes, M.; Kummer, M.P.; Orlandi, M.; Heneka, M.T. PPARγ and RXRγ ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J. Neurochem. 2009, 109, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Elstner, E.; McQueen, T.J.; Tsao, T.; Sudarikov, A.; Hu, W.; Schober, W.D.; Wang, R.-Y.; Chism, D.; Kornblau, S.M.; et al. Peroxisome proliferator-activated receptor γ and retinoid X receptor ligands are potent inducers of differentiation and apoptosis in leukemias. Mol. Cancer Ther. 2004, 3, 1249–1262. [Google Scholar] [PubMed]
- Thiounn, N.; Pages, F.; Flam, T.; Tartour, E.; Mosseri, V.; Zerbib, M.; Beuzeboc, P.; Deneux, L.; Fridman, W.H.; Debré, B. IL-6 is a survival prognostic factor in renal cell carcinoma. Immunol. Lett. 1997, 58, 121–124. [Google Scholar] [CrossRef]
- Tilg, H.; Vogel, W.; Dinarello, C.A. Interferon-α induces circulating tumor necrosis factor receptor p55 in humans. Blood 1995, 85, 433–435. [Google Scholar] [CrossRef]
- Jabs, W.J.; Busse, M.; Krüger, S.; Jocham, D.; Steinhoff, J.; Doehn, C. Expression of C-reactive protein by renal cell carcinomas and unaffected surrounding renal tissue. Kidney Int. 2005, 68, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Buer, J.; Probst, M.; Ganser, A.; Atzpodien, J. Response to 13-cis-retinoic acid plus interferon alfa-2a in two patients with therapy-refractory advanced renal cell carcinoma. J. Clin. Oncol. 1995, 13, 2679–2680. [Google Scholar] [CrossRef] [PubMed]
- Aviles, A.; Neri, N.; Fernandez-Diez, J.; Silva, L.; Nambo, M.-J. Interferon and low doses of methotrexate versus interferon and retinoids in the treatment of refractory/relapsed cutaneous T-cell lymphoma. Hematology 2015, 20, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Finch, E.R.; Tukaramrao, D.B.; Goodfield, L.L.; Quickel, M.D.; Paulson, R.F.; Prabhu, K.S. Activation of PPARγ by endogenous prostaglandin J2 mediates the antileukemic effect of selenium in murine leukemia. Blood 2017, 129, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.-H.; Chan, H.-H.; Kuo, H.-M.; Liu, L.-F.; Hu, T.-H.; Sun, C.-K.; Kung, M.-L.; Lin, S.-W.; Wang, E.-M.; Ma, Y.-L.; et al. Celecoxib suppresses hepatoma stemness and progression by up-regulating PTEN. Oncotarget 2014, 5, 1475–1490. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Han, C.; Lim, K.; Wu, T. Cross-talk between peroxisome proliferator-activated receptor delta and cytosolic phospholipase A(2)α/cyclooxygenase-2/prostaglandin E(2) signaling pathways in human hepatocellular carcinoma cells. Cancer Res. 2006, 66, 11859–11868. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhou, L.; Gao, S.; Yang, Z.; Yao, J.; Zheng, S. Prognostic role of C-reactive protein in hepatocellular carcinoma: A systematic review and meta-analysis. Int. J. Med. Sci. 2013, 10, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.; Kruger, W.D. Suppression of tumor formation by a cyclooxygenase-2 inhibitor and a peroxisome proliferator-activated receptor γ agonist in an in vivo mouse model of spontaneous breast cancer. Clin. Cancer Res. 2008, 14, 4935–4942. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mai, K.; Zhang, Y.; Xu, W.; Ai, Q. Tumour necrosis factor-α inhibits hepatic lipid deposition through GSK-3β/β-catenin signaling in juvenile turbot (Scophthalmus maximus L.). Gen. Comp. Endocrinol. 2015, 228, 1–8. [Google Scholar] [CrossRef] [PubMed]
- DeCicco, K.L.; Tanaka, T.; Andreola, F.; Luca, L.M. The effect of thalidomide on non-small cell lung cancer (NSCLC) cell lines: Possible involvement in the PPARγ pathway. Carcinogenesis 2004, 25, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Roman, J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARγ-dependent and PPARγ-independent signal pathways. Mol. Cancer Ther. 2006, 5, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Reichle, A.; Vogt, T. New indications for established drugs: Combined tumor-stroma-targeted cancer therapy with PPARγ agonists, COX-2 inhibitors, mTOR antagonists and metronomic chemotherapy. CCDT 2005, 5, 393–419. [Google Scholar] [CrossRef]
- Reichle, A. Evolution-adjusted Tumor Pathophysiology; Springer: Dordrecht, The Netherlands, 2013. [Google Scholar]
- Muqaku, B.; Eisinger, M.; Meier, S.M.; Tahir, A.; Pukrop, T.; Haferkamp, S.; Slany, A.; Reichle, A.; Gerner, C. Multi-omics Analysis of Serum Samples Demonstrates Reprogramming of Organ Functions Via Systemic Calcium Mobilization and Platelet Activation in Metastatic Melanoma. Mol. Cell. Proteom. 2017, 16, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Hanash, S.M.; Pitteri, S.J.; Faca, V.M. Mining the plasma proteome for cancer biomarkers. Nature 2008, 452, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A. Tumor Systems Need to be Rendered Usable for a New Action-Theoretical Abstraction: The Starting Point for Novel Therapeutic Options. In From Molecular to Modular Tumor Therapy; Reichle, A., Ed.; Springer Netherlands: Dordrecht, The Netherlands, 2010; pp. 9–28. [Google Scholar]



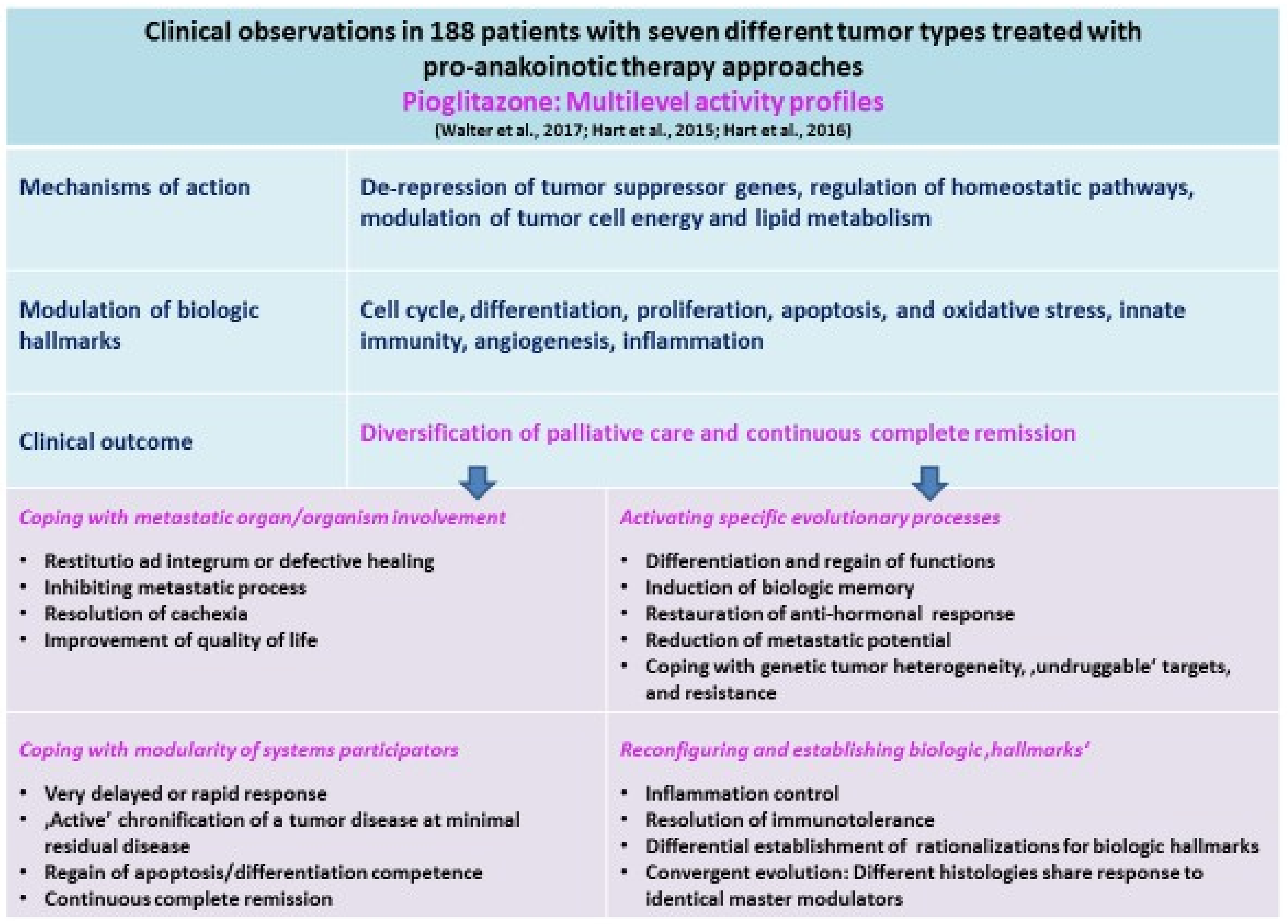{kind=link}
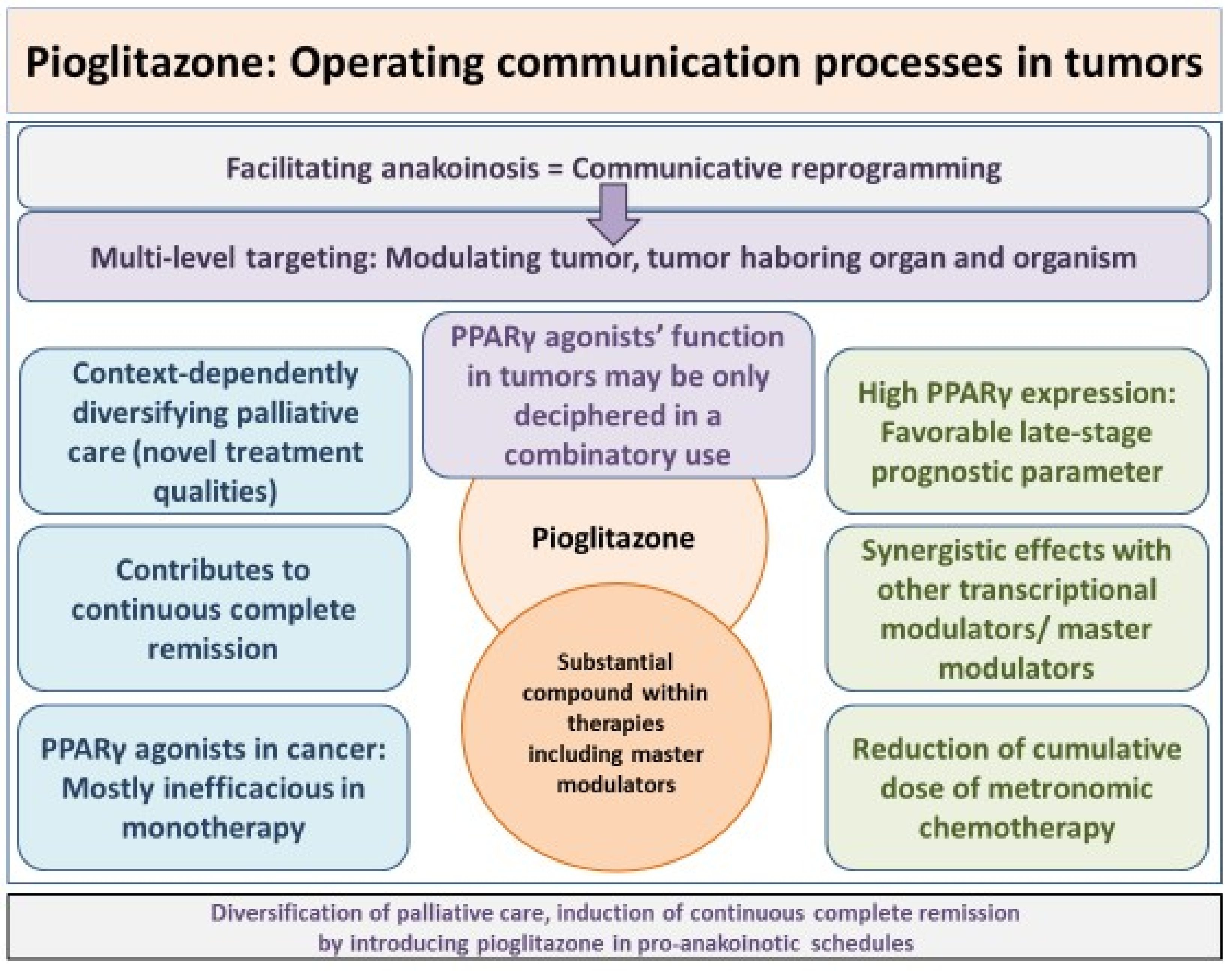{kind=link}
| Communication-Associated Terms | Explanation |
|---|---|
| Anakoinosis | Anakoinosis is a novel paradigm for cancer treatment based on a key role for communicative reprogramming of tumor systems. Building on a systems biology approach to cancer, anakoinosis utilizes a range of non-cancer and cancer drugs in combination to treat advanced tumor disease, such as pioglitazone. In contrast to standard therapies, anakoinosis protocols are characterized by low toxicity and a good safety profile, with encouraging responses in a number of clinical trials to date. The use of drug repurposing, that is the use of non-cancer drugs as cancer treatments, is especially a notable feature of this approach. |
| Pro-anakoinotic therapeutic tools (examples) | Transcriptional modulators, nuclear receptor agonists and antagonists, metronomic low-dose chemotherapy, cyclooxygenase-2 inhibitors, IMiDs, arsenic trioxide, liposomal encapsulated small oligonucleotide encoding small activating RNAs, etc. |
| Metronomic tumor therapy | Metronomic tumor therapy may be defined as the frequent administration of (repurposed) drugs at doses significantly below the maximum tolerated dose with no prolonged drug-free breaks, or as the minimum biologically effective dose of an agent given as a continuous dosing regimen with no prolonged drug-free breaks that still leads to anti-tumor activity. |
| Rationalizations | Describe the physical organization of tumor-associated normative notions (e.g. hallmarks of cancer); are to some degree histology- and genotype-independent; may be re-directed and reorganized by anakoinosis. |
| Metabolism of evolution | The sum of extrinsically, i.e., therapeutically, and intrinsically inducible evolutionary processes within the tumor environment (tumor stroma, hosting organ, distant organ sites). |
| Modularity | Modularity describes the degree and specificity to which systems’ objects, i.e. cells, pathways, molecules, therapeutic targets etc. may be communicatively rededicated by anakoinosis. |
| Validity and denotation | Validity of systems objects, functions and hubs: Availability on demand at distinct systems stages; denotation: Current functional impact at a distinct systems stage, e.g. of potentially tumor-promoting pathways. In the bio-world, presence and functioning of an object (e.g., an enzyme), respectively. |
| Glitazones in Refractory Tumors or Hematologic Neoplasia | ||||||
|---|---|---|---|---|---|---|
| Neoplasia | No pts | Chemotherapy (* = Metronomic) | Transcriptional Modulators | Small Molecule | Best Response | Reference |
| Sarcomas | ||||||
| Liposarcomas, intermediate to high-grade (case reports) | - | - | Troglitazone | - | Histological and biochemical differentiation | [45] |
| Liposarcoma | 3 | Trofosfamide * | Troglitazone | - | Lineage-appropriate differentiation can be induced pharmacologically in a human solid tumor. | [46] |
| Liposarcoma (Phase II study) | 12 | - | Rosiglitazone | - | Rosiglitazone is not effective as an antitumoral drug in the treatment of liposarcomas | [47] |
| Kaposi sarcoma, refractory | 1 | Trofosfamide * | Pioglitazone | COX-2 inhibitor | Partial remission | [48] |
| (Hem-)angiosarcomas | 12 | Trofosfamide * | Pioglitazone | COX-2 inhibitor | Continuos complete remission | [49] |
| Breast cancer | ||||||
| Refractory breast cancer (Phase II study) | 22 | - | Troglitazone | - | No significant effect | [50] |
| Melanoma | ||||||
| Melanoma III (versus DTIC), phase II Clinical Trials.gov:NCT01614301 | 6 | Trofosfamide * | Pioglitazone | Temsirolimus COX-2 inhibitor | Partial remission, Resolution of cachexia | [51] |
| Melanoma (randomized) | ||||||
| Melanoma II Arm M | 35 | Trofosfamide * | Pioglitazone | - | Stable disease | [52] |
| Arm A/M | 32 | Trofosfamide * | Pioglitazone | COX-2 inhibitor | Partial remission | |
| Hepatocellular carcinoma | ||||||
| Hepatocellular carcinoma | 38 | Capecitabine * | Pioglitazone | COX-2 inhibitor | Partial remission | [4] |
| Cholangiocellular carcinoma | ||||||
| Cholangiocellular carcinoma | 21 | Trofosfamide * | Pioglitazone | COX-2 inhibitor | Partial remission | [18] |
| Colorectal cancer | ||||||
| Chemotherapy-resistant metastatic colorectal cancer (phase II study) | 25 | - | Troglitazone | - | Not active for the treatment of metastatic colorectal cancer | [53] |
| Renal clear cell carcinoma (historic comparison) | ||||||
| Renal clear cell carcinoma, relapsed | 18 | Capecitabine * | Pioglitazone | COX-2 inhibitor | Partial remission | [54] |
| Renal clear cell carinoma, relapsed | 33 | Capecitabine * | Pioglitazone Interferon-alpha | COX-2 inhibitor | Continuous complete remission | [5] |
| Prostate cancer | ||||||
| Prostate cancer | 41 | - | Troglitazone | - | Lengthened stabilisation of prostate-specific antigen | [55] |
| Castration-resistant prostate cancer | 61 | Treosulfan * | Pioglitazone, Dexamethasone | COX-2 inhibitor Imatinib | Long-term tumor control at minimal disease | [56] |
| Castration-resistant prostate cancer | 36 | Capecitabine * | Pioglitazone, Dexamethasone | COX-2 inhibitor | Long-term tumor control | [57,58] |
| Prostate carcinoma (randomized) | ||||||
| Rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy | 106 | - | Rosiglitazone Versus Placebo | Rosiglitazone did not increase PSA doubling time or prolong the time to disease progression | [59] | |
| Gastric cancer (randomized) | ||||||
| Gastric cancer Arm A/M | 21 | Capecitabine * | Pioglitazone | COX-2 inhibitor | Partial remission | [60] |
| Arm M | 21 | Capecitabine * | Pioglitazone no impact | |||
| Glioblastoma | ||||||
| Glioblastoma, refractory | 14 | Capecitabine * | Pioglitazone | COX-2 inhibitor | Disease stabilization | [61] |
| Multiple myeloma | ||||||
| Multiple myeloma, third-line Clinicaltrials.gov, NCT001010243 | 6 | Treosulfan * | Pioglitazone, Dexamethasone | Lenalidomide | Complete remission | [62] |
| Langerhans cell histiocytosis | ||||||
| Langerhans cell histiocytosis, refractory | 2 + 7 | Trofosfamide * | Pioglitazone Dexamethasone | COX-2 inhibitor | Continuous complete remission | [13,63,64] |
| Hodgkin‘s lymphoma | ||||||
| Hodgkin lymphoma, refractory | 3 | Treosulfan * | Pioglitazone, Dexamethasone | COX-2 inhibitor Everolimus | Continuous complete remission | [65] |
| Chronic myelocytic leukemia | ||||||
| Chronic myelocytic leukemia without moleclar CR | 24 | - | Pioglitazone | Imatinib | Molecular complete remission (54%) | [66] |
| Acute myelocytic leukemia | ||||||
| Acute myelocytic leukemia Refractory (on-going trial) | 5 + 7 | Azacitidine | Pioglitazone All-trans retinoic acid | Molecular complete remission Myelodysplastic synrome with phagocytically active blasts | [67,68] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heudobler, D.; Rechenmacher, M.; Lüke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. Int. J. Mol. Sci. 2018, 19, 3540. https://doi.org/10.3390/ijms19113540
Heudobler D, Rechenmacher M, Lüke F, Vogelhuber M, Pukrop T, Herr W, Ghibelli L, Gerner C, Reichle A. Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. International Journal of Molecular Sciences. 2018; 19(11):3540. https://doi.org/10.3390/ijms19113540
Chicago/Turabian StyleHeudobler, Daniel, Michael Rechenmacher, Florian Lüke, Martin Vogelhuber, Tobias Pukrop, Wolfgang Herr, Lina Ghibelli, Christopher Gerner, and Albrecht Reichle. 2018. "Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue" International Journal of Molecular Sciences 19, no. 11: 3540. https://doi.org/10.3390/ijms19113540
APA StyleHeudobler, D., Rechenmacher, M., Lüke, F., Vogelhuber, M., Pukrop, T., Herr, W., Ghibelli, L., Gerner, C., & Reichle, A. (2018). Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. International Journal of Molecular Sciences, 19(11), 3540. https://doi.org/10.3390/ijms19113540