Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes—Mechanisms for Viral Survivability and Proliferation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
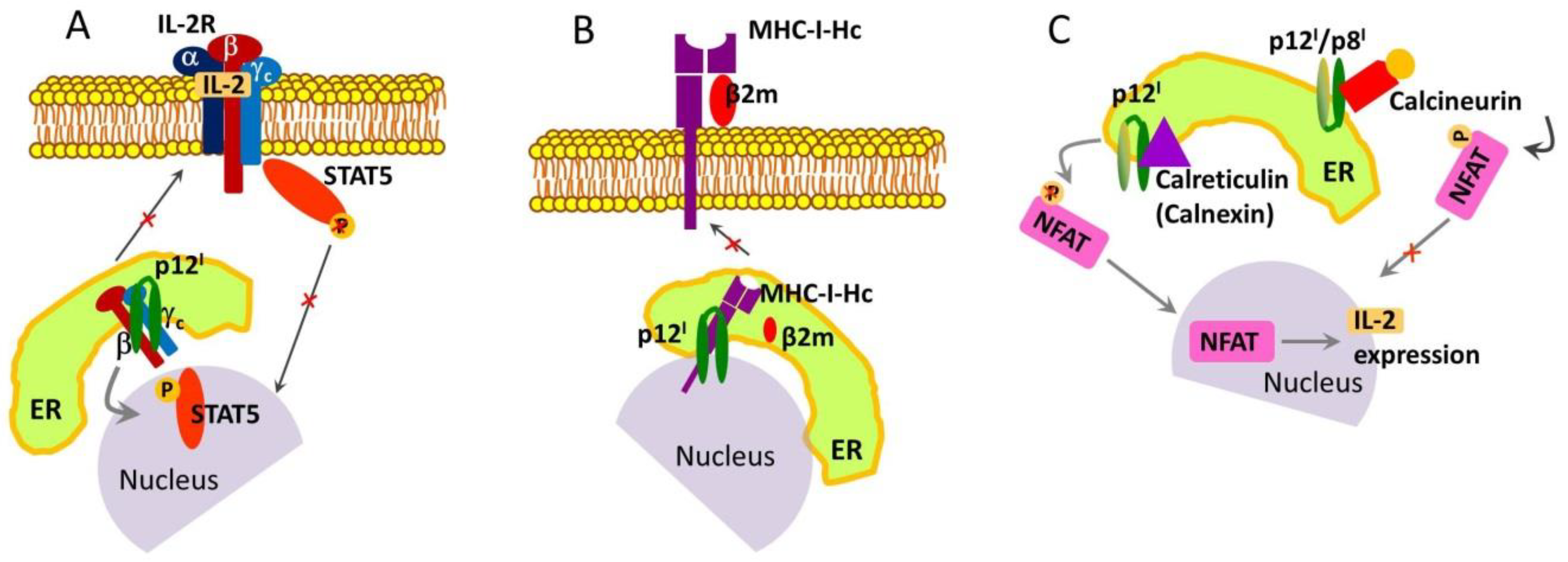
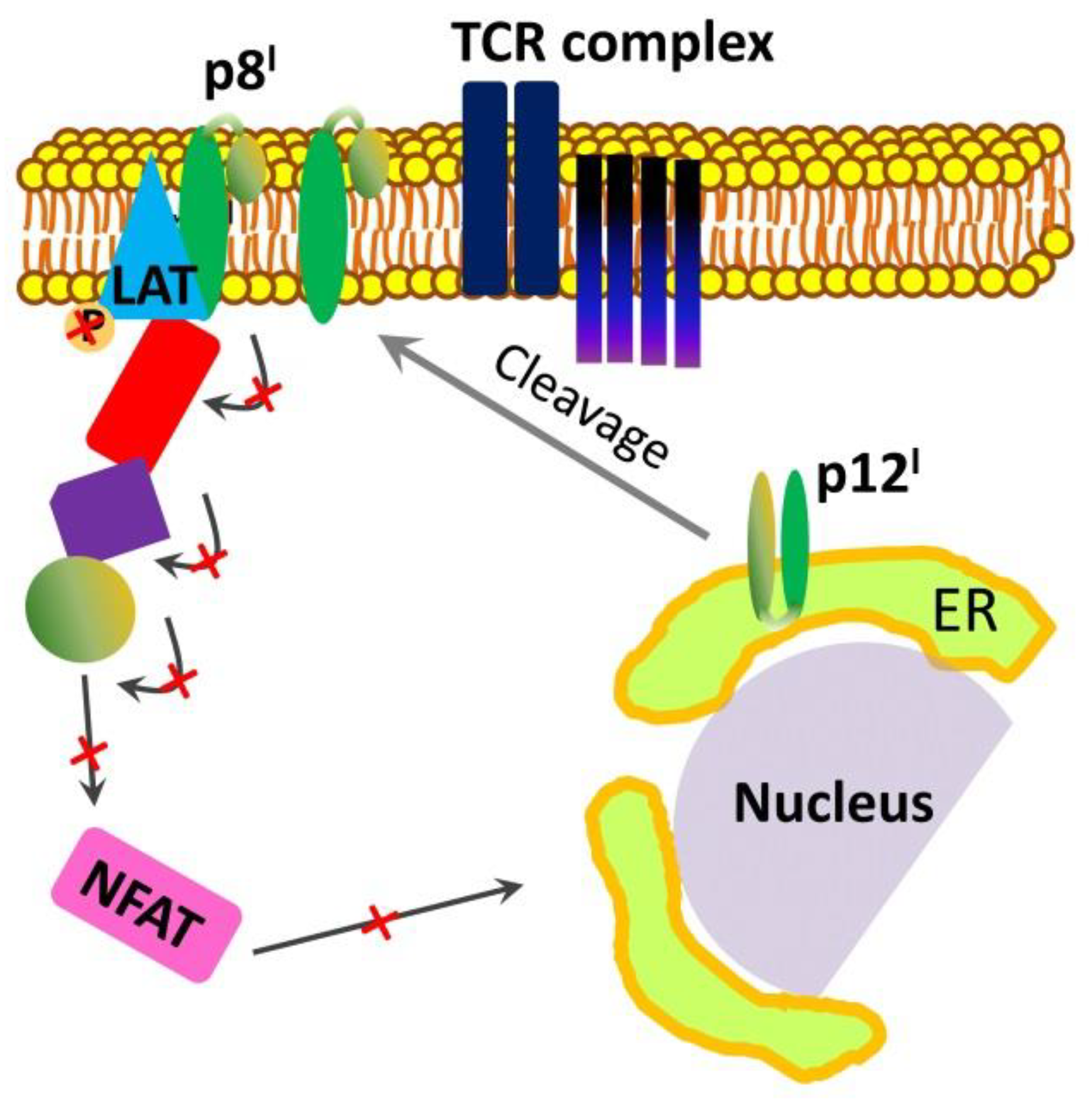
2. p12I and p8I Proteins and Their Roles in HTLV-1 Adaptation and Proliferation in the Host
2.1. Roles and Functional Mechanisms of p12I in the ER
2.2. Roles and Functional Mechanisms of p8I in the Plasma Membrane
3. p13II Protein and Its Role in the Control of Mitochondrial Apoptosis
4. Roles of Tax, HBZ, and p30II NSPs in HTLV-1 Life Cycle
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallo, R.C. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M. Discovery of HTLV-1, the first human retrovirus, its unique regulatory mechanisms, and insights into pathogenesis. Oncogene 2005, 24, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Barmak, K.; Harhaj, E.; Grant, C.; Alefantis, T.; Wigdahl, B. Human T cell leukemia virus type I-induced disease: Pathways to cancer and neurodegeneration. Virology 2003, 308, 1–12. [Google Scholar] [CrossRef]
- Haabeth, O.A.; Tveita, A.A.; Fauskanger, M.; Schjesvold, F.; Lorvik, K.B.; Hofgaard, P.O.; Omholt, H.; Munthe, L.A.; Dembic, Z.; Corthay, A.; et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front. Immunol. 2014, 5, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [PubMed]
- Ratner, L. Adult T cell leukemia lymphoma. Front. Biosci. 2004, 9, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.H.; Edwards, A.J.; Cruickshank, J.K.; Rudge, P.; Dalgleish, A.G. In vivo cellular tropism of human T-cell leukemia virus type 1. J. Virol. 1990, 64, 5682–5687. [Google Scholar] [PubMed]
- Taylor, G.P.; Matsuoka, M. Natural history of adult T-cell leukemia/lymphoma and approaches to therapy. Oncogene 2005, 24, 6047–6057. [Google Scholar] [CrossRef] [PubMed]
- Verdonck, K.; Gonzalez, E.; Van Dooren, S.; Vandamme, A.M.; Vanham, G.; Gotuzzo, E. Human T-lymphotropic virus 1: Recent knowledge about an ancient infection. Lancet Infect. Dis. 2007, 7, 266–281. [Google Scholar] [CrossRef]
- Cruickshank, J.K.; Rudge, P.; Dalgleish, A.G.; Newton, M.; McLean, B.N.; Barnard, R.O.; Kendall, B.E.; Miller, D.H. Tropical spastic paraparesis and human T cell lymphotropic virus type 1 in the United Kingdom. Brain 1989, 112 Pt 4, 1057–1090. [Google Scholar] [CrossRef]
- Godoy, A.J.; Kira, J.; Hasuo, K.; Goto, I. Characterization of cerebral white matter lesions of HTLV-I-associated myelopathy/tropical spastic paraparesis in comparison with multiple sclerosis and collagen-vasculitis: A semiquantitative MRI study. J. Neurol. Sci. 1995, 133, 102–111. [Google Scholar] [CrossRef]
- Levin, M.C.; Jacobson, S. HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP): A chronic progressive neurologic disease associated with immunologically mediated damage to the central nervous system. J. Neurovirol. 1997, 3, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Dourado, L. Endocrine and metabolic disorders in HTLV-1 infected patients. Braz. J. Infect. Dis. 2010, 14, 613–620. [Google Scholar] [CrossRef]
- Miyoshi, I.; Kubonishi, I.; Yoshimoto, S.; Akagi, T.; Ohtsuki, Y.; Shiraishi, Y.; Nagata, K.; Hinuma, Y. Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature 1981, 294, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed]
- Inaba, S.; Sato, H.; Okochi, K.; Fukada, K.; Takakura, F.; Tokunaga, K.; Kiyokawa, H.; Maeda, Y. Prevention of transmission of human T-lymphotropic virus type 1 (HTLV-1) through transfusion, by donor screening with antibody to the virus. One-year experience. Transfusion 1989, 29, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, K.; Kinoshita, K.; Moriuchi, R.; Katamine, S.; Amagasaki, T.; Ikeda, S.; Ichimaru, M.; Miyamoto, T.; Hino, S. Oral transmission of human T-cell leukemia virus type-I into a common marmoset (Callithrix jacchus) as an experimental model for milk-borne transmission. Jpn. J. Cancer Res. 1985, 76, 481–487. [Google Scholar] [PubMed]
- Hino, S.; Yamaguchi, K.; Katamine, S.; Sugiyama, H.; Amagasaki, T.; Kinoshita, K.; Yoshida, Y.; Doi, H.; Tsuji, Y.; Miyamoto, T. Mother-to-child transmission of human T-cell leukemia virus type-I. Jpn. J. Cancer Res. 1985, 76, 474–480. [Google Scholar] [PubMed]
- Kalyanaraman, V.S.; Sarngadharan, M.G.; Robert-Guroff, M.; Miyoshi, I.; Golde, D.; Gallo, R.C. A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia. Science 1982, 218, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Calattini, S.; Chevalier, S.A.; Duprez, R.; Bassot, S.; Froment, A.; Mahieux, R.; Gessain, A. Discovery of a new human T-cell lymphotropic virus (HTLV-3) in Central Africa. Retrovirology 2005, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, N.D.; Heneine, W.; Carr, J.K.; Garcia, A.D.; Shanmugam, V.; Tamoufe, U.; Torimiro, J.N.; Prosser, A.T.; Lebreton, M.; Mpoudi-Ngole, E.; et al. Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc. Natl. Acad. Sci. USA 2005, 102, 7994–7999. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.C.; Montagnier, L. The discovery of HIV as the cause of AIDS. N. Engl. J. Med. 2003, 349, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.L.; Fridey, J.; Smith, J.W.; Engstrom, J.; Sacher, R.A.; Miller, K.; Gibble, J.; Stevens, J.; Thomson, R.; Hansma, D.; et al. HTLV-associated myelopathy in a cohort of HTLV-I and HTLV-II-infected blood donors. The REDS investigators. Neurology 1997, 48, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.; Hall, W.W. Human T-lymphotropic virus type II and neurological disease. Ann. Neurol. 2004, 56, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Okada, M.; Koyanagi, Y.; Kannagi, M.; Hinuma, Y. Transformation of human leukocytes by cocultivation with an adult T cell leukemia virus producer cell line. Science 1982, 217, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.S.; Quan, S.G.; Golde, D.W. Human T-cell leukemia virus type II transforms normal human lymphocytes. Proc. Natl. Acad. Sci. USA 1983, 80, 7006–7009. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, R. Human T-lymphotropic virus type 1 non-structural proteins: Requirements for latent infection. Cancer Sci. 2013, 104, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Edlich, R.F.; Arnette, J.A.; Williams, F.M. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I). J. Emerg. Med. 2000, 18, 109–119. [Google Scholar] [CrossRef]
- Willems, L.; Hasegawa, H.; Accolla, R.; Bangham, C.; Bazarbachi, A.; Bertazzoni, U.; Carneiro-Proietti, A.B.; Cheng, H.; Chieco-Bianchi, L.; Ciminale, V.; et al. Reducing the global burden of HTLV-1 infection: An agenda for research and action. Antivir. Res. 2017, 137, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T. Human T cell leukemia virus type I (HTLV-I) and human diseases. Annu. Rev. Immunol. 1997, 15, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Proietti, F.A.; Carneiro-Proietti, A.B.; Catalan-Soares, B.C.; Murphy, E.L. Global epidemiology of HTLV-I infection and associated diseases. Oncogene 2005, 24, 6058–6068. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Bangham, C.R.M. HTLV-1: Regulating the Balance between Proviral Latency and Reactivation. Front. Microbiol. 2018, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Biron, C.A. Here today—Not gone tomorrow: Roles for activating receptors in sustaining NK cells during viral infections. Eur. J. Immunol. 2010, 40, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.C.; Theorell, J.; Entesarian, M.; Meeths, M.; Mastafa, M.; Al-Herz, W.; Frisk, P.; Gilmour, K.C.; Ifversen, M.; Langenskiold, C.; et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 2013, 121, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Mortreux, F.; Gabet, A.S.; Wattel, E. Molecular and cellular aspects of HTLV-1 associated leukemogenesis in vivo. Leukemia 2003, 17, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R. CTL quality and the control of human retroviral infections. Eur. J. Immunol. 2009, 39, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, K.; Rowan, A.G.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. CADM1/TSLC1 Identifies HTLV-1-Infected Cells and Determines Their Susceptibility to CTL-Mediated Lysis. PLoS Pathog. 2016, 12, e1005560. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R. HTLV-1 infection: Role of CTL efficiency. Blood 2008, 112, 2176–2177. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.T.; Nicot, C. Overview on HTLV-1 p12, p8, p30, p13: Accomplices in persistent infection and viral pathogenesis. Front. Microbiol. 2012, 3, 400. [Google Scholar] [CrossRef] [PubMed]
- Van Prooyen, N.; Gold, H.; Resen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Silverman, L.; Phipps, A.J.; Hiraragi, H.; Ratner, L.; Lairmore, M.D. Human T-lymphotropic virus type-1 p30 alters cell cycle G2 regulation of T lymphocytes to enhance cell survival. Retrovirology 2007, 4, 49. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, B.; D’Souza, C.D.; Ding, W.; Tridandapani, S.; Coggeshall, K.M.; Lairmore, M.D. Activation of nuclear factor of activated T cells by human T-lymphotropic virus type 1 accessory protein p12(I). J. Virol. 2002, 76, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Feuer, G.; Barker, E. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and -2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J. Virol. 2007, 81, 9707–9717. [Google Scholar] [PubMed]
- Azran, I.; Schavinsky-Khrapunsky, Y.; Aboud, M. Role of Tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology 2004, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, R.; Andresen, V.; Bialuk, I.; Cecchinato, V.; Walser, J.C.; Valeri, V.W.; Nauroth, J.M.; Gessain, A.; Nicot, C.; Franchini, G. In vivo genetic mutations define predominant functions of the human T-cell leukemia/lymphoma virus p12I protein. Blood 2009, 113, 3726–3734. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Fullen, J.; Franchini, G. The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments. J. Virol. 1993, 67, 2360–2366. [Google Scholar] [PubMed]
- Van Prooyen, N.; Andresen, V.; Gold, H.; Bialuk, I.; Pise-Masison, C.; Franchini, G. Hijacking the T-cell communication network by the human T-cell leukemia/lymphoma virus type 1 (HTLV-1) p12 and p8 proteins. Mol. Asp. Med. 2010, 31, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Valeri, V.W.; Hryniewicz, A.; Resen, V.; Jones, K.; Fenizia, C.; Bialuk, I.; Chung, H.K.; Fukumoto, R.; Parks, R.W.; Ferrari, M.G.; et al. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood 2010, 116, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.D.; Newbound, G.C.; Albrecht, B.; Beard, J.L.; Ratner, L.; Lairmore, M.D. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood 1998, 91, 4701–4707. [Google Scholar] [PubMed]
- Albrecht, B.; Collins, N.D.; Burniston, M.T.; Nisbet, J.W.; Ratner, L.; Green, P.L.; Lairmore, M.D. Human T-lymphotropic virus type 1 open reading frame I p12(I) is required for efficient viral infectivity in primary lymphocytes. J. Virol. 2000, 74, 9828–9835. [Google Scholar] [CrossRef] [PubMed]
- Franchini, G. Molecular mechanisms of human T-cell leukemia/lymphotropic virus type I infection. Blood 1995, 86, 3619–3639. [Google Scholar] [PubMed]
- Feller, S.M.; Ren, R.; Hanafusa, H.; Baltimore, D. SH2 and SH3 domains as molecular adhesives: The interactions of Crk and Abl. Trends Biochem. Sci. 1994, 19, 453–458. [Google Scholar] [CrossRef]
- Koch, C.A.; Erson, D.; Moran, M.F.; Ellis, C.; Pawson, T. SH2 and SH3 domains: Elements that control interactions of cytoplasmic signaling proteins. Science 1991, 252, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Trovato, R.; Mulloy, J.C.; Johnson, J.M.; Takemoto, S.; de Oliveira, M.P.; Franchini, G. A lysine-to-arginine change found in natural alleles of the human T-cell lymphotropic/leukemia virus type 1 p12(I) protein greatly influences its stability. J. Virol. 1999, 73, 6460–6467. [Google Scholar] [PubMed]
- Edwards, D.; Fukumoto, R.; de Castro-Amarante, M.F.; Alcantara, L.C.; Galvao-Castro, B.; Washington Parks, R.; Pise-Masison, C.; Franchini, G. Palmitoylation and p8-mediated human T-cell leukemia virus type 1 transmission. J. Virol. 2014, 88, 2319–2322. [Google Scholar] [CrossRef] [PubMed]
- Pise-Masison, C.A.; de Castro-Amarante, M.F.; Enose-Akahata, Y.; Buchmann, R.C.; Fenizia, C.; Parks, R.W.; Edwards, D.; Fiocchi, M.; Alcantara, L.C., Jr.; Bialuk, I.; et al. Co-dependence of HTLV-1 p12 and p8 functions in virus persistence. PLoS Pathog. 2014, 10, e1004454. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, R.; Dundr, M.; Nicot, C.; Adams, A.; Valeri, V.W.; Samelson, L.E.; Franchini, G. Inhibition of T-cell receptor signal transduction and viral expression by the linker for activation of T cells-interacting p12(I) protein of human T-cell leukemia/lymphoma virus type 1. J. Virol. 2007, 81, 9088–9099. [Google Scholar] [CrossRef] [PubMed]
- Nicot, C.; Mulloy, J.C.; Ferrari, M.G.; Johnson, J.M.; Fu, K.; Fukumoto, R.; Trovato, R.; Fullen, J.; Leonard, W.J.; Franchini, G. HTLV-1 p12(I) protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood 2001, 98, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, J.C.; Crownley, R.W.; Fullen, J.; Leonard, W.J.; Franchini, G. The human T-cell leukemia/lymphotropic virus type 1 p12I proteins bind the interleukin-2 receptor beta and gammac chains and affects their expression on the cell surface. J. Virol. 1996, 70, 3599–3605. [Google Scholar] [PubMed]
- Gaffen, S.L.; Liu, K.D. Overview of interleukin-2 function, production and clinical applications. Cytokine 2004, 28, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Jacobson, E.L.; Emert, R.; Giordano, M.; Kovacs, E.; Mumneh, N.; Pilaro, F.; Sohn, T.; Warren, D. Restoration of immunity with interleukin-2 therapy. AIDS Read. 1999, 9, 563–572. [Google Scholar] [PubMed]
- Takemoto, S.; Mulloy, J.C.; Cereseto, A.; Migone, T.S.; Patel, B.K.; Matsuoka, M.; Yamaguchi, K.; Takatsuki, K.; Kamihira, S.; White, J.D.; et al. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 13897–13902. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Cumaraswamy, A.A.; Gunning, P.T. Progress towards direct inhibitors of Stat5 protein. Horm. Mol. Biol. Clin. Investig. 2012, 10, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.M.; Nicot, C.; Fullen, J.; Ciminale, V.; Casareto, L.; Mulloy, J.C.; Jacobson, S.; Franchini, G. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J. Virol. 2001, 75, 6086–6094. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.J.; Travers, P.; Walport, M.; Shlomchik, M.J. (Eds.) The Major Histocompatibility Complex and Its Functions; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Alvaro-Benito, M.; Stolzenberg, S.; Noe, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Chirathaworn, C.; Kohlmeier, J.E.; Tibbetts, S.A.; Rumsey, L.M.; Chan, M.A.; Benedict, S.H. Stimulation through intercellular adhesion molecule-1 provides a second signal for T cell activation. J. Immunol. 2002, 168, 5530–5537. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Fukudome, K.; Hayashi, M.; Takagi, S.; Yoshie, O. Induction of ICAM-1 and LFA-3 by Tax1 of human T-cell leukemia virus type 1 and mechanism of down-regulation of ICAM-1 or LFA-1 in adult-T-cell-leukemia cell lines. Int. J. Cancer 1995, 60, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Fukudome, K.; Furuse, M.; Fukuhara, N.; Orita, S.; Imai, T.; Takagi, S.; Nagira, M.; Hinuma, Y.; Yoshie, O. Strong induction of ICAM-1 in human T cells transformed by human T-cell-leukemia virus type 1 and depression of ICAM-1 or LFA-1 in adult T-cell-leukemia-derived cell lines. Int. J. Cancer 1992, 52, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Albrecht, B.; Kelley, R.E.; Muthusamy, N.; Kim, S.J.; Altschuld, R.A.; Lairmore, M.D. Human T-cell lymphotropic virus type 1 p12(I) expression increases cytoplasmic calcium to enhance the activation of nuclear factor of activated T cells. J. Virol. 2002, 76, 10374–10382. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.W.; Rincon, M.; Davis, R.J. Requirement for transcription factor NFAT in interleukin-2 expression. Mol. Cell. Biol. 1999, 19, 2300–2307. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Albrecht, B.; Luo, R.; Zhang, W.; Stanley, J.R.; Newbound, G.C.; Lairmore, M.D. Endoplasmic reticulum and cis-Golgi localization of human T-lymphotropic virus type 1 p12(I): Association with calreticulin and calnexin. J. Virol. 2001, 75, 7672–7682. [Google Scholar] [CrossRef] [PubMed]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar] [PubMed]
- Kim, S.J.; Ding, W.; Albrecht, B.; Green, P.L.; Lairmore, M.D. A conserved calcineurin-binding motif in human T lymphotropic virus type 1 p12I functions to modulate nuclear factor of activated T cell activation. J. Biol. Chem. 2003, 278, 15550–15557. [Google Scholar] [CrossRef] [PubMed]
- Rothermel, B.; Vega, R.B.; Yang, J.; Wu, H.; Bassel-Duby, R.; Williams, R.S. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J. Biol. Chem. 2000, 275, 8719–8725. [Google Scholar] [CrossRef] [PubMed]
- Miskin, J.E.; Abrams, C.C.; Dixon, L.K. African swine fever virus protein A238L interacts with the cellular phosphatase calcineurin via a binding domain similar to that of NFAT. J. Virol. 2000, 74, 9412–9420. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, F.; Kondo, E.; Akagi, T.; McKeon, F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature 1997, 386, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Foyouzi-Youssefi, R.; Arnaudeau, S.; Borner, C.; Kelley, W.L.; Tschopp, J.; Lew, D.P.; Demaurex, N.; Krause, K.H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 5723–5728. [Google Scholar] [CrossRef] [PubMed]
- Wulfing, C.; Davis, M.M. A receptor/cytoskeletal movement triggered by costimulation during T cell activation. Science 1998, 282, 2266–2269. [Google Scholar] [CrossRef] [PubMed]
- Manz, B.N.; Groves, J.T. Spatial organization and signal transduction at intercellular junctions. Nat. Rev. Mol. Cell Biol. 2010, 11, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Brown, M.; Nejmeddine, M.; Kim, K.J.; Ratner, L.; Lairmore, M.; Nicot, C. Novel role for interleukin-2 receptor-Jak signaling in retrovirus transmission. J. Virol. 2009, 83, 11467–11476. [Google Scholar] [CrossRef] [PubMed]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Biasiotto, R.; Resen, V.; Franchini, G.; D’Agostino, D.M.; Ciminale, V. HTLV-1 p13, a small protein with a busy agenda. Mol. Asp. Med. 2010, 31, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Ghorbel, S.; Sinha-Datta, U.; Dundr, M.; Brown, M.; Franchini, G.; Nicot, C. Human T-cell leukemia virus type I p30 nuclear/nucleolar retention is mediated through interactions with RNA and a constituent of the 60 S ribosomal subunit. J. Biol. Chem. 2006, 281, 37150–37158. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, D.M.; Ciminale, V.; Zotti, L.; Rosato, A.; Chieco-Bianchi, L. The human T-cell lymphotropic virus type 1 Tof protein contains a bipartite nuclear localization signal that is able to functionally replace the amino-terminal domain of Rex. J. Virol. 1997, 71, 75–83. [Google Scholar] [PubMed]
- Andresen, V.; Pise-Masison, C.A.; Sinha-Datta, U.; Bellon, M.; Valeri, V.; Washington Parks, R.; Cecchinato, V.; Fukumoto, R.; Nicot, C.; Franchini, G. Suppression of HTLV-1 replication by Tax-mediated rerouting of the p13 viral protein to nuclear speckles. Blood 2011, 118, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Currer, R.; Van Duyne, R.; Jaworski, E.; Guendel, I.; Sampey, G.; Das, R.; Narayanan, A.; Kashanchi, F. HTLV tax: A fascinating multifunctional co-regulator of viral and cellular pathways. Front. Microbiol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Biasiotto, R.; Aguiari, P.; Rizzuto, R.; Pinton, P.; D’Agostino, D.M.; Ciminale, V. The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake. Biochim. Biophys. Acta 2010, 1797, 945–951. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, D.M.; Ranzato, L.; Arrigoni, G.; Cavallari, I.; Belleudi, F.; Torrisi, M.R.; Silic-Benussi, M.; Ferro, T.; Petronilli, V.; Marin, O.; et al. Mitochondrial alterations induced by the p13II protein of human T-cell leukemia virus type 1. Critical role of arginine residues. J. Biol. Chem. 2002, 277, 34424–34433. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Cannizzaro, E.; Venerando, A.; Cavallari, I.; Petronilli, V.; La Rocca, N.; Marin, O.; Chieco-Bianchi, L.; Di Lisa, F.; D’Agostino, D.M.; et al. Modulation of mitochondrial K(+) permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1. Biochim. Biophys. Acta 2009, 1787, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Marin, O.; Biasiotto, R.; D’Agostino, D.M.; Ciminale, V. Effects of human T-cell leukemia virus type 1 (HTLV-1) p13 on mitochondrial K+ permeability: A new member of the viroporin family? FEBS Lett. 2010, 584, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Hiraragi, H.; Kim, S.J.; Phipps, A.J.; Silic-Benussi, M.; Ciminale, V.; Ratner, L.; Green, P.L.; Lairmore, M.D. Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13(II) is required for viral infectivity in vivo. J. Virol. 2006, 80, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- Omura, T. Mitochondria-targeting sequence, a multi-role sorting sequence recognized at all steps of protein import into mitochondria. J. Biochem. 1998, 123, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Sinet, F. Complex Splicing in the Human T-cell Leukemia Virus (HTLV) Family of Retroviruses: A Number of Proteins Play a Key Role in the Establishment and Maintenance of Viral Infection. BioSciences Master Reviews Ecole Normale Supérieure de Lyon. July 2013, pp. 1–10. Available online: http://biologie.ens-lyon.fr/ressources/bibliographies (accessed on 8 November 2018).
- Franchini, G.; Nicot, C.; Johnson, J.M. Seizing of T cells by human T-cell leukemia/lymphoma virus type 1. Adv. Cancer Res. 2003, 89, 69–132. [Google Scholar] [PubMed]
- D’Agostino, D.M.; Silic-Benussi, M.; Hiraragi, H.; Lairmore, M.D.; Ciminale, V. The human T-cell leukemia virus type 1 p13II protein: Effects on mitochondrial function and cell growth. Cell Death Differ. 2005, 12 (Suppl. 1), 905–915. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. II. Steroid effects. Biochim. Biophys. Acta 1966, 122, 167–174. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [PubMed]
- Costantini, P.; Chernyak, B.V.; Petronilli, V.; Bernardi, P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 1996, 271, 6746–6751. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Cavallari, I.; Vajente, N.; Vidali, S.; Chieco-Bianchi, L.; Di Lisa, F.; Saggioro, D.; D’Agostino, D.M.; Ciminale, V. Redox regulation of T-cell turnover by the p13 protein of human T-cell leukemia virus type 1: Distinct effects in primary versus transformed cells. Blood 2010, 116, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P. Mitochondria, from cell death to proliferation. Nat. Genet. 2002, 30, 352–353. [Google Scholar] [CrossRef] [PubMed]
- Hiraragi, H.; Michael, B.; Nair, A.; Silic-Benussi, M.; Ciminale, V.; Lairmore, M. Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13II sensitizes Jurkat T cells to Ras-mediated apoptosis. J. Virol. 2005, 79, 9449–9457. [Google Scholar] [CrossRef] [PubMed]
- Mathias, S.; Pena, L.A.; Kolesnick, R.N. Signal transduction of stress via ceramide. Biochem. J. 1998, 335 Pt 3, 465–480. [Google Scholar] [CrossRef]
- Nagata, S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999, 33, 29–55. [Google Scholar] [CrossRef] [PubMed]
- Giam, C.Z.; Semmes, O.J. HTLV-1 Infection and Adult T-Cell Leukemia/Lymphoma-A Tale of Two Proteins: Tax and HBZ. Viruses 2016, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Yasunaga, J.; Matsuoka, M. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 2016, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Watanabe, T. HTLV-1 Rex: The courier of viral messages making use of the host vehicle. Front. Microbiol. 2012, 3, 330. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Bellon, M.; Nicot, C. HTLV-1 Yin and Yang: Rex and p30 master regulators of viral mRNA trafficking. AIDS Rev. 2008, 10, 195–204. [Google Scholar] [PubMed]
- Bai, X.T.; Baydoun, H.H.; Nicot, C. HTLV-I p30: A versatile protein modulating virus replication and pathogenesis. Mol. Asp. Med. 2010, 31, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Shibata, H.; Fujisawa, J.I.; Inoue, H.; Hakura, A.; Tsukahara, T.; Fujii, M. Human T-cell leukemia virus type 1 Tax protein transforms rat fibroblasts via two distinct pathways. J. Virol. 1997, 71, 4445–4451. [Google Scholar] [PubMed]
- Tanaka, A.; Takahashi, C.; Yamaoka, S.; Nosaka, T.; Maki, M.; Hatanaka, M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 1071–1075. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgieva, E.R. Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes—Mechanisms for Viral Survivability and Proliferation. Int. J. Mol. Sci. 2018, 19, 3508. https://doi.org/10.3390/ijms19113508
Georgieva ER. Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes—Mechanisms for Viral Survivability and Proliferation. International Journal of Molecular Sciences. 2018; 19(11):3508. https://doi.org/10.3390/ijms19113508
Chicago/Turabian StyleGeorgieva, Elka R. 2018. "Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes—Mechanisms for Viral Survivability and Proliferation" International Journal of Molecular Sciences 19, no. 11: 3508. https://doi.org/10.3390/ijms19113508
APA StyleGeorgieva, E. R. (2018). Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes—Mechanisms for Viral Survivability and Proliferation. International Journal of Molecular Sciences, 19(11), 3508. https://doi.org/10.3390/ijms19113508