Prediction of the Toxicity of Binary Mixtures by QSAR Approach Using the Hypothetical Descriptors



Abstract
:1. Introduction
2. Results and Discussion
2.1. MLR Results
2.2. Model Applicability Domain Analysis and Improved MLR Model
2.3. RBFNN Results
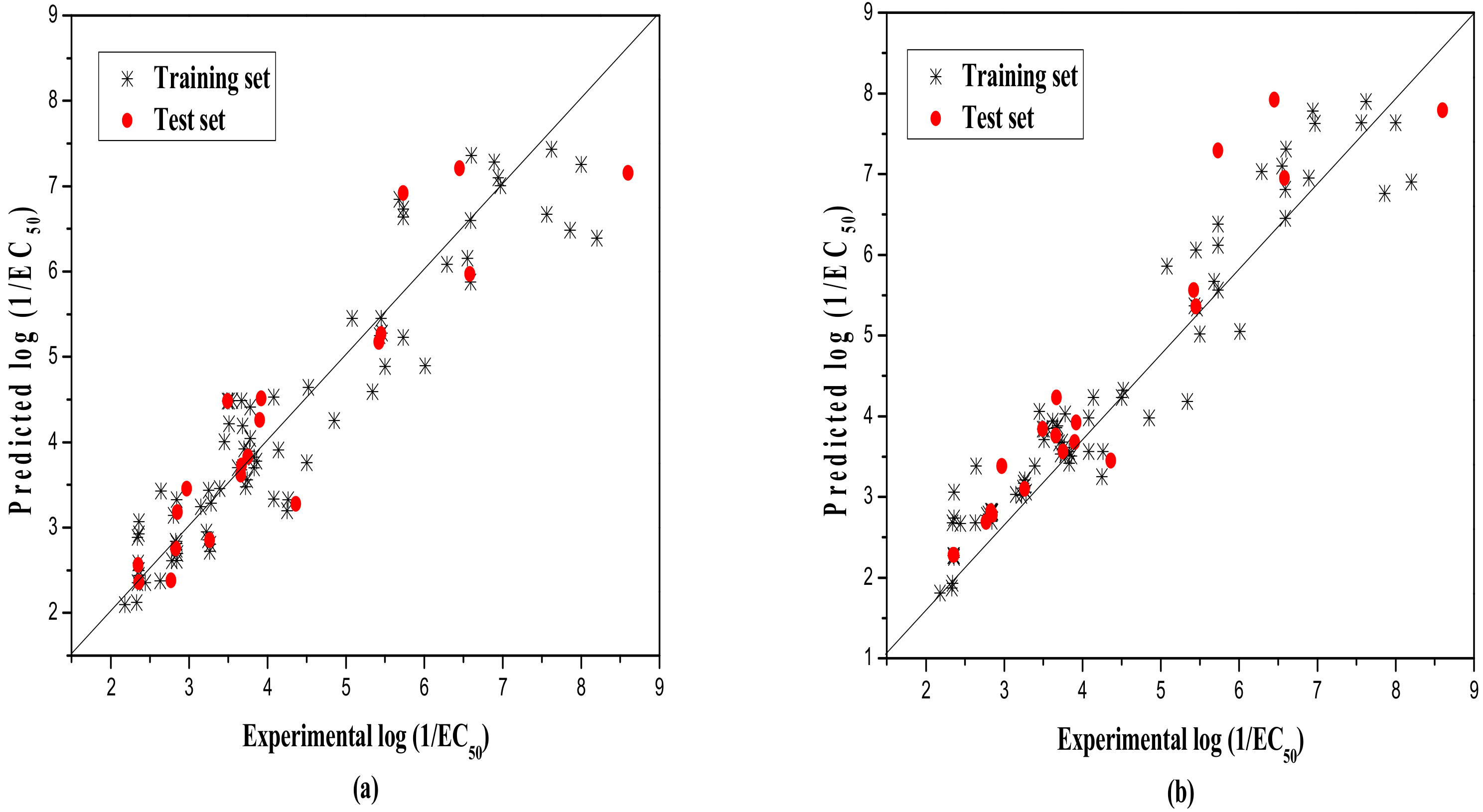
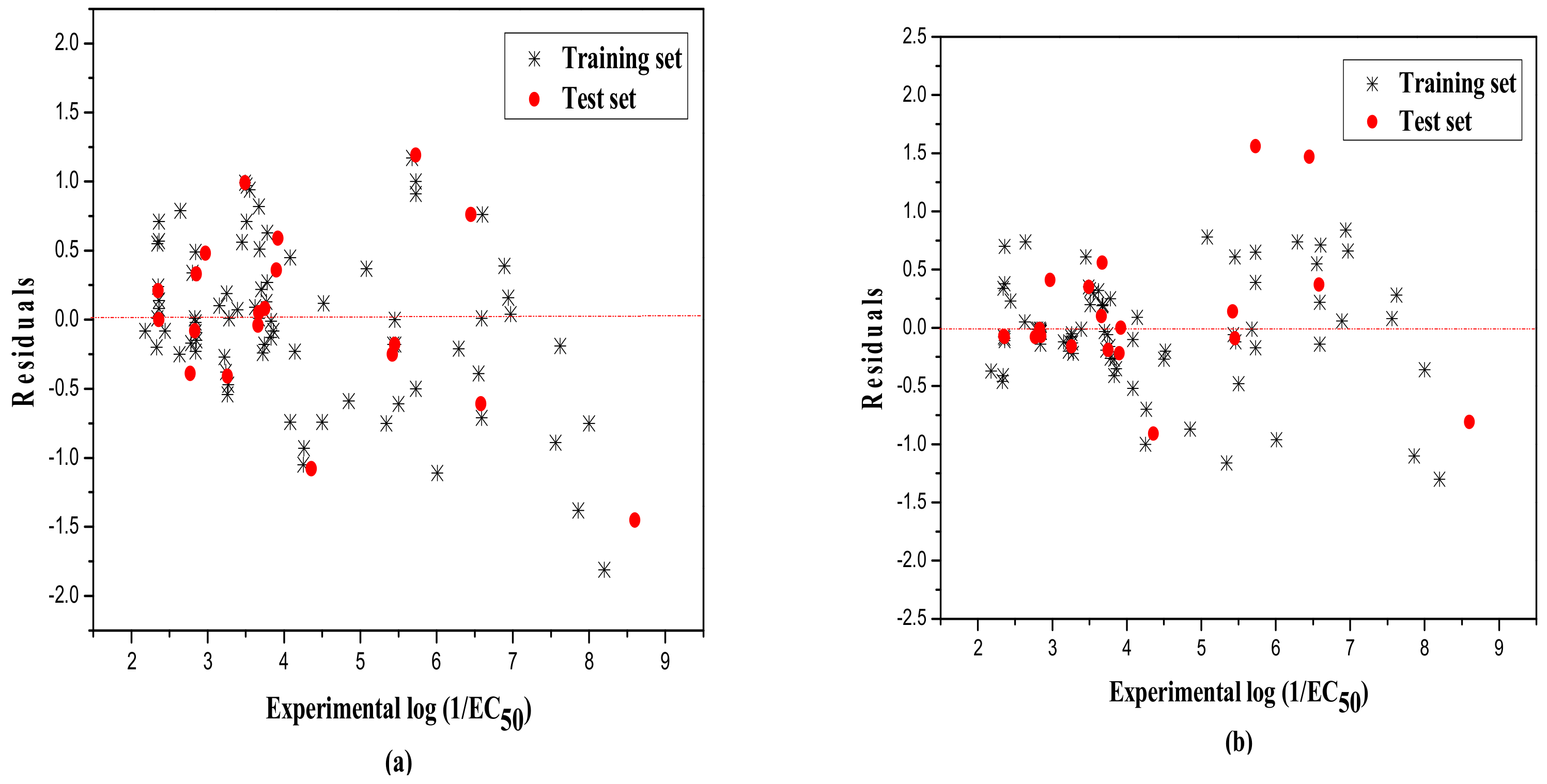
2.4. Validation Results of the Models
2.5. Interpretation of Model Descriptors
3. Materials and Methods
3.1. Datasets
3.2. Molecular Descriptors Generation and Selection
3.3. Multiple Linear Regressions (MLR)
3.4. Radial Basis Function Neural Networks (RBFNNs)
3.5. Applicability Domain (AD) of the Model
3.6. Validation of QSAR Models
3.6.1. Y-Randomization Test
3.6.2. Leave Many Out Cross-Validation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, T.; Zhou, X.H.; Wang, D.L.; Yin, D.Q.; Lin, Z.F. Using molecular docking between organic chemicals and lipid membrane to revise the well-known octanol-water partition coefficient of the mixture. Environ. Toxicol. Pharmacol. 2012, 34, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Villa, S.; Vighi, M.; Finizio, A. Experimental and predicted acute toxicity of antibacterial compounds and their mixtures using the luminescent bacterium Vibrio fischeri. Chemosphere 2014, 108, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Escher, B.I.; Baumer, A.; Bittermann, K.; Henneberger, L.; König, M.; Kühnert, C.; Klüver, N. General baseline toxicity QSAR for nonpolar, polar and ionisable chemicals and their mixtures in the bioluminescence inhibition assay with Aliivibrio fischeri. Environ. Sci. Process Impacts 2017, 19, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Teuschler, L.K.; Hertzberg, R.C. Current and future risk assessment guidelines, policy, and methods development for chemical mixtures. Toxicology 1995, 105, 137–144. [Google Scholar]
- Logan, D.T.; Wilson, H.T. An ecological risk assessment method for species exposed to contaminant mixtures. Environ. Toxicol. Chem. 1995, 14, 351–359. [Google Scholar] [CrossRef]
- de Bruijn, J.; Hansen, B.G.; Johansson, S.; Luotamo, M.; Munn, S.J.; Musset, C.; Olsen, S.I.; Olsson, H.; Paya-perez, A.B.; Pedersen, F.; et al. Technical Guidance Document on risk Assessment. Part 1. Part 2. 2003. Available online: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/technical-guidance-document-risk-assessment-part-1-part-2 (accessed on 30 October 2018).
- Altenburger, R.; Backhaus, T.; Boedeker, W.; Faust, M.; Scholze, M.; Grimme, L.H. Predictability of the toxicity of multiple chemical mixtures to Vibrio fischeri: Mixtures composed of similarly acting chemicals. Environ. Toxicol. Chem. 2010, 19, 2341–2347. [Google Scholar] [CrossRef]
- Lin, Z.; Yu, H.; Wei, D.; Wang, G.; Feng, J.; Wang, L. Prediction of mixture toxicity with its total hydrophobicity. Chemosphere 2002, 46, 305–310. [Google Scholar] [CrossRef]
- Wang, B.; Yu, G.; Zhang, Z.; Hu, H.; Wang, L. Quantitative structure-activity relationship and prediction of mixture toxicity of alkanols. Chin. Sci. Bull. 2006, 51, 2717–2723. [Google Scholar] [CrossRef]
- Luan, F.; Xu, X.; Liu, H.T.; Cordeiro, M.N.D.S. Prediction of the baseline toxicity of nonpolar narcotic chemical mixtures by QSAR approach. Chemosphere 2013, 90, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Lin, Z.; Ding, J.; Yin, D.; Zhang, Y. Application of the similarity parameter (λ) to prediction of the joint effects of nonequitoxic mixtures. Arch. Environ. Contam. Toxicol. 2011, 62, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Lin, Z.; Deng, Z.; Yin, D.; Zhang, Y. The joint effects of sulfonamides and their potentiator on photobacterium phosphoreum: Differences between the acute and chronic mixture toxicity mechanisms. Chemosphere 2012, 86, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Toropova, A.P.; Toropov, A.A.; Benfenati, E.; Gini, G.; Leszczynska, D.; Leszczynski, J. Coral: Models of toxicity of binary mixtures. Chemometr. Intell. Lab. Syst. 2012, 119, 39–43. [Google Scholar] [CrossRef]
- Tang, J.Y.M.; Mccarty, S.; Glenn, E.; Neale, P.A.; Warne, M.S.J.; Escher, B.I. Mixture effects of organic micropollutants present in water: Towards the development of effect-based water quality trigger values for baseline toxicity. Water Res. 2013, 47, 3300–3314. [Google Scholar] [PubMed]
- Yao, Z.; Lin, Z.; Wang, T.; Tian, D.; Zou, X.; Gao, Y. Using molecular docking-based binding energy to predict toxicity of binary mixture with different binding sites. Chemosphere 2013, 92, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, D.; Lin, Z.; An, Q.; Yin, C.; Huang, O. Prediction of mixture toxicity from the hormesis of a single chemical: A case study of combinations of antibiotics and quorum-sensing inhibitors with gram-negative bacteria. Chemosphere 2016, 150, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Topliss, J.G.; Edwards, R.P. Chance factors in studies of quantitative structure-activity relationships. J. Med. Chem. 1979, 22, 1238–1244. [Google Scholar] [PubMed]
- García, J.; Duchowicz, P.R.; Rozas, M.F.; Caram, J.A.; Mirífico, M.V.; Fernández, F.M.; Castro, E.A. A comparative QSAR on 1, 2, 5-thiadiazolidin-3-one 1, 1-dioxide compounds as selective inhibitors of human serine proteinases. J. Mol. Graph. Model. 2011, 31, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Riahi, S.; Pourbasheer, E.; Dinarvand, R.; Ganjali, M.R.; Norouzi, P. QSAR study of 2-(1-Propylpiperidin-4-yl)-1H-Benzimidazole-4-Carboxamide as PARP inhibitors for treatment of cancer. Chem. Biol. Drug Des. 2008, 72, 575–584. [Google Scholar] [CrossRef] [PubMed]
- ISIS Draw2.3; MDL Information Systems, Inc.: San Ramon, CA, USA, 1990–2000.
- HyperChem 6.01; Hypercube, Inc.: Waterloo, ON, Canada, 2000.
- Dewar, M.J.S.; Storch, D.M. Development and use of quantum molecular models. 75. Comparative tests of theoretical procedures for studying chemical reactions. J. Am. Chem. Soc. 1985, 107, 3898–3902. [Google Scholar] [CrossRef]
- Stewart, J.P.P. MOPAC 6.0, Quantum Chemistry Program Exchange, No. 455; Indiana University: Bloomington, IN, USA, 1989. [Google Scholar]
- Katritzky, A.R.; Lobanov, V.S.; Karelson, M. CODESSA 2.63: Training Manual; University of Florida: Gainesville, FL, USA, 1995. [Google Scholar]
- Luan, F.; Wang, X.H.; Liu, H.T.; Gao, Y.; Guo, Y.; Xie, Z.Y.; Zhang, X.Y. Studies on the quantitative relationship between the olfactory thresholds of pyrazine derivatives and their molecular structures. Flavour Frag. J. 2009, 24, 62–68. [Google Scholar] [CrossRef]
- Luan, F.; Kleandrova, V.V.; González-Díaz, H.; Ruso, J.M.; Melo, A.; Speck-Planche, A.; Cordeiro, M.N.D. Computer-aided nanotoxicology: Assessing cytotoxicity of nanoparticles under diverse experimental conditions by using a novel QSTR-perturbation approach. Nanoscale 2014, 6, 10623–10630. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. Mol. Inform. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- O’brien, R.M. A caution regarding rules of thumb for variance inflation factors. Qual. Quant. 2007, 41, 673–690. [Google Scholar] [CrossRef]
- Adimi, M.; Salimi, M.; Nekoei, M.; Pourbasheer, E.; Beheshti, A. A quantitative structure-activity relationship study on histamine receptor antagonists using the genetic algorithm multi-parameter linear regression method. J. Serb. Chem. Soc. 2012, 77, 639–650. [Google Scholar] [CrossRef]
- Haykin, S. Neural Networks: A Comprehensive Foundation, 3rd ed; Prentice-Hall, Inc: Upper Saddle River, NJ, USA, 2007; pp. 256–258. ISBN 0131471392. [Google Scholar]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Atkinson, A.C. Plots, transformations, and regression. An introduction to graphical methods of diagnostic regression analysis. J. R. Stat. Soc. 1985, 52, 1927–1934. [Google Scholar]
- Gadaleta, D.; Mangiatordi, G.F.; Catto, M.; Carotti, A.; Nicolotti, O. Applicability domain for QSAR models: Where theory meets reality. IJQSPR 2016, 1, 45–63. [Google Scholar] [CrossRef]
- Rücker, C.; Rücker, G.; Meringer, M. Y-randomization–a useful tool in QSAR validation, or folklore? J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| No. | Single Chemicals | CAS | −logEC50 (mol/L) | Residual | |
|---|---|---|---|---|---|
| Experimental | Predicted | ||||
| 1# | Acetaldehyde | 75-07-0 | 2.36 | 3.177 | 0.817 |
| 2# | Propanal | 123-38-6 | 2.72 | 3.212 | 0.492 |
| 3# | Butyraldehyde | 123-72-8 | 3.25 | 3.224 | −0.0265 |
| 4# | Valeraldehyde | 110-62-3 | 3.27 | 3.628 | 0.358 |
| 5# | Benzaldehyde | 100-52-7 | 3.43 | 4.552 | 1.122 |
| 6# | p-Nitrobenzaldehyde | 555-16-8 | 4.28 | 3.634 | −0.646 |
| 7# | p-Terephthaldehyde | 623-27-8 | 4.07 | 4.880 | 0.810 |
| 8# | p-Chlorobenzaldehyde | 104-88-1 | 3.97 | 3.876 | −0.094 |
| 9# | p-Bromobenzaldehyde | 1122-91-4 | 4.3 | 3.861 | −0.437 |
| 10# | p-Hydroxybenzaldehyde | 123-08-0 | 4.54 | 3.777 | −0.763 |
| 11# | p-Methyl benzaldehyde | 104-87-0 | 3.82 | 4.030 | 0.210 |
| 12# | p-Methoxybenzaldehyde | 123-11-5 | 4.03 | 3.985 | −0.0448 |
| 13# | p-Dimethylaminobenzaldehyde | 100-10-7 | 5.4 | 4.622 | −0.778 |
| 14# | Malononitrile | 109-77-3 | 2.55 | 1.783 | −0.767 |
| 15# | Glycolonitrile | 107-16-4 | 2.98 | 2.141 | −0.839 |
| 16# | α-Hydroxyisobutyronitrile | 75-86-5 | 3.61 | 3.834 | 0.227 |
| 17# | Allyl cyanide | 109-75-1 | 2.06 | 1.507 | −0.553 |
| 18# | Benzonitrile | 100-47-0 | 3.48 | 3.456 | −0.0237 |
| 19# | Benzyl cyanide | 140-29-4 | 4.23 | 2.963 | −1.267 |
| 20# | Acetonitrile | 1975-5-8 | 0.75 | 2.023 | 1.273 |
| 21# | Acrylonitrile | 107-13-1 | 1.51 | 1.467 | −0.0414 |
| 22# | Succinonitrile | 110-61-2 | 0.36 | 2.401 | 2.042 |
| 23# | Phthalonitrile | 91-15-6 | 3.51 | 2.622 | −0.888 |
| 24# | Lactonitrile | 78-97-7 | 2.01 | 2.440 | 0.430 |
| 25# | Atrazine | 1912-24-9 | 6.68 | 7.543 | 0863 |
| 26# | Prometryn | 7287-19-6 | 8.07 | 6.457 | −1.613 |
| 27# | Simetryn | 1014-70-6 | 6.29 | 5.565 | −0.725 |
| 28# | Prometone | 1610-18-0 | 8.99 | 7.801 | −1.182 |
| 29# | Simazine | 122-34-9 | 5.43 | 6.892 | 1.462 |
| 30# | Metribuzin | 21087-64-9 | 5.7 | 6.873 | 1.173 |
| 31# | Cyanazine | 21725-46-2 | 6.61 | 6.631 | 0.0212 |
| 32# | Terbutryn | 886-50-0 | 7.95 | 6.131 | −1.819 |
| 33# | Terbutylazine | 5915-41-3 | 6.93 | 8.225 | 1.295 |
| 34# | Ametryn | 834-12-8 | 6.56 | 5.516 | −1.044 |
| 35# | Diuron | 330-54-1 | 7.72 | 6.065 | −1.655 |
| 36# | Chlorotoluron | 15545-48-9 | 8.4 | 5.898 | −2.502 |
| 37# | Monolinuron | 1746-81-2 | 7.33 | 6.384 | −0.946 |
| 38# | Monuron | 150-68-5 | 6.3 | 6.254 | −0.0460 |
| 39# | Methabenzthiazuron | 18691-97-9 | 7.02 | 6.022 | −0.998 |
| 40# | Isoproturon | 34123-59-6 | 7.18 | 6.554 | −0.627 |
| 41# | Fenuron | 101-42-8 | 7.33 | 6.665 | −0.665 |
| 42# | Ethametsulfuron | 111353-84-5 | 4.13 | 6.302 | 2.172 |
| 43# | Chlorsulfuron | 64902-72-3 | 6.42 | 6.337 | −0.0833 |
| 44# | Metsulfuron | 79510-48-8 | 6.29 | 6.245 | −0.0447 |
| 45# | Sulfamethazine | 57-68-1 | 4.08 | 5.506 | 1.426 |
| 46# | Sulfapyridine | 144-83-2 | 3.84 | 3.407 | −0.433 |
| 47# | Sulfamethoxazole | 723-46-6 | 4.45 | 4.511 | 0.0609 |
| 48# | Sulfadiazine | 68-35-9 | 4.5 | 5.021 | 0.521 |
| 49# | Sulfisoxazole | 127-69-5 | 4.43 | 5.506 | 1.076 |
| 50# | Sulfamonomethoxine | 1220-83-3 | 5.05 | 4.535 | −0.515 |
| 51# | Sulfachloropyridazine | 80-32-0 | 4.78 | 5.117 | 0.337 |
| 52# | Sulfachinoxalin | 59-40-5 | 4.53 | 5.203 | 0.673 |
| 53# | Sulfamethoxydiazine | 651-06-9 | 4.41 | 5.050 | 0.640 |
| 54# | Sulfamethoxypyridazine | 80-35-3 | 4.36 | 4.934 | 0.579 |
| 55# | Trimethoprim | 738-70-5 | 3.22 | 5.209 | 1.989 |
| Coefficients | Standard Error | t-Test | Descriptors | VIF | MF |
|---|---|---|---|---|---|
| −0.405 | 0.463 | −0.874 | Intercept | ||
| −0.688 | 0.322 | −2.137 | Number of triple bonds (NTB) | 2.210 | −0.051 |
| 1.847 | 0.255 | 7.257 | Average Complementary Information content (order 2) (ACIC2) | 1.109 | 0.401 |
| 63.611 | 7.542 | 8.434 | Max partial charge for a C atom (Zefirov’s PC) () | 1.002 | 0.650 |
| ACIC2 | NTB | ||
|---|---|---|---|
| ACIC2 | 1.000 | ||
| NTB | −0.314 | 1.000 | |
| 0.740 | 0.013 | 1.000 |
| MLR | RBFNN | |
|---|---|---|
| R2 | 0.853 | 0.896 |
| 0.825 | 0.890 | |
| 0.849 | 0.896 | |
| (R2−)/R2 | 0.005 | 0.000 |
| k | 0.983 | 1.030 |
| F | 30.861 | 155.424 |
| RMS | 0.691 | 0.547 |
| No. | Chemicals in the Mixtures | The Ratio of Toxic Unit | Experimental−log(EC50mix) (mol/L) | MLR | RBFNN | |||
|---|---|---|---|---|---|---|---|---|
| Predicted−log(EC50mix) (mol/L) | Residual | Predicted−log(EC50mix) (mol/L) | Residual | Set* | ||||
| 1 | 1#:14# | 1:1 | 2.44 | 2.36 | −0.08 | 2.67 | 0.23 | A |
| 2 | 2#:14# | 1:1 | 2.63 | 2.38 | −0.25 | 2.68 | 0.05 | B |
| 3* | 3#:14# | 1:1 | 2.77 | 2.38 | −0.39 | 2.69 | −0.08 | T |
| 4 | 4#:14# | 1:1 | 2.78 | 2.61 | −0.17 | 2.77 | −0.01 | C |
| 5 | 5#:14# | 1:1 | 2.8 | 3.14 | 0.34 | 2.79 | −0.01 | D |
| 6 | 6#:14# | 1:1 | 2.84 | 2.61 | −0.23 | 2.80 | −0.04 | A |
| 7 | 7#:14# | 1:1 | 2.84 | 3.33 | 0.49 | 2.70 | −0.14 | B |
| 8* | 8#:14# | 1:1 | 2.83 | 2.75 | −0.08 | 2.82 | −0.01 | T |
| 9 | 9#:14# | 1:1 | 2.84 | 2.75 | −0.09 | 2.81 | −0.03 | C |
| 10 | 10#:14# | 1:1 | 2.85 | 2.70 | −0.15 | 2.81 | −0.04 | D |
| 11 | 11#:14# | 1:1 | 2.83 | 2.84 | 0.01 | 2.82 | −0.01 | A |
| 12 | 12#:14# | 1:1 | 2.84 | 2.82 | −0.02 | 2.82 | −0.02 | B |
| 13* | 13#:14# | 1:1 | 2.85 | 3.18 | 0.33 | 2.78 | −0.07 | T |
| 14 | 5#:15# | 1:1 | 3.15 | 3.25 | 0.10 | 3.03 | −0.12 | C |
| 15 | 6#:15# | 1:1 | 3.26 | 2.72 | −0.54 | 3.21 | −0.05 | D |
| 16 | 7#:15# | 1:1 | 3.25 | 3.44 | 0.19 | 3.17 | −0.08 | A |
| 17 | 8#:15# | 1:1 | 3.24 | 2.86 | −0.38 | 3.11 | −0.13 | B |
| 18* | 9#:15# | 1:1 | 3.26 | 2.85 | −0.41 | 3.10 | −0.16 | T |
| 19 | 10#:15# | 1:1 | 3.27 | 2.80 | −0.47 | 3.14 | −0.13 | C |
| 20 | 11#:15# | 1:1 | 3.22 | 2.95 | −0.27 | 3.02 | −0.20 | D |
| 21 | 13#:15# | 1:1 | 3.28 | 3.29 | 0.01 | 3.06 | −0.22 | A |
| 22 | 1#:16# | 1:1 | 2.64 | 3.43 | 0.79 | 3.38 | 0.74 | B |
| 23* | 2#:16# | 1:1 | 2.97 | 3.45 | 0.48 | 3.38 | 0.41 | T |
| 24 | 3#:16# | 1:1 | 3.39 | 3.46 | 0.07 | 3.38 | −0.01 | C |
| 25 | 5#:16# | 1:1 | 3.51 | 4.22 | 0.71 | 3.71 | 0.20 | D |
| 26 | 6#:16# | 1:1 | 3.83 | 3.70 | −0.13 | 3.42 | −0.41 | A |
| 27 | 7#:16# | 1:1 | 3.78 | 4.41 | 0.63 | 3.52 | −0.26 | B |
| 28* | 8#:16# | 1:1 | 3.75 | 3.83 | 0.08 | 3.56 | −0.19 | T |
| 29 | 9#:16# | 1:1 | 3.83 | 3.82 | −0.01 | 3.56 | −0.27 | C |
| 30 | 10#:16# | 1:1 | 3.86 | 3.78 | −0.08 | 3.51 | −0.35 | D |
| 31 | 11#:16# | 1:1 | 3.7 | 3.92 | 0.22 | 3.67 | −0.03 | A |
| 32 | 12#:16# | 1:1 | 3.77 | 3.90 | 0.13 | 3.61 | −0.16 | B |
| 33* | 13#:16# | 1:1 | 3.9 | 4.26 | 0.36 | 3.68 | −0.22 | T |
| 34 | 1#:17# | 1:1 | 2.18 | 2.10 | −0.08 | 1.81 | −0.37 | C |
| 35 | 3#:17# | 1:1 | 2.33 | 2.13 | −0.20 | 1.87 | −0.46 | D |
| 36 | 4#:17# | 1:1 | 2.34 | 2.35 | 0.01 | 1.93 | −0.41 | A |
| 37 | 5#:17# | 1:1 | 2.34 | 2.89 | 0.55 | 2.68 | 0.34 | B |
| 38* | 6#:17# | 1:1 | 2.36 | 2.36 | 0.00 | 2.28 | −0.08 | T |
| 39 | 7#:17# | 1:1 | 2.36 | 3.07 | 0.71 | 3.06 | 0.70 | C |
| 40 | 8#:17# | 1:1 | 2.36 | 2.50 | 0.14 | 2.27 | −0.09 | D |
| 41 | 10#:17# | 1:1 | 2.36 | 2.44 | 0.08 | 2.25 | −0.11 | A |
| 42 | 11#:17# | 1:1 | 2.35 | 2.59 | 0.24 | 2.27 | −0.08 | B |
| 43* | 12#:17# | 1:1 | 2.35 | 2.56 | 0.21 | 2.28 | −0.07 | T |
| 44 | 13#:17# | 1:1 | 2.36 | 2.93 | 0.57 | 2.74 | 0.38 | C |
| 45 | 5#:18# | 1:1 | 3.45 | 4.01 | 0.56 | 4.06 | 0.61 | D |
| 46 | 6#:18# | 1:1 | 3.72 | 3.48 | −0.24 | 3.53 | −0.19 | A |
| 47 | 7#:18# | 1:1 | 3.68 | 4.19 | 0.51 | 3.88 | 0.20 | B |
| 48* | 8#:18# | 1:1 | 3.66 | 3.62 | −0.04 | 3.76 | 0.10 | T |
| 49 | 10#:18# | 1:1 | 3.74 | 3.56 | −0.18 | 3.68 | −0.06 | C |
| 50 | 11#:18# | 1:1 | 3.62 | 3.71 | 0.09 | 3.94 | 0.32 | D |
| 51 | 12#:18# | 1:1 | 3.67 | 3.68 | 0.01 | 3.86 | 0.19 | A |
| 52 | 13#:18# | 1:1 | 3.78 | 4.05 | 0.27 | 4.03 | 0.25 | B |
| 53* | 5#:19# | 1:1 | 3.67 | 3.72 | 0.05 | 4.23 | 0.56 | T |
| 54 | 6#:19# | 1:1 | 4.25 | 3.20 | −1.05 | 3.25 | −1.00 | C |
| 55 | 7#:19# | 1:1 | 4.14 | 3.91 | −0.23 | 4.23 | 0.09 | D |
| 56 | 8#:19# | 1:1 | 4.08 | 3.34 | −0.74 | 3.56 | −0.52 | A |
| 67 | 9#:19# | 1:1 | 4.26 | 3.33 | −0.93 | 3.56 | −0.70 | B |
| 58* | 10#19# | 1:1 | 4.36 | 3.28 | −1.08 | 3.45 | −0.91 | T |
| 59 | 13#:19# | 1:1 | 4.5 | 3.76 | −0.74 | 4.23 | −0.27 | C |
| 60 | 25#:35# | 1:1 | 6.94 | 7.10 | 0.16 | 7.78 | 0.84 | D |
| 61 | 25#:36# | 1:1 | 6.97 | 7.01 | 0.04 | 7.63 | 0.66 | A |
| 62 | 25#:37# | 1:1 | 6.89 | 7.28 | 0.39 | 6.95 | 0.06 | B |
| 63* | 25#:38# | 1:1 | 6.45 | 7.21 | 0.76 | 7.92 | 1.47 | T |
| 64 | 26#:35# | 1:1 | 7.86 | 6.48 | −1.38 | 6.76 | −1.10 | C |
| 65 | 26#:36# | 1:1 | 8.2 | 6.39 | −1.81 | 6.90 | −1.30 | D |
| 66 | 26#:37# | 1:1 | 7.56 | 6.67 | −0.89 | 7.64 | 0.08 | A |
| 67 | 26#:38# | 1:1 | 6.59 | 6.60 | 0.01 | 6.45 | −0.14 | B |
| 68* | 27#:35# | 1:1 | 6.58 | 5.97 | −0.61 | 6.95 | 0.37 | T |
| 69 | 27#:36# | 1:1 | 6.59 | 5.88 | −0.71 | 6.81 | 0.22 | C |
| 70 | 27#:37# | 1:1 | 6.55 | 6.16 | −0.39 | 7.10 | 0.55 | D |
| 71 | 27#:38# | 1:1 | 6.29 | 6.08 | −0.21 | 7.03 | 0.74 | A |
| 72 | 28#:35# | 1:1 | 8 | 7.25 | −0.75 | 7.64 | −0.36 | B |
| 73* | 28#:36# | 1:1 | 8.6 | 7.15 | −1.45 | 7.79 | −0.81 | T |
| 74 | 28#:37# | 1:1 | 7.62 | 7.43 | −0.19 | 7.90 | 0.28 | C |
| 75 | 28#:38# | 1:1 | 6.6 | 7.36 | 0.76 | 7.31 | 0.71 | D |
| 76 | 29#:35# | 1:1 | 5.73 | 6.73 | 1.00 | 6.12 | 0.39 | A |
| 77 | 29#:36# | 1:1 | 5.73 | 6.64 | 0.91 | 6.38 | 0.65 | B |
| 78* | 29#:37# | 1:1 | 5.73 | 6.92 | 1.19 | 7.29 | 1.56 | T |
| 79 | 29#:38# | 1:1 | 5.68 | 6.85 | 1.17 | 5.67 | −0.01 | C |
| 80 | 45#:55# | 1:1 | 5.08 | 5.45 | 0.37 | 5.86 | 0.78 | D |
| 81 | 46#:55# | 1:1 | 4.85 | 4.26 | −0.59 | 3.98 | −0.87 | A |
| 82 | 47#:55# | 1:1 | 5.5 | 4.89 | −0.61 | 5.02 | −0.48 | B |
| 83* | 48#:55# | 1:1 | 5.42 | 5.17 | −0.25 | 5.56 | 0.14 | T |
| 84 | 49#:55# | 1:1 | 5.45 | 5.45 | 0.00 | 6.06 | 0.61 | C |
| 85 | 50#:55# | 1:1 | 6.01 | 4.90 | −1.11 | 5.05 | −0.96 | D |
| 86 | 51#:55# | 1:1 | 5.73 | 5.23 | −0.50 | 5.56 | −0.17 | A |
| 87 | 47#:55# | 13396:1 | 3.49 | 4.48 | 0.99 | 3.84 | 0.35 | B |
| 88* | 47#:55# | 8587:1 | 3.49 | 4.48 | 0.99 | 3.84 | 0.35 | T |
| 89 | 47#:55# | 2747:1 | 3.49 | 4.48 | 0.99 | 3.84 | 0.35 | C |
| 90 | 47#:55# | 858:1 | 3.51 | 4.48 | 0.97 | 3.84 | 0.33 | D |
| 91 | 47#:55# | 274:1 | 3.55 | 4.49 | 0.94 | 3.85 | 0.30 | A |
| 92 | 47#:55# | 85:1 | 3.67 | 4.49 | 0.82 | 3.86 | 0.19 | B |
| 93* | 47#:55# | 27:1 | 3.92 | 4.51 | 0.59 | 3.92 | 0.00 | T |
| 94 | 47#:55# | 15:1 | 4.08 | 4.53 | 0.45 | 3.98 | −0.10 | C |
| 95 | 47#:55# | 4:1 | 4.52 | 4.64 | 0.12 | 4.32 | −0.20 | D |
| 96 | 47#:55# | 1:6 | 5.34 | 4.59 | −0.75 | 4.18 | −1.16 | A |
| 97 | 47#:55# | 1:21 | 5.43 | 5.25 | −0.18 | 5.37 | −0.06 | B |
| 98* | 47#:55# | 1:37 | 5.45 | 5.27 | −0.18 | 5.36 | −0.09 | T |
| 99 | 47#:55# | 1:116 | 5.46 | 5.28 | −0.18 | 5.34 | −0.12 | C |
| No. | MLR | RBFNN | ||
|---|---|---|---|---|
| R2 | LOOq2 | R2 | LOOq2 | |
| 1 | 0.028 | 0.016 | 0.019 | 0.006 |
| 2 | 0.013 | 0.000 | 0.027 | 0.014 |
| 3 | 0.02 | 0.007 | 0.017 | 0.004 |
| 4 | 0.047 | 0.035 | 0.035 | 0.022 |
| 5 | 0.03 | 0.017 | 0.034 | 0.022 |
| 6 | 0.014 | 0.002 | 0.024 | 0.011 |
| 7 | 0.034 | 0.022 | 0.017 | 0.004 |
| 8 | 0.013 | 0.001 | 0.04 | 0.028 |
| 9 | 0.018 | 0.005 | 0.024 | 0.012 |
| 10 | 0.049 | 0.036 | 0.014 | 0.001 |
| Average | 0.0266 | 0.0141 | 0.0251 | 0.0124 |
| Training Set | R2 | F | RMS | Test Set | R2 | F | RMS |
|---|---|---|---|---|---|---|---|
| A+B+C+D | 0.869 | 165.290 | 0.599 | T | 0.853 | 30.861 | 0.691 |
| B+C+D+T | 0.857 | 150.314 | 0.633 | A | 0.904 | 50.357 | 0.527 |
| A+C+D+T | 0.864 | 159.122 | 0.611 | B | 0.883 | 40.100 | 0.611 |
| A+B+D+T | 0.867 | 162.819 | 0.600 | C | 0.866 | 32.528 | 0.674 |
| A+B+C+T | 0.868 | 167.263 | 0.594 | D | 0.856 | 29.716 | 0.713 |
| Average | 0.865 | 160.962 | 0.607 | 0.872 | 36.712 | 0.643 |
| Training Set | R2 | F | RMS | Test Set | R2 | F | RMS |
|---|---|---|---|---|---|---|---|
| A+B+C+D | 0.925 | 950.686 | 0.447 | T | 0.896 | 155.424 | 0.547 |
| B+C+D+T | 0.915 | 827.525 | 0.494 | A | 0.932 | 247.421 | 0.418 |
| A+C+D+T | 0.910 | 778.942 | 0.519 | B | 0.954 | 371.815 | 0.361 |
| A+B+D+T | 0.915 | 824.679 | 0.500 | C | 0.931 | 244.294 | 0.455 |
| A+B+C+T | 0.921 | 911.329 | 0.472 | D | 0.900 | 152.589 | 0.559 |
| Average | 0.917 | 858.632 | 0.486 | 0.923 | 234.309 | 0.468 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Tang, L.; Luan, F.; Cordeiro, M.N.D.S. Prediction of the Toxicity of Binary Mixtures by QSAR Approach Using the Hypothetical Descriptors. Int. J. Mol. Sci. 2018, 19, 3423. https://doi.org/10.3390/ijms19113423
Wang T, Tang L, Luan F, Cordeiro MNDS. Prediction of the Toxicity of Binary Mixtures by QSAR Approach Using the Hypothetical Descriptors. International Journal of Molecular Sciences. 2018; 19(11):3423. https://doi.org/10.3390/ijms19113423
Chicago/Turabian StyleWang, Ting, Lili Tang, Feng Luan, and M. Natália D. S. Cordeiro. 2018. "Prediction of the Toxicity of Binary Mixtures by QSAR Approach Using the Hypothetical Descriptors" International Journal of Molecular Sciences 19, no. 11: 3423. https://doi.org/10.3390/ijms19113423
APA StyleWang, T., Tang, L., Luan, F., & Cordeiro, M. N. D. S. (2018). Prediction of the Toxicity of Binary Mixtures by QSAR Approach Using the Hypothetical Descriptors. International Journal of Molecular Sciences, 19(11), 3423. https://doi.org/10.3390/ijms19113423