Composition and Genetic Diversity of the Nicotiana tabacum Microbiome in Different Topographic Areas and Growth Periods



Abstract
1. Introduction
2. Results
2.1. Sequencing Results
2.2. Overview of Fungal Taxonomic Composition
2.3. Dissimilarity of Fungal Communities at Different Growth Stages
2.4. Dissimilarity of Fungal Communities at Different Altitudes
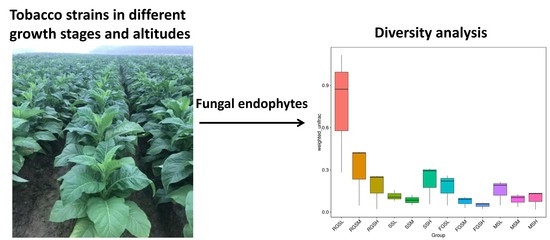
2.5. Alpha Diversity
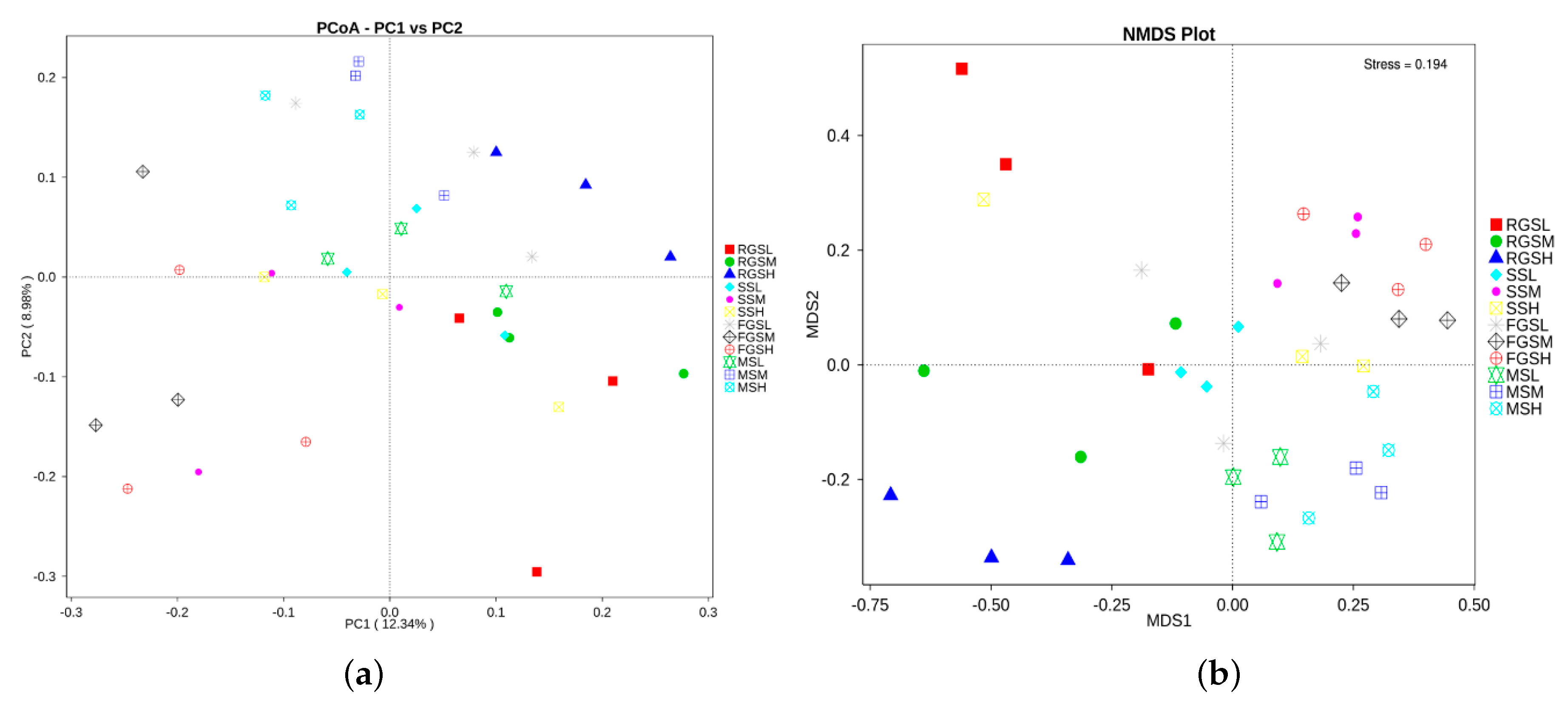
2.6. Beta Diversity
3. Discussion
4. Materials and Methods
4.1. Material Collection
4.2. DNA Extraction and PCR Amplification
4.3. Library Construction and Sequencing
4.4. Data Processing
4.5. OTU Clustering and Annotation
4.6. Alpha Diversity
4.7. Beta Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hyde, K.D.; Soytong, K. The fungal endophyte dilemma. Fungal Divers. 2008, 33, 163–173. [Google Scholar]
- Saikkonen, K.; Faeth, S.H.; Helander, M.; Sullivan, T.J. Fungal endophytes: A continuum of interactions with host plants. Ann. Rev. Ecol. Syst. 1998, 29, 319–343. [Google Scholar] [CrossRef]
- Spurr, H.W. Characterization of endophytic fungi in healthy leaves of nicotiana spp. Phytopathology 1975, 65, 417. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Zhao, K.; Wang, R.; Zhang, X. Diversity of the rhizosphere soil culture-dependent fungi of mature tobacco. Am. J. Microbiol. 2011, 2, 9–14. [Google Scholar]
- Norse, D. Fungi isoalted from surface-sterilized toabcco leaves. Transact. Brit. Mycol. Soc. 1972, 58, 515–518. [Google Scholar] [CrossRef]
- Guo, L.D.; Hyde, K.D.; Liew, E.C.Y. Identification of endophytic fungi from Livistona chinensis based on morphology and rDNA sequences. New Phytol. 2010, 147, 617–630. [Google Scholar] [CrossRef]
- Welty, R.E.; Lucas, G.B.; Fletcher, J.T.; Yang, H. Fungi isolated from tobacco leaves and brown-spot lesions before and after flue-curing. Appl. Microbiol. 1968, 16, 1309. [Google Scholar] [PubMed]
- English, J.T.; Mitchell, D.J. Development of microbial communities associated with tobacco root systems. Soil Biol. Biochem. 1988, 20, 137–144. [Google Scholar] [CrossRef]
- Makimura, K.; Tamura, Y.; Mochizuki, T.; Hasegawa, A.; Tajiri, Y.; Hanazawa, R.; Uchida, K.; Saito, H.; Yamaguchi, H. Phylogenetic classification and species identification of dermatophyte strains based on DNA sequences of nuclear ribosomal internal transcribed spacer 1 regions. J. Clin. Microbiol. 1999, 37, 920–924. [Google Scholar] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Hyde, K.D.; Pawłowska, J.; Ryberg, M.; Tedersoo, L.; Aas, A.B.; Alias, S.A.; Alves, A.; Anderson, C.L.; Antonelli, A. Improving ITS sequence data for identification of plant pathogenic fungi. Fungal Divers. 2014, 67, 11–19. [Google Scholar] [CrossRef]
- Gargas, A.; Taylor, J.W. Polymerase chain reaction (PCR) primers for amplifying and sequencing nuclear 18s rDNA from lichenized fungi. Mycologia 1992, 84, 589–592. [Google Scholar] [CrossRef]
- Bachy, C.; Dolan, J.R.; Deschamps, P.; Moreira, D. Accuracy of protist diversity assessments: Morphology compared with cloning and direct pyrosequencing of 18s rRNA genes and ITS regions using the conspicuous tintinnid ciliates as a case study. ISME J. 2013, 7, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Pryce, T.M.; Palladino, S.; Kay, I.D.; Coombs, G.W. Rapid identification of fungi by sequencing the ITS1 and ITS2 regions using an automated capillary electrophoresis system. Med. Mycol. 2003, 41, 369. [Google Scholar] [CrossRef] [PubMed]
- Brookman, J.L.; Mennim, G.; Trinci, A.P.J.; Theodorou, M.K.; Tuckwell, D.S. Identification and characterization of anaerobic gut fungi using molecular methodologies based on ribosomal ITS1 and 18s rRNA. Microbiology 2000, 146, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.M.; Kauserud, H. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 2013, 13, 218. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.N.; Burton, O.T.; Wise, P.; Zhang, Y.Q.; Hobson, S.A.; Lloret, M.G.; Chehoud, C.; Kuczynski, J.; Desantis, T.; Warrington, J. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J. Allergy Clin. Immunol. 2013, 131, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Mishra, A.K.; Tiwari, S.; Singh, H.N.; Kalra, A. Enhanced tolerance of Mentha arvensis against Meloidogyne incognita (Kofoid and White) Chitwood through mutualistic endophytes and PGPRs. J. Plant Interact. 2011, 6, 247–253. [Google Scholar] [CrossRef]
- Sherameti, I.; Shahollari, B.; Venus, Y.; Altschmied, L.; Varma, A.; Oelmüller, R. The endophytic fungus Piriformospora indica stimulates the expression of nitrate reductase and the starch-degrading enzyme glucan-water dikinase in tobacco and Arabidopsis roots through a homeodomain transcription factor that binds to a conserved motif in their promoters. J. Biol. Chem. 2005, 280, 26241–26247. [Google Scholar] [PubMed]
- Pei, Z.; Zhang, M. Distribution characteristics of endophytic fungi in tobacco. J. Henan Agric. Sci. 2009, 6, 97–99. [Google Scholar]
- Novotný, Č.; Erbanova, P.; Cajthaml, T.; Rothschild, N.; Dosoretz, C.; Šašek, V. Irpex lacteus, a white rot fungus applicable to water and soil bioremediation. Appl. Microbiol. Biotechnol. 2000, 54, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Baborová, P.; Möder, M.; Baldrian, P.; Cajthamlová, K.; Cajthaml, T. Purification of a new manganese peroxidase of the white-rot fungus Irpex lacteus, and degradation of polycyclic aromatic hydrocarbons by the enzyme. Res. Microbiol. 2006, 157, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.F.; Xing, H.H.; Wang, Y.D.; Yu, Z.H.; Yan, K.L.; Zhou, K.; Du, G. Prenylated isocoumarins from the fermentation products of the endophytic fungus Aspergillus versicolor and their anti-tobacco mosaic virus activities. Chem. Nat. Compd. 2017, 53, 436–439. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, M.; Hu, L.; Liu, G.; Zhou, K.; Gu, L.; Zhang, Z. Function of endophytes and their corresponding functions on tobacco. Tobacco Sci. Technol. 2017, 49, 33–41. [Google Scholar]
- Jing, Y.D.; He, Z.L.; Yang, X.E. Role of soil rhizobacteria in phytoremediation of heavy metal contaminated soils. J. Zhejiang Univ. Sci. B 2007, 8, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Houlden, A.; Timms-Wilson, T.M.; Day, M.J.; Bailey, M.J. Influence of plant developmental stage on microbial community structure and activity in the rhizosphere of three field crops. FEMS Microbiol. Ecol. 2008, 65, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, R.B.; Whittaker, R.H. Altitudinally coordinated patterns of soils and vegetation in-the San Jacinto mountains, California. Soil Sci. 1976, 121, 114–124. [Google Scholar] [CrossRef]
- Bangroo, S.A.; Najar, G.R.; Rasool, A. Effect of altitude and aspect on soil organic carbon and nitrogen stocks in the Himalayan Mawer Forest Range. Catena 2017, 158, 63–68. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, D.; Sun, D.; Wang, X.; Smith, J.L.; Du, G. Impacts of altitude and position on the rates of soil nitrogen mineralization and nitrification in alpine meadows on the eastern Qinghai–Tibetan Plateau, China. Biol. Fert. Soils 2012, 48, 393–400. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, A.; Yang, W.; Xu, Z.; Wu, F.; Tan, B.; Liu, Y.; Chen, L. Soil microbial abundance and community structure vary with altitude and season in the coniferous forests, China. J. Soils Sediments 2016, 17, 1–11. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E. Chimeric 16s rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtssonpalme, J.; Callaghan, T.M. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Observed Species | Shannon | Simpson | Chao1 | ACE | Good’s Coverage | PD_Whole Tree |
|---|---|---|---|---|---|---|---|
| SSL | 161.33 ± 11.59 | 1.24 ± 0.23 | 0.31 ± 0.06 | 188.74 ± 6.69 | 202.87 ± 18.15 | 0.99 ± 0 | 48.24 ± 2.25 |
| SSM | 108.67 ± 15.95 | 0.98 ± 0.36 | 0.26 ± 0.10 | 120.22 ± 22.99 | 126.28 ± 27.77 | 0.99 ± 0 | 39.10 ± 6.89 |
| SSH | 161.67 ± 24.08 | 1.15 ± 0.54 | 0.29 ± 0.14 | 183.57 ± 23.35 | 193.26 ± 18.65 | 0.99 ± 0 | 49.96 ± 9.17 |
| RGSL | 204.54 ± 69.00 | 2.21 ± 1.14 | 0.52 ± 0.28 | 232.33 ± 65.59 | 246.19 ± 76.29 | 0.99 ± 0 | 56.10 ± 11.99 |
| RGSM | 196.33 ± 36.56 | 1.75 ± 0.88 | 0.40 ± 0.17 | 231.17 ± 54.97 | 247.21 ± 51.28 | 0.99 ± 0 | 52.26± 4.58 |
| RGSH | 238.33 ± 61.61 | 1.28 ± 0.60 | 0.29 ± 0.12 | 261.76 ± 59.95 | 268.72 ± 60.71 | 0.99 ± 0 | 65.16 ± 8.26 |
| FGSL | 162.67 ± 14.57 | 1.29 ± 0.30 | 0.33 ± 0.09 | 196.60 ± 28.75 | 198.75 ± 18.52 | 0.99 ± 0 | 50.46 ± 5.90 |
| FGSM | 134.13 ± 18.52 | 1.08 ± 0.34 | 0.29 ± 0.09 | 159.54 ± 29.82 | 169.47 ± 30.37 | 0.99 ± 0 | 51.84 ± 3.10 |
| FGSH | 127.67 ± 19.30 | 1.05 ± 0.40 | 0.28 ± 0.09 | 136.94 ± 21.49 | 141.81 ± 20.66 | 1.00 ± 0 | 48.14 ± 6.40 |
| MSL | 164.67 ± 44.77 | 1.20 ± 0.42 | 0.30 ± 0.12 | 188.69 ± 60.70 | 196.37 ± 57.17 | 0.99 ± 0 | 54.48 ± 7.73 |
| MSM | 88.67 ± 12.66 | 0.94 ± 0.27 | 0.25 ± 0.06 | 99.51 ± 17.63 | 105.90 ± 23.83 | 1.00 ± 0 | 31.27 ± 5.26 |
| MSH | 102.33 ± 26.08 | 0.90 ± 0.27 | 0.25 ± 0.07 | 122.94 ± 29.65 | 127.68 ± 34.26 | 0.99 ± 0 | 35.47 ± 6.91 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.-L.; Cao, M.; Liu, X.-M.; Du, Y.-M.; Shen, G.-M.; Zhang, Z.-F.; Li, J.-H.; Zhang, P. Composition and Genetic Diversity of the Nicotiana tabacum Microbiome in Different Topographic Areas and Growth Periods. Int. J. Mol. Sci. 2018, 19, 3421. https://doi.org/10.3390/ijms19113421
Yuan X-L, Cao M, Liu X-M, Du Y-M, Shen G-M, Zhang Z-F, Li J-H, Zhang P. Composition and Genetic Diversity of the Nicotiana tabacum Microbiome in Different Topographic Areas and Growth Periods. International Journal of Molecular Sciences. 2018; 19(11):3421. https://doi.org/10.3390/ijms19113421
Chicago/Turabian StyleYuan, Xiao-Long, Min Cao, Xin-Min Liu, Yong-Mei Du, Guo-Ming Shen, Zhong-Feng Zhang, Jin-Hai Li, and Peng Zhang. 2018. "Composition and Genetic Diversity of the Nicotiana tabacum Microbiome in Different Topographic Areas and Growth Periods" International Journal of Molecular Sciences 19, no. 11: 3421. https://doi.org/10.3390/ijms19113421
APA StyleYuan, X.-L., Cao, M., Liu, X.-M., Du, Y.-M., Shen, G.-M., Zhang, Z.-F., Li, J.-H., & Zhang, P. (2018). Composition and Genetic Diversity of the Nicotiana tabacum Microbiome in Different Topographic Areas and Growth Periods. International Journal of Molecular Sciences, 19(11), 3421. https://doi.org/10.3390/ijms19113421