Global Analysis of WOX Transcription Factor Gene Family in Brassica napus Reveals Their Stress- and Hormone-Responsive Patterns


Abstract
1. Introduction
2. Results
2.1. Identification of 58 BnWOX Genes and Their Physicochemical Properties
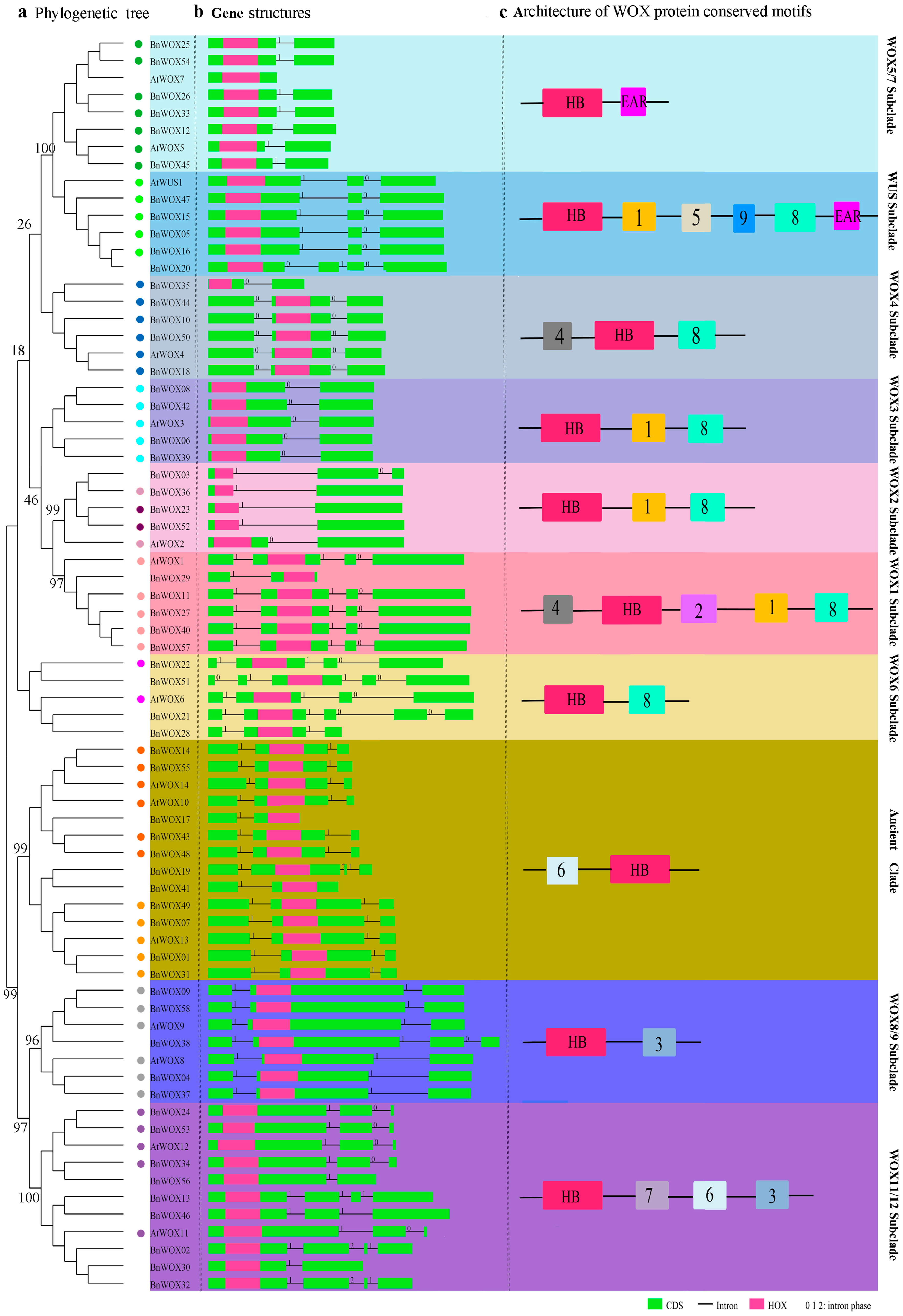
2.2. Phylogenetic Analysis of the BnWOX Gene Family
2.3. Sequence Analysis of B. napus WOX Domains
2.4. Gene Structure and Intron Pattern Analysis
2.5. Conserved Motif Analysis of B. napus WOX Proteins
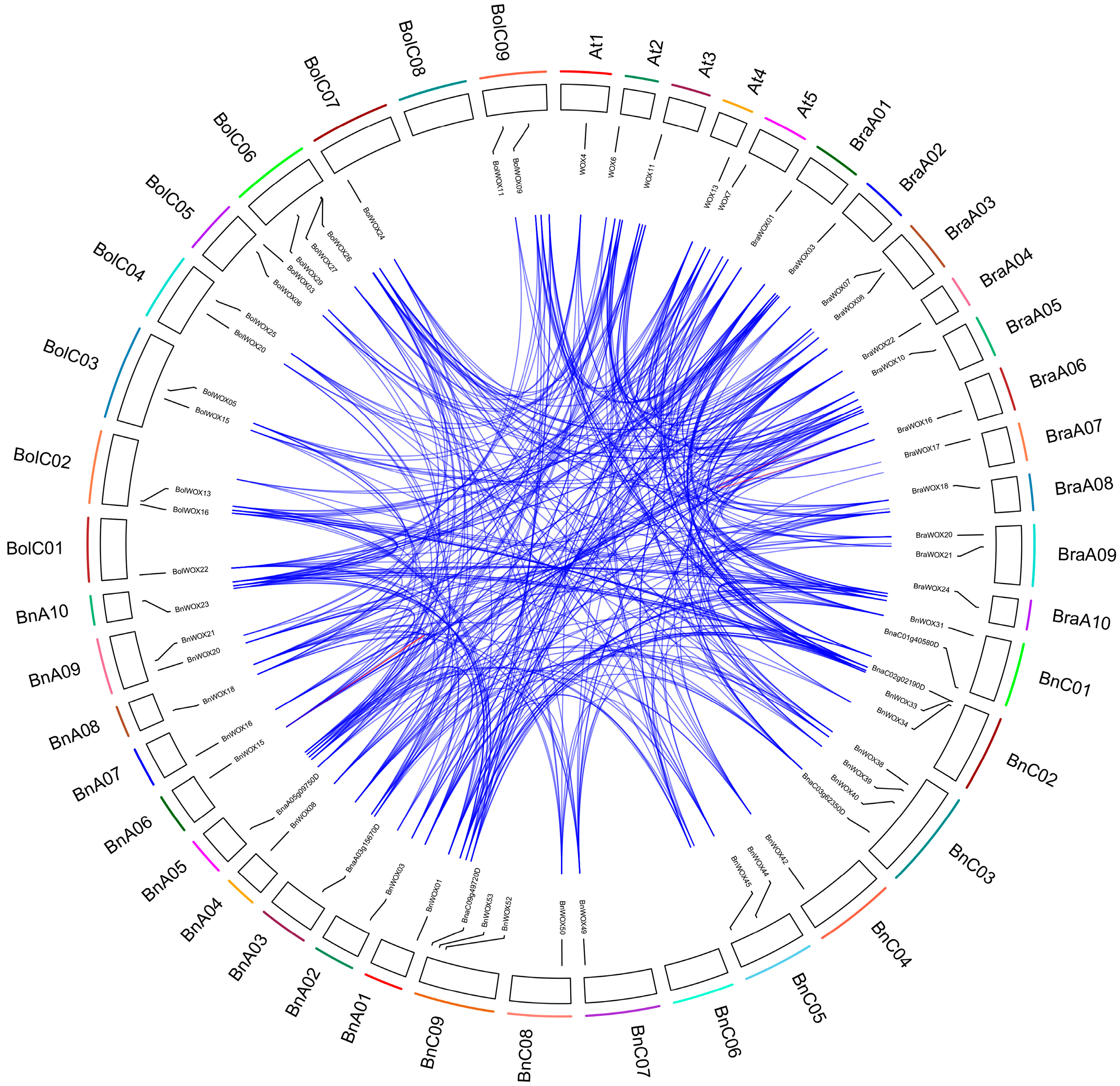
2.6. Chromosomal Distribution and Duplication of B. napus WOX Genes
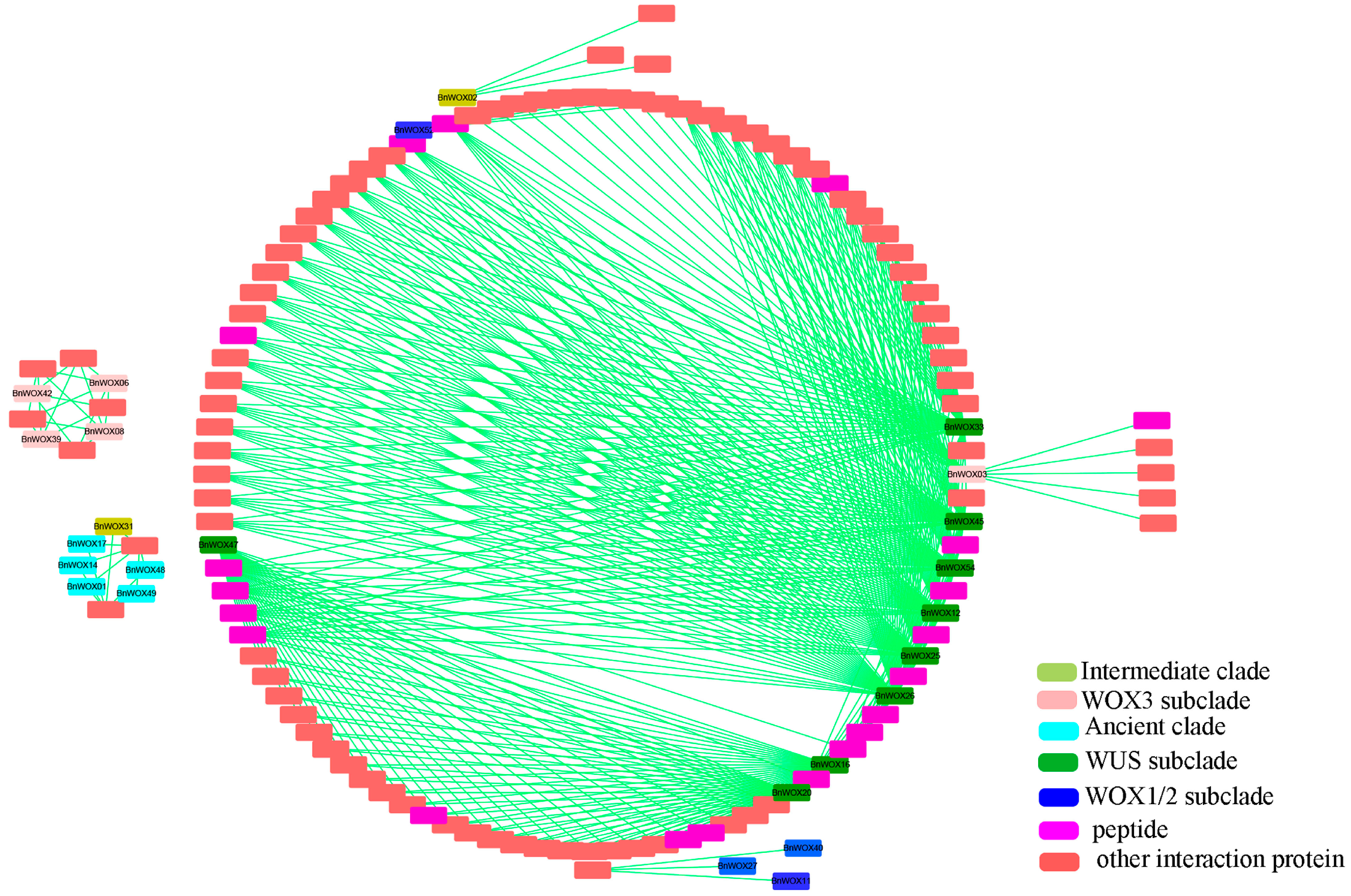
2.7. Prediction of BnWOX Protein Interaction Networks in B. napus
2.8. Expression Analysis of WOX Genes at Different Developmental Stages in B. napus
2.9. Responses of BnWOX Genes to Environmental Stresses and Phytohormone Induction
3. Discussion
3.1. Structural and Functional Conservation of the Plant WOX Gene Family
3.2. Conservation of Expression Profiles in Different Plants Supports Their Functional Conservation
4. Materials and Methods
4.1. Identification of WOX Proteins and Phylogenetic Analysis of the B. napus Genome
4.2. Sequence, Gene Structure and Conserved Motif Analysis and Construction of the Protein Interaction Network
4.3. Chromosomal Location and Synteny Analysis
4.4. Analysis of Expression Profiles of BnWOXs at Different Developmental Stages and After Hormone and Stress Treatment
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABA | abscisic acid |
| ACC | 1-aminocyclopropane-1-carboxylic acid |
| CLV3 | CLAVATA3 |
| CLE40 | CLAVATA3/ESR-RELATED 40 |
| FPKM | fragments per kilobase of transcript per million mapped reads |
| GA | gibberellic acid |
| GSDS | gene structure display server |
| HB domain | homeodomain |
| IAA | indole-3-acetic acid |
| ORF | open reading frame |
| PEG | polyethylene glycol |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| RAM | root apical meristem |
| RNA-seq | RNA sequencing |
| SAM | shoot apical meristem |
| TPL | TOPLESS |
| WOX | WUSCHEL-related homeobox |
| ZS11 | ”Zhongshuang 11” |
References
- Mayer, K.F.; Schoof, H.; Haecker, A.; Lenhard, M.; Jürgens, G.; Laux, T. Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 1998, 95, 805. [Google Scholar] [CrossRef]
- Vandenbussche, M.; Horstman, A.; Zethof, J.; Koes, R.; Rijpkema, A.S.; Gerats, T. Differential recruitment of WOX transcription factors for lateral development and organ fusion in petunia and Arabidopsis. Plant Cell 2009, 21, 2269–2283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zong, J.; Liu, J.; Yin, J.; Zhang, D. Genome-wide analysis of WOX gene family in rice, sorghum, maize, Arabidopsis and poplar. J. Integr. Plant Biol. 2010, 52, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Haecker, A.; Gross-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Niu, L.; McHale, N.A.; Ohme-Takagi, M.; Mysore, K.S.; Tadege, M. Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Plant Signal. Behav. 2013, 110, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.K.; Luijten, M.; Miyashima, S.; Lenhard, M.; Hashimoto, T.; Nakajima, K.; Scheres, B.; Heidstra, R.; Laux, T.; Sarkar, A.K.; et al. Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature 2007, 446, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef]
- Breuninger, H.; Rikirsch, E.; Hermann, M.; Ueda, M.; Laux, T. Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev. Cell 2008, 14, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dabi, T.; Weigel, D. Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr. Biol. 2005, 15, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Etchells, J.P.; Provost, C.M.; Mishra, L.; Turner, S.R. WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organisation. Development 2013, 140, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Nagasaki, H.; Morikami, A.; Sato, Y.; Matsuoka, M. Isolation and characterization of a rice WUSCHEL-type homeobox gene that is specifically expressed in the central cells of a quiescent center in the root apical meristem. Plant J. 2003, 35, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Nardmann, J.; Werr, W. The shoot stem cell niche in angiosperms: Expression patterns of WUS orthologues in rice and maize imply major modifications in the course of mono- and dicot evolution. Mol. Biol. Evol. 2006, 23, 2492–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Moschou, P.N.; Alvarez, J.M.; Sohlberg, J.J.; Arnold, S.V. WUSCHEL-RELATED HOMEOBOX 8/9 is important for proper embryo patterning in the gymnosperm Norway spruce. J. Exp. Bot. 2014, 65, 6543–6552. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Cheng, F.; Li, Y.; Wang, X.; Parkin, I.A.; Chalhoub, B.; Liu, S.; Bancroft, I. Construction of Brassica A and C genome-based ordered pan-transcriptomes for use in rapeseed genomic research. Data Brief 2015, 4, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef] [PubMed]
- Stéphane, G.; Frédéric, D.; Jean-François, D.; Olivier, G. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [CrossRef]
- Cao, Y.; Han, Y.; Meng, D.; Li, G.; Li, D.; Abdullah, M.; Jin, Q.; Lin, Y.; Cai, Y. Genome-wide analysis suggests the relaxed purifying selection affect the evolution of WOX genes in Pyrus bretschneideri, Prunus persica, Prunus mume, and Fragaria vesca. Front. Genet. 2017, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Nardmann, J.; Werr, W. Symplesiomorphies in the WUSCHEL clade suggest that the last common ancestor of seed plants contained at least four independent stem cell niches. New Phytol. 2013, 199, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Gong, Q.; Qin, W.; Yang, Z.; Cheng, Y.; Lu, L.; Ge, X.; Zhang, C.; Wu, Z.; Li, F. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Ding, Z.; Wang, Q.; Zhang, D.; Xu, J. Origins and Evolution of WUSCHEL-related homeobox protein family in plant kingdom. Sci. World J. 2014, 2014, 534140. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Hu, B.; Li, J.; Zhang, G.; Ying, R.; Huang, H.; Wang, H.; Xu, L. Stem cell lineage in body layer specialization and vascular patterning of rice root and leaf. Sci. Bull. 2016, 61, 847–858. [Google Scholar] [CrossRef]
- Hedman, H.; Zhu, T.; Arnold, S.V.; Sohlberg, J.J. Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in the conifer picea abies reveals extensive conservation as well as dynamic patterns. BMC Plant Biol. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2014, 31, 1296. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ohme-Takagi, M. Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 2009, 21, 3493–3505. [Google Scholar] [CrossRef] [PubMed]
- Paponov, I.A.; Teale, W.; Lang, D.; Paponov, M.; Reski, R.; Rensing, S.A.; Palme, K. The evolution of nuclear auxin signalling. BMC Evol. Biol. 2009, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hamyat, M.; Liu, C.; Salman, A.; Gao, X.; Guo, C.; Wang, Y.; Guo, Y. Identification and characterization of the WOX family genes in five Solanaceae Species reveal their conserved roles in peptide signaling. Genes 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.C.; Brand, U.; Running, M.P.; Simon, R.; Meyerowitz, E.M. Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science 1999, 283, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y. posttranslationally modified small-peptide signals in plants. Annu. Rev. Plant Biol. 2014, 65, 385–413. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.F.; Saiga, S.; Abe, M.; Laux, T. OBE3 and WUS interaction in shoot meristem stem cell regulation. PLoS ONE 2016, 11, e0155657. [Google Scholar] [CrossRef] [PubMed]
- Della, R.F.; Fattorini, L.; D’Angeli, S.; Veloccia, A.; Del, D.S.; Cai, G.; Falasca, G.; Altamura, M.M. Arabidopsis SHR and SCR transcription factors and AUX1 auxin influx carrier control the switch between adventitious rooting and xylogenesis in planta and in vitro cultured thin cell layers. Ann. Bot. 2015, 115, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chory, J.; Weigel, D. Combinations of WOX activities regulate tissue proliferation during Arabidopsis embryonic development. Dev. Biol. 2007, 309, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Huang, Y.; Zhu, N.; Zhao, Y. The rice WUSCHEL-related homeobox genes are involved in reproductive organ development, hormone signaling and abiotic stress response. Gene 2014, 549, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sheng, L.; Xu, Y.; Li, J.; Yang, Z.; Huang, H.; Xu, L. WOX11 and 12 are involved in the first-step cell fate transition during de novo root organogenesis in Arabidopsis. Plant Cell 2014, 26, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Dolzblasz, A.; Nardmann, J.; Clerici, E.; Causier, B.; van der Graaff, E.; Chen, J.; Davies, B.; Werr, W.; Laux, T. Stem cell regulation by Arabidopsis WOX genes. Mol. Plant 2016, 9, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Y.; Li, G.; Tang, Y.; Kramer, E.M.; Tadege, M. STENOFOLIA recruits TOPLESS to repress ASYMMETRIC LEAVES2 at the leaf margin and promote leaf blade outgrowth in Medicago truncatula. Plant Cell 2014, 26, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, Y.; Dai, M.; Huang, L.; Zhou, D.X. The WUSCHEL-related homeobox gene WOX11 is required to activate shoot-borne crown root development in rice. Plant Cell 2009, 21, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the "gold standard" annotated reference Plant Genome. Genesis 2015, 53, 474. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2007, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, S.; Li, H.; Du, H.; Huang, H.; Li, Y.; Hu, Y.; Liu, H.; Liu, Y.; Yu, G. Identification of transcription factors ZmMYB111 and ZmMYB148 involved in phenylpropanoid metabolism. Front. Plant Sci. 2016, 7, 148. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Genome ID | Chromosome | Protein Length (Amino Acids) | cDNA Length (bp) | Introns/Exons | pI | Molecular Weight (Da) | Subcellular Localization |
|---|---|---|---|---|---|---|---|---|
| BnWOX01 | BnaA01g01910D | chrA01 | 256 | 940 | 2/3 | 5.72 | 28,458.4 | Nucleus |
| BnWOX02 | BnaA01g34040D | chrA01 | 289 | 870 | 3/4 | 6.48 | 31,778.6 | Nucleus |
| BnWOX03 | BnaA02g07100D | chrA02 | 175 | 528 | 2/3 | 6.36 | 19,236.2 | Nucleus |
| BnWOX04 | BnaA02g24210D | chrA02 | 320 | 1053 | 2/3 | 6.96 | 35,591.2 | Nucleus |
| BnWOX05 | BnaA02g36550D | chrA02 | 302 | 909 | 2/3 | 7.09 | 34,348.7 | Nucleus |
| BnWOX06 | BnaA03g22070D | chrA03 | 226 | 681 | 1/2 | 9.33 | 26,087 | Nucleus |
| BnWOX07 | BnaA03g53400D | chrA03 | 263 | 1036 | 2/3 | 5.3 | 29,170.3 | Nucleus |
| BnWOX08 | BnaA04g16520D | chrA04 | 234 | 705 | 1/2 | 9.32 | 27,143.2 | Nucleus |
| BnWOX09 | BnaA05g09770D | chrA05 | 390 | 1173 | 2/3 | 8.35 | 43,341.9 | Nucleus |
| BnWOX10 | BnaA05g18600D | chrA05 | 251 | 756 | 2/3 | 9.63 | 28,555.1 | Nucleus |
| BnWOX11 | BnaA05g22250D | chrA05 | 351 | 1287 | 3/4 | 8.59 | 40,110.9 | Nucleus |
| BnWOX12 | BnaA05g27750D | chrA05 | 191 | 718 | 1/2 | 8.87 | 22,361 | Nucleus |
| BnWOX13 | BnaA05g32800D | chrA05 | 326 | 981 | 3/4 | 6.63 | 35,661 | Nucleus |
| BnWOX14 | BnaA06g14590D | chrA06 | 203 | 768 | 2/3 | 5.43 | 22,850.2 | Nucleus |
| BnWOX15 | BnaA06g25450D | chrA06 | 294 | 885 | 2/3 | 7.81 | 33,475.7 | Nucleus |
| BnWOX16 | BnaA07g02390D | chrA07 | 302 | 909 | 2/3 | 6.85 | 34,305.6 | Nucleus |
| BnWOX17 | BnaA07g11310D | chrA07 | 133 | 402 | 1/2 | 7.89 | 15,260.3 | Nucleus |
| BnWOX18 | BnaA08g04100D | chrA08 | 256 | 942 | 2/3 | 9.31 | 29,143.6 | Nucleus |
| BnWOX19 | BnaA08g14960D | chrA08 | 239 | 720 | 3/4 | 5.73 | 27,383.7 | Nucleus |
| BnWOX20 | BnaA09g09400D | chrA09 | 316 | 1022 | 3/4 | 6.89 | 35,862 | Nucleus |
| BnWOX21 | BnaA09g18650D | chrA09 | 267 | 804 | 4/5 | 6.41 | 30,920.3 | Nucleus |
| BnWOX22 | BnaA09g45340D | chrA09 | 275 | 901 | 3/4 | 6.28 | 31,962 | Nucleus |
| BnWOX23 | BnaA10g12360D | chrA10 | 210 | 633 | 1/2 | 6.83 | 23,085.3 | Nucleus |
| BnWOX24 | BnaA10g16970D | chrA10 | 274 | 949 | 2/3 | 6.34 | 30,342.5 | Nucleus |
| BnWOX25 | BnaA10g24790D | chrA10 | 192 | 579 | 1/2 | 9.17 | 22,114.9 | Nucleus |
| BnWOX26 | BnaAnng01480D | chrAnn_random | 191 | 576 | 1/2 | 6.66 | 21,832.3 | Nucleus |
| BnWOX27 | BnaAnng11050D | chrAnn_random | 353 | 1062 | 3/4 | 6.97 | 40,197.6 | Nucleus |
| BnWOX28 | BnaAnng35720D | chrAnn_random | 166 | 501 | 2/3 | 8.99 | 19,173.4 | Nucleus |
| BnWOX29 | BnaAnng36010D | chrAnn_random | 121 | 366 | 1/2 | 9.61 | 13,962.8 | Nucleus |
| BnWOX30 | BnaAnng41310D | chrAnn_random | 247 | 847 | 1/2 | 8.77 | 27,574.8 | Nucleus |
| BnWOX31 | BnaC01g03050D | chrC01 | 265 | 1016 | 2/3 | 5.67 | 29,250.4 | Nucleus |
| BnWOX32 | BnaC01g40590D | chrC01 | 289 | 972 | 3/4 | 6.48 | 31,778.6 | Nucleus |
| BnWOX33 | BnaC02g02200D | chrC02 | 191 | 576 | 1/2 | 5.18 | 46,938.7 | Nucleus |
| BnWOX34 | BnaC02g07490D | chrC02 | 283 | 852 | 2/3 | 5.08 | 71,825.3 | Nucleus |
| BnWOX35 | BnaC02g08530D | chrC02 | 121 | 366 | 1/2 | 6.09 | 31,334.6 | Nucleus |
| BnWOX36 | BnaC02g10040D | chrC02 | 199 | 600 | 1/2 | 5.95 | 21,671.7 | Nucleus |
| BnWOX37 | BnaC02g32050D | chrC02 | 320 | 1277 | 2/3 | 6.96 | 35,542.2 | Nucleus |
| BnWOX38 | BnaC03g18850D | chrC03 | 397 | 1303 | 3/4 | 6.57 | 44,157.5 | Nucleus |
| BnWOX39 | BnaC03g26450D | chrC03 | 222 | 669 | 1/2 | 9.26 | 25,657.6 | Nucleus |
| BnWOX40 | BnaC03g40380D | chrC03 | 350 | 1053 | 3/4 | 7.72 | 40,024.6 | Nucleus |
| BnWOX41 | BnaC03g62360D | chrC03 | 171 | 641 | 1/2 | 7.65 | 19,697.3 | Nucleus |
| BnWOX42 | BnaC04g39860D | chrC04 | 236 | 711 | 1/2 | 9.3 | 27,260.4 | Nucleus |
| BnWOX43 | BnaC04g46990D | chrC04 | 195 | 588 | 2/3 | 5.51 | 21,785.3 | Nucleus |
| BnWOX44 | BnaC05g25380D | chrC05 | 251 | 908 | 2/3 | 9.58 | 28,526 | Nucleus |
| BnWOX45 | BnaC05g41930D | chrC05 | 191 | 718 | 1/2 | 8.87 | 22,485.1 | Nucleus |
| BnWOX46 | BnaC05g48100D | chrC05 | 339 | 1020 | 3/4 | 5.98 | 37,085.8 | Nucleus |
| BnWOX47 | BnaC07g06960D | chrC07 | 302 | 2162 | 2/3 | 7.36 | 34,407.9 | Nucleus |
| BnWOX48 | BnaC07g15200D | chrC07 | 195 | 588 | 2/3 | 5.35 | 21,686.1 | Nucleus |
| BnWOX49 | BnaC07g45680D | chrC07 | 261 | 1022 | 2/3 | 5.23 | 29,076.2 | Nucleus |
| BnWOX50 | BnaC08g04810D | chrC08 | 255 | 938 | 2/3 | 9.43 | 28,912.4 | Nucleus |
| BnWOX51 | BnaC08g39150D | chrC08 | 290 | 873 | 4/5 | 5.93 | 33,988.5 | Nucleus |
| BnWOX52 | BnaC09g34650D | chrC09 | 210 | 633 | 1/2 | 6.58 | 23,144.4 | Nucleus |
| BnWOX53 | BnaC09g40100D | chrC09 | 276 | 1794 | 2/3 | 5.97 | 30,547.8 | Nucleus |
| BnWOX54 | BnaC09g49730D | chrC09 | 192 | 579 | 1/2 | 8.99 | 22,099.8 | Nucleus |
| BnWOX55 | BnaCnng49040D | chrCnn_random | 209 | 579 | 2/3 | 5.48 | 23,619.1 | Nucleus |
| BnWOX56 | BnaCnng49570D | chrCnn_random | 280 | 843 | 1/2 | 5.97 | 31,047.2 | Nucleus |
| BnWOX57 | BnaCnng51820D | chrCnn_random | 351 | 1314 | 3/4 | 8.4 | 40,001.6 | Nucleus |
| BnWOX58 | BnaCnng62530D | chrCnn_random | 390 | 1954 | 2/3 | 8.35 | 43,232.7 | Nucleus |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.-M.; Liu, M.-M.; Ran, F.; Guo, P.-C.; Ke, Y.-Z.; Wu, Y.-W.; Wen, J.; Li, P.-F.; Li, J.-N.; Du, H. Global Analysis of WOX Transcription Factor Gene Family in Brassica napus Reveals Their Stress- and Hormone-Responsive Patterns. Int. J. Mol. Sci. 2018, 19, 3470. https://doi.org/10.3390/ijms19113470
Wang M-M, Liu M-M, Ran F, Guo P-C, Ke Y-Z, Wu Y-W, Wen J, Li P-F, Li J-N, Du H. Global Analysis of WOX Transcription Factor Gene Family in Brassica napus Reveals Their Stress- and Hormone-Responsive Patterns. International Journal of Molecular Sciences. 2018; 19(11):3470. https://doi.org/10.3390/ijms19113470
Chicago/Turabian StyleWang, Mang-Mang, Ming-Ming Liu, Feng Ran, Peng-Cheng Guo, Yun-Zhuo Ke, Yun-Wen Wu, Jing Wen, Peng-Feng Li, Jia-Na Li, and Hai Du. 2018. "Global Analysis of WOX Transcription Factor Gene Family in Brassica napus Reveals Their Stress- and Hormone-Responsive Patterns" International Journal of Molecular Sciences 19, no. 11: 3470. https://doi.org/10.3390/ijms19113470
APA StyleWang, M.-M., Liu, M.-M., Ran, F., Guo, P.-C., Ke, Y.-Z., Wu, Y.-W., Wen, J., Li, P.-F., Li, J.-N., & Du, H. (2018). Global Analysis of WOX Transcription Factor Gene Family in Brassica napus Reveals Their Stress- and Hormone-Responsive Patterns. International Journal of Molecular Sciences, 19(11), 3470. https://doi.org/10.3390/ijms19113470