Exogenous Cripto-1 Suppresses Self-Renewal of Cancer Stem Cell Model

 ,
,  , , , ,
, , , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Expression of Cr-1 and Related Molecules in miPS-LLCcm Cells
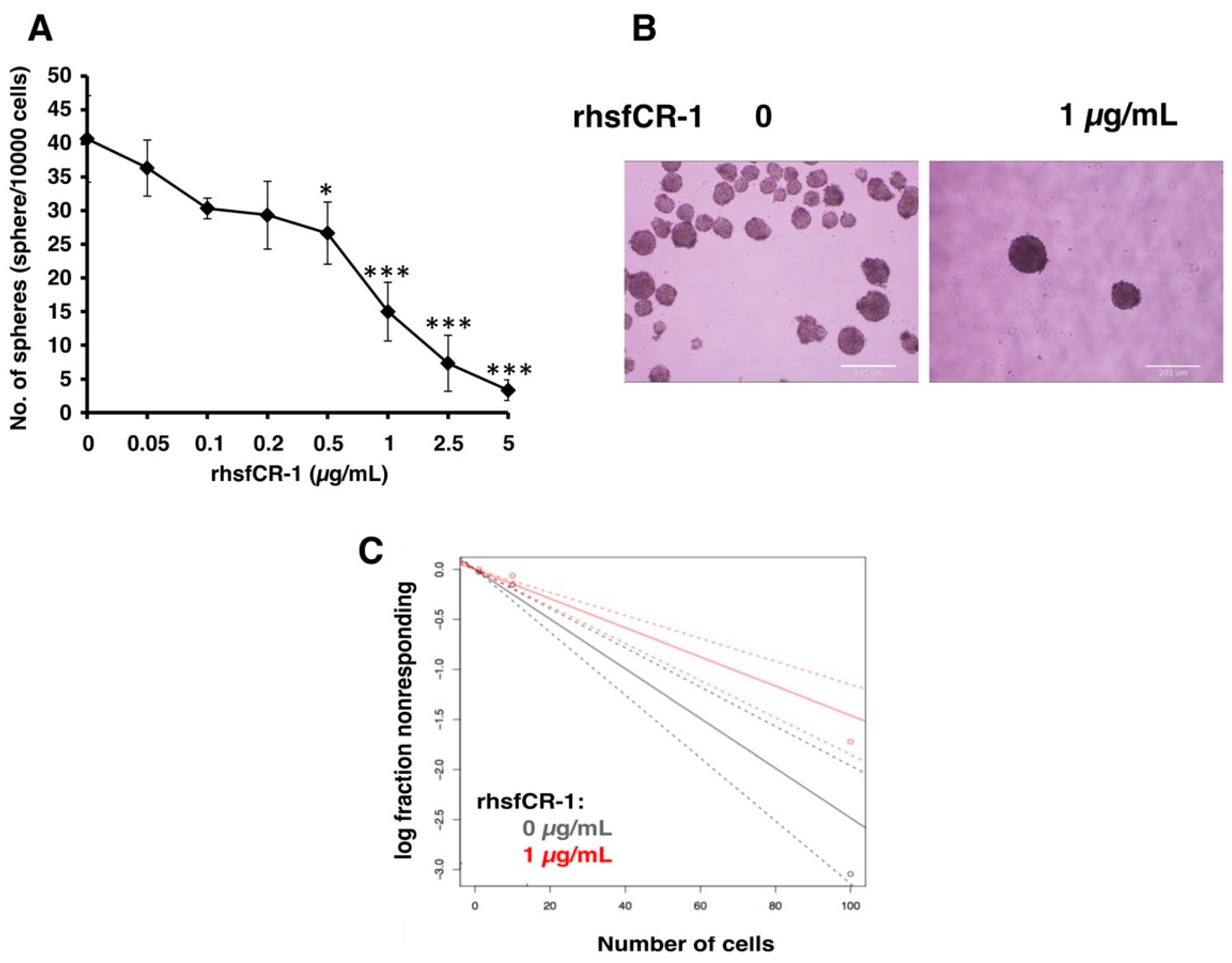
2.2. rhsfCR-1 Suppressed Differentiation, Proliferation and Sphere Formation of miPS-LLCcm Cells
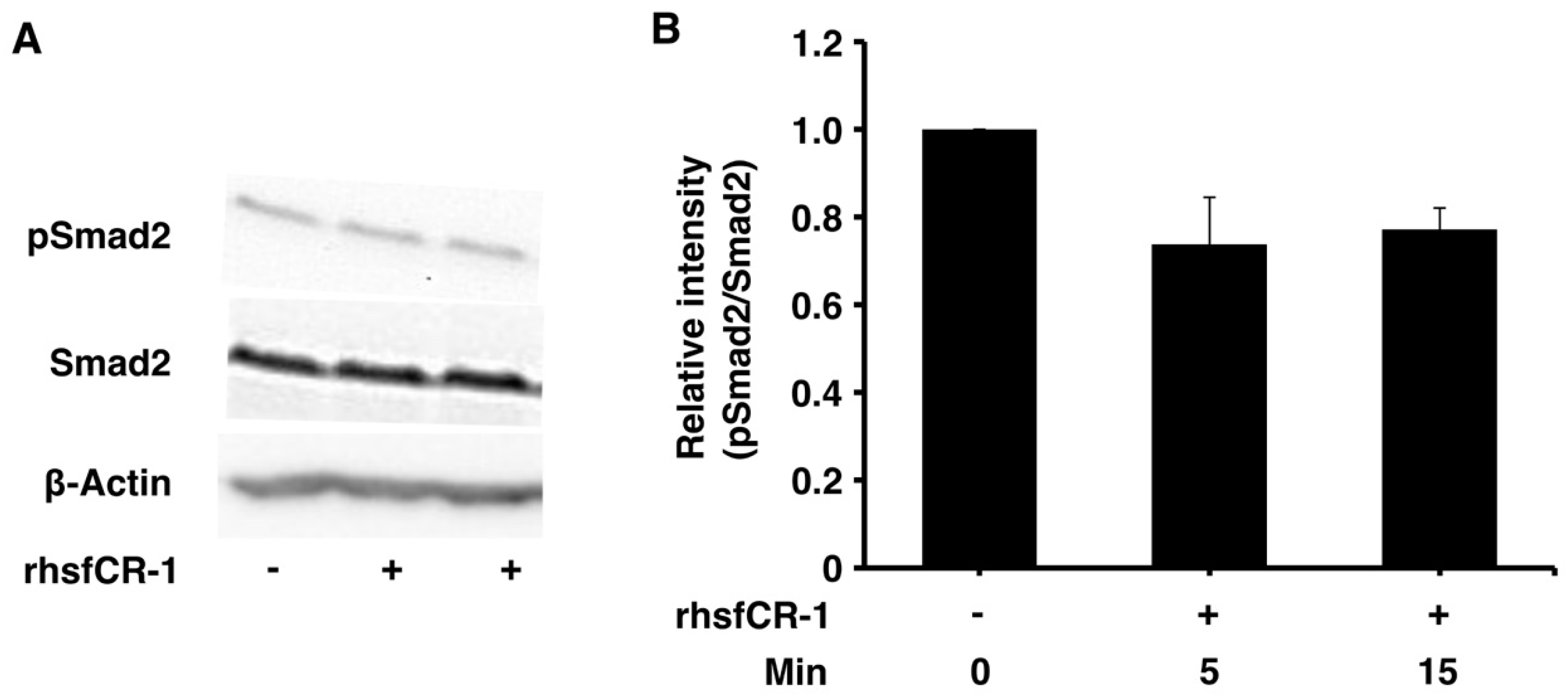
2.3. rhsfCR-1 Suppressed Phosphorylation of Smad2 in miPS-LLCcm Cells
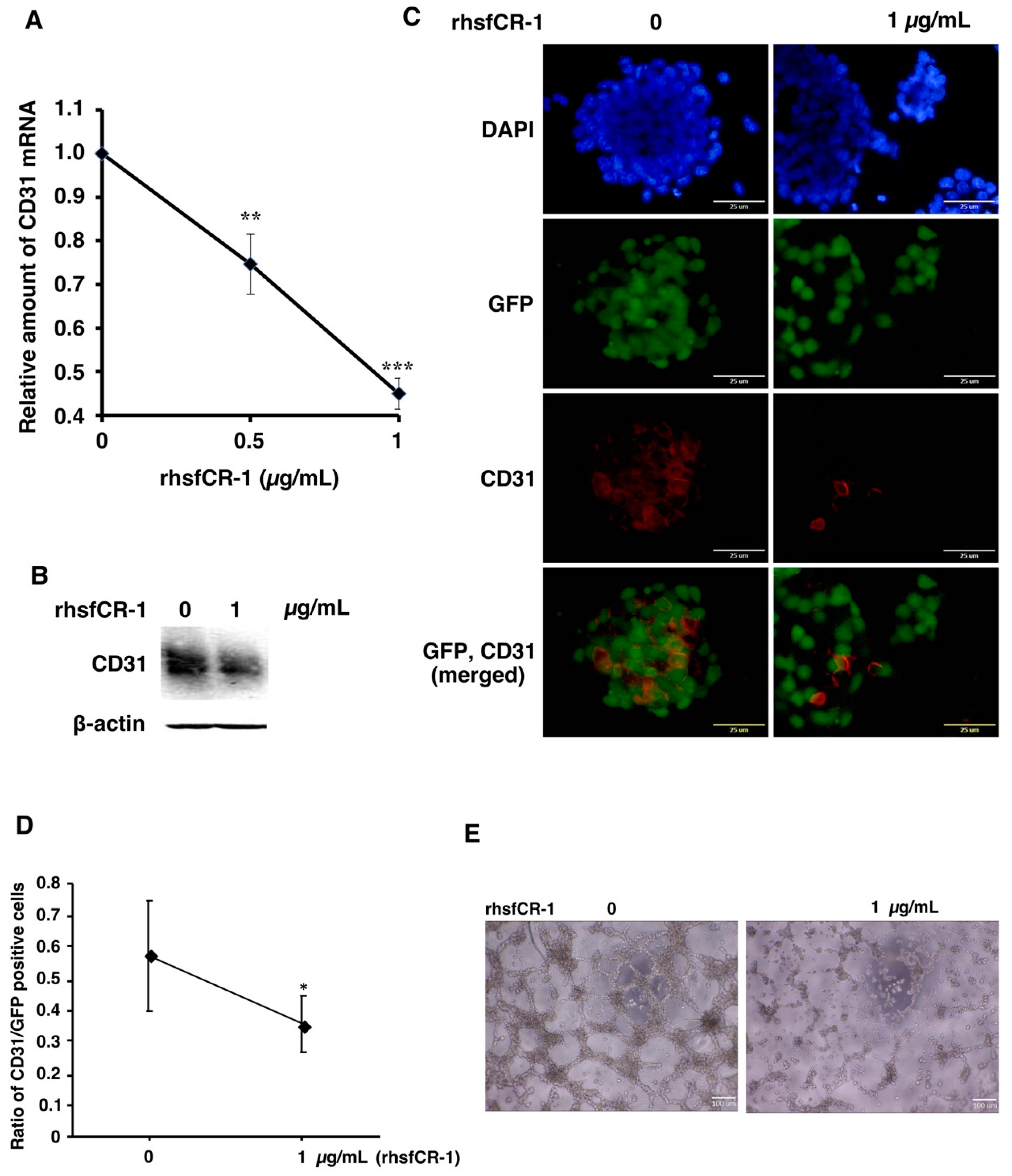
2.4. rhsfCR-1 Suppressed Endothelial Cell Tube Formation by Differentiated miPS-LLCcm
2.5. rhsfCR-1 Enhanced the Expression of Klf4 and c-Myc in miPS-LLCcm Cells
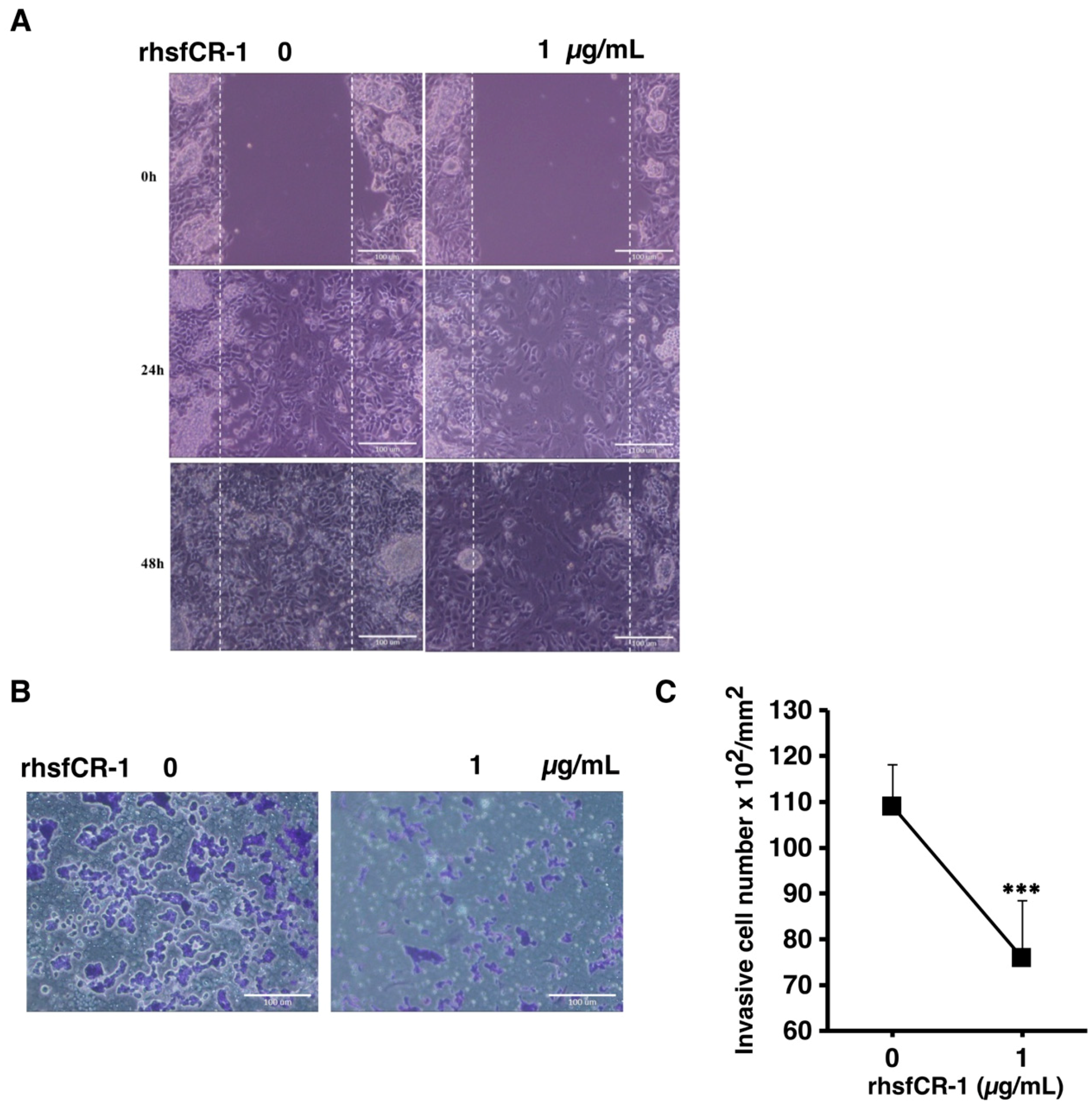
2.6. rhsfCR-1 Suppressed Migration and Invasion of miPS-LLCcm Cells
3. Discussion
4. Materials and Methods
4.1. Mutagenesis, Expression and Purification of Truncated Soluble rhsfCR-1
4.2. Refolding and Dialysis of rhsfCR-1 Protein
4.3. SDS-PAGE and Estimation of Protein
4.4. Cell Culture
4.5. Cell Proliferation and Viability Assays
4.6. Apoptosis and Cell Cycle Analysis
4.7. Sphere Formation and Extreme Limiting Dilution Analysis
4.8. In Vitro Tube Formation Assay
4.9. Immunofluorescence Staining
4.10. RNA Extraction and Quantitative Reverse Transcription PCR (rt-qPCR)
4.11. In Vitro Migration and Invasion Assays
4.12. Western Blotting
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dono, R.; Scalera, L.; Pacifico, F.; Acampora, D.; Persico, M.G.; Simeone, A. The murine cripto gene: Expression during mesoderm induction and early heart morphogenesis. Development 1993, 118, 1157–1168. [Google Scholar] [PubMed]
- Shen, M.M.; Wang, H.; Leder, P. A differential display strategy identifies Cryptic, a novel EGF-related gene expressed in the axial and lateral mesoderm during mouse gastrulation. Development 1997, 124, 429–442. [Google Scholar] [PubMed]
- Colas, J.F.; Schoenwolf, G.C. Subtractive hybridization identifies chick-cripto, a novel EGF-CFC ortholog expressed during gastrulation, neurulation and early cardiogenesis. Gene 2000, 255, 205–217. [Google Scholar] [CrossRef]
- Kinoshita, N.; Minshull, J.; Kirschner, M.W. The identification of two novel ligands of the fgf receptor by a yeast screening method and their activity in xenopus development. Cell 1995, 83, 621–630. [Google Scholar] [CrossRef]
- Zhang, J.; Talbot, W.S.; Schier, A.F. Positional cloning identifies zebrafish one-eyed pinhead as a permissive EGF-related ligand required during gastrulation. Cell 1998, 92, 241–251. [Google Scholar] [CrossRef]
- Ciccodicola, A.; Dono, R.; Obici, S.; Simeone, A.; Zollo, M.; Persico, M.G. Molecular characterization of a gene of the “EGF family” expressed in undifferentiated human NTERA2 teratocarcinoma cells. EMBO J. 1989, 8, 1987–1991. [Google Scholar] [CrossRef] [PubMed]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Strizzi, L.; Bianco, C.; Normanno, N.; Salomon, D. Cripto-1: A multifunctional modulator during embryogenesis and oncogenesis. Oncogene 2005, 24, 5731–5741. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Yang, L.; Yan, Y.T.; Chen, A.; Desai, N.; Wynshaw-Boris, A.; Shen, M.M. Cripto is required for correct orientation of the anterior-posterior axis in the mouse embryo. Nature 1998, 395, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.F.; Liscia, D.S.; Normanno, N.; Merlo, G.; Johnson, G.R.; Gullick, W.J.; Ciardiello, F.; Saeki, T.; Brandt, R.; Kim, N. Expression of transforming growth factor α, amphiregulin and cripto-1 in human breast carcinomas. Br. J. Cancer 1994, 69, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Kim, N.; Saeki, T.; Dono, R.; Persico, M.G.; Plowman, G.D.; Garrigues, J.; Radke, S.; Todaro, G.J.; Salomon, D.S. Differential expression of epidermal growth factor-related proteins in human colorectal tumors. Proc. Natl. Acad. Sci. USA 1991, 88, 7792–7796. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Yoshida, K.; Yokozaki, H.; Yasui, W.; Ito, H.; Toge, T.; Ciardiello, F.; Persico, M.G.; Saeki, T.; Salomon, D.S.; et al. Expression of cripto, a novel gene of the epidermal growth factor family, in human gastrointestinal carcinomas. Jpn. J. Cancer Res. 1991, 82, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, G.; Romano, A.; Armenante, F.; Rambaldi, M.; Paoletti, I.; Sandomenico, C.; Pepe, S.; Staibano, S.; Salvatore, G.; De Rosa, G.; et al. Expression of teratocarcinoma-derived growth factor-1 (TDGF-1) in testis germ cell tumors and its effects on growth and differentiation of embryonal carcinoma cell line NTERA2/D1. Oncogene 1997, 15, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Stromberg, K.; Johnson, G.R.; O’Connor, D.M.; Sorensen, C.M.; Gullick, W.J.; Kannan, B. Frequent immunohistochemical detection of egf supergene family members in ovarian carcinogenesis. Int. J. Gynecol. Pathol. 1994, 13, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Fontanini, G.; Boldrini, L.; Vignati, S.; Chinè, S.; Basolo, F.; Silvestri, V.; Lucchi, M.; Mussi, A.; Angeletti, C.A.; Bevilacqua, G. Bcl2 and p53 regulate vascular endothelial growth factor (VEGF)-mediated angiogenesis in non-small cell lung carcinoma. Eur. J. Cancer 1998, 34, 718–723. [Google Scholar] [CrossRef]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Kobrin, M.S.; Tahara, E.; Korc, M. Cripto, a member of the epidermal growth factor family, is over-expressed in human pancreatic cancer and chronic pancreatitis. Int. J. Cancer 1994, 56, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Welss, T.; Papoutsaki, M.; Michel, G.; Reifenberger, J.; Chimenti, S.; Ruzicka, T.; Abts, H.F. Molecular basis of basal cell carcinoma: Analysis of differential gene expression by differential display PCR and expression array. Int. J. Cancer 2003, 104, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.L.; Autzen, P.; Birch, P.; Robinson, M.C.; Gullick, W.J.; Neal, D.E.; Hamdy, F.C. The immunohistochemical detection of cripto-1 in benign and malignant human bladder. J. Pathol. 1998, 185, 108–111. [Google Scholar] [CrossRef]
- Minchiotti, G.; Parisi, S.; Liguori, G.; Signore, M.; Lania, G.; Adamson, E.D.; Lago, C.T.; Persico, M.G. Membrane-anchorage of Cripto protein by glycosylphosphatidylinositol and its distribution during early mouse development. Mech. Dev. 2000, 90, 133–142. [Google Scholar] [CrossRef]
- Reissmann, E.; Jörnvall, H.; Blokzijl, A.; Andersson, O.; Chang, C.; Minchiotti, G.; Persico, M.G.; Ibáñez, C.F.; Brivanlou, A.H. The orphan receptor ALK7 and the Activin receptor ALK4 mediate signaling by Nodal proteins during vertebrate development. Genes Dev. 2001, 15, 2010–2022. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.Y.; Whitman, M. Nodal signals to Smads through Cripto-dependent and Cripto-independent mechanisms. Mol. Cell 2001, 7, 949–957. [Google Scholar] [CrossRef]
- Bianco, C.; Adkins, H.B.; Wechselberger, C.; Seno, M.; Normanno, N.; De Luca, A.; Sun, Y.; Khan, N.; Kenney, N.; Ebert, A.; et al. Cripto-1 activates nodal- and ALK4-dependent and -independent signaling pathways in mammary epithelial Cells. Mol. Cell. Biol. 2002, 22, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.K.; Olale, F.; Bennett, J.T.; Brivanlou, A.H.; Schier, A.F. EGF-CFC proteins are essential coreceptors for the TGF-β signals VG1 and GDF1. Genes Dev. 2003, 17, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Bianco, C.; Strizzi, L.; Hamada, S.; Mancino, M.; Bailly, V.; Mo, W.; Wen, D.; Miatkowski, K.; Gonzales, M.; et al. Growth factor induction of cripto-1 shedding by glycosylphosphatidylinositol-phospholipase D and enhancement of endothelial cell migration. J. Biol. Chem. 2007, 282, 31643–31655. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Wechselberger, C.; Ebert, A.; Khan, N.I.; Sun, Y.; Salomon, D.S. Identification of cripto-1 in human milk. Breast Cancer Res. Treat. 2001, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Mancino, M.; Rehman, A.; Hamada, S.; Watanabe, K.; De Luca, A.; Jones, B.; Balogh, G.; Russo, J.; et al. Identification of Cripto-1 as a novel serologic marker for breast and colon cancer. Clin. Cancer Res. 2006, 12, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Normannos, N.; Gullickll, W.J.; Lin, J.; Harkins, R.; Schneider, D.; Jones, B.; Ciardielloss, F.; Persico, M.G.; Armenantego, F.; et al. Identification and Biological Characterization of an Epidermal. J. Biol. Chem. 1994, 269, 17320–17328. [Google Scholar] [PubMed]
- Yan, Y.; Liu, J.; Luo, Y.; Chaosu, E.; Robert, S.; Abate-shen, C.; Shen, M.M.; Haltiwanger, R.S. Dual Roles of Cripto as a Ligand and Coreceptor in the Nodal Signaling Pathway Dual Roles of Cripto as a Ligand and Coreceptor in the Nodal Signaling Pathway. Mol. Cell. Biol. 2002, 22, 4439–4449. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.L.; Kannan, S.; Smith, G.H.; Seno, M.; Bianco, C.; Kim, N.; Martinez-Lacaci, I.; Wallace-Jones, B.; Salomon, D.S. Cripto-1 inhibits β-casein expression in mammary epithelial cells through a p21ras-and phosphatidylinositol 3’-kinase-dependent pathway. Cell Growth Differ. 1997, 8, 1257–1266. [Google Scholar] [PubMed]
- Kannan, S.; De Santis, M.; Lohmeyer, M.; Riese, D.J.; Smith, G.H.; Hynes, N.; Seno, M.; Brandt, R.; Bianco, C.; Persico, G.; et al. Cripto enhances the tyrosine phosphorylation of Shc and activates mitogen-activated protein kinase (MAPK) in mammary epithelial cells. J. Biol. Chem. 1997, 272, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Wechselberger, C.; Frank, S.; Wallace-Jones, B.; Seno, M.; Martinez-Lacaci, I.; Bianco, C.; De Santis, M.; Weitzel, H.K.; Salomon, D.S. Cripto-1 induces phosphatidylinositol 3’-kinase-dependent phosphorylation of AKT and glycogen synthase kinase 3β in human cervical carcinoma cells. Cancer Res. 1999, 59, 4502–4505. [Google Scholar] [PubMed]
- Bianco, C.; Normanno, N.; De Luca, A.; Maiello, M.R.; Wechselberger, C.; Sun, Y.; Khan, N.; Adkins, H.; Sanicola, M.; Vonderhaar, B.; et al. Detection and localization of cripto-1 binding in mouse mammary epithelial cells and in the mouse mammary gland using an immunoglobulin—Cripto-1 fusion protein. J. Cell. Physiol. 2002, 190, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Rehman, A.; Normanno, N.; Wechselberger, C.; Sun, Y.; Khan, N.; Hirota, M.; Adkins, H.; Williams, K.; et al. A Nodal- and ALK4-independent signaling pathway activated by Cripto-1 through Glypican-1 and c-Src. Cancer Res. 2003, 63, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Kannan, S.; De Santis, M.; Seno, M.; Tang, C.K.; Martinez-Lacaci, I.; Kim, N.; Wallace-Jones, B.; Lippman, M.E.; Ebert, A.D.; et al. Cripto-1 indirectly stimulates the tyrosine phosphorylation of erb B-4 through a novel receptor. J. Biol. Chem. 1999, 274, 8624–8629. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS ONE 2012, 7, e33544. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Yan, T.; Mizutani, A.; Sota, T.; Hiramoto, Y.; Prieto-Vila, M.; Chen, L.; Satoh, A.; Kudoh, T.; Kasai, T.; et al. Cancer stem cells maintain a hierarchy of differentiation by creating their niche. Int. J. Cancer 2014, 135, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Yan, T.; Calle, A.S.; Nair, N.; Hurley, L.; Kasai, T.; Kakuta, H.; Masuda, J.; Murakami, H.; Mizutani, A.; et al. iPSC-derived cancer stem cells provide a model of tumor vasculature. Am. J. Cancer Res. 2016, 6, 1906–1921. [Google Scholar] [PubMed]
- Chen, L.; Mizutani, A.; Kasai, T.; Yan, T.; Jin, G.; Vaidyanath, A.; El-Aarag, B.Y.; Liu, Y.; Kudoh, T.; Salomon, D.S.; et al. Mouse induced pluripotent stem cell microenvironment generates epithelial-mesenchymal transition in mouse Lewis lung cancer cells. Am. J. Cancer Res. 2014, 4, 80–88. [Google Scholar] [PubMed]
- Yan, T.; Mizutani, A.; Chen, L.; Takaki, M.; Hiramoto, Y.; Matsuda, S.; Shigehiro, T.; Kasai, T.; Kudoh, T.; Murakami, H.; et al. Characterization of cancer stem-like cells derived from mouse induced pluripotent stem cells transformed by tumor-derived extracellular vesicles. J. Cancer 2014, 5, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Oo, A.K.K.; Calle, A.S.; Nair, N.; Mahmud, H.; Vaidyanath, A.; Yamauchi, J.; Khayrani, A.C.; Du, J.; Alam, M.J.; Seno, A.; et al. Up-Regulation of PI 3-Kinases and the Activation of PI3K-Akt Signaling Pathway in Cancer Stem-Like Cells Through DNA Hypomethylation Mediated by the Cancer Microenvironment. Transl. Oncol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Seno, M.; DeSantis, M.; Kannan, S.; Bianco, C.; Tada, H.; Kim, N.; Kosaka, M.; Gullick, W.J.; Yamada, H.; Salomon, D.S. Purification and characterization of a recombinant human cripto-1 protein. Growth Factors 1998, 15, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Hamada, S.; Bianco, C.; Mancino, M.; Nagaoka, T.; Gonzales, M.; Bailly, V.; Strizzi, L.; Salomon, D.S. Requirement of glycosylphosphatidylinositol anchor of Cripto-1 for trans activity as a nodal co-receptor. J. Biol. Chem. 2007, 282, 35772–35786. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Sun, B.; Sun, H.; Zhao, X.; Zhang, D.; Liu, T.; Zhao, N.; Gu, Q.; Dong, X.; Liu, F. Nodal signaling activates the Smad2/3 pathway to regulate stem cell-like properties in breast cancer cells. Am. J. Cancer Res. 2017, 7, 503–517. [Google Scholar] [PubMed]
- Lonardo, E.; Hermann, P.C.; Mueller, M.T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cui, X.; Yu, X.; Bian, B.-S.-J.; Qian, F.; Hu, X.; Ji, C.; Yang, L.; Ren, Y.; Cui, W.; et al. Cripto-1 acts as a functional marker of cancer stem-like cells and predicts prognosis of the patients in esophageal squamous cell carcinoma. Mol. Cancer 2017, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Das, A.B.; Loying, P.; Bose, B. Human recombinant Cripto-1 increases doubling time and reduces proliferation of HeLa cells independent of pro-proliferation pathways. Cancer Lett. 2012, 318, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Strizzi, L.; Ebert, A.; Chang, C.; Rehman, A.; Normanno, N.; Guedez, L.; Salloum, R.; Ginsburg, E.; Sun, Y.; et al. Role of human cripto-1 in tumor angiogenesis. J. Natl. Cancer Inst. 2005, 97, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Walsh, L.A.; Zhang, G.; Findlay, S.D.; Moreno, J.; Fung, L.; Ablack, A.; Lewis, J.D.; Done, S.J.; Hess, D.A.; et al. Embryonic protein nodal promotes breast cancer vascularization. Cancer Res. 2012, 72, 3851–3863. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Shi, Z.; Xu, H.; Chen, R.; Xue, S.; Sun, X. Knockdown of Cripto-1 inhibits the proliferation, migration, invasion, and angiogenesis in prostate carcinoma cells. J. Biosci. 2017, 42, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Jan, H.-J.; Lai, J.-H.; Ma, H.-I.; Hueng, D.-Y.; Lee, Y.-C.G.; Cheng, Y.-Y.; Liu, L.-W.; Wei, H.-W.; Lee, H.-M. Nodal promotes growth and invasion in human gliomas. Oncogene 2010, 29, 3110–3123. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.K.K.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.S.; Joly, M.M.; Allen-Petersen, B.L.; Worth, P.J.; Lanciault, C.; Sauer, D.; Link, J.; Pelz, C.; Heiser, L.M.; Morton, J.P.; et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, O.; Imamura, H.; Shimizu, T.; Kinoshita, J.; Okabe, T.; Hirano, A.; Yoshimatsu, K.; Konno, S.; Aiba, M.; Ogawa, K. Expression of twist and Wnt in human breast cancer. Anticancer Res. 2004, 24, 3851–3856. [Google Scholar] [PubMed]
- Shigehiro, T.; Kasai, T.; Murakami, M.; Sekhar, S.C.; Tominaga, Y.; Okada, M.; Kudoh, T.; Mizutani, A.; Murakami, H.; Salomon, D.S.; et al. Efficient Drug Delivery of Paclitaxel Glycoside: A Novel Solubility Gradient Encapsulation into Liposomes Coupled with Immunoliposomes Preparation. PLoS ONE 2014, 9, e107976. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, M.J.; Takahashi, R.; Afify, S.M.; Oo, A.K.K.; Kumon, K.; Nawara, H.M.; Khayrani, A.C.; Du, J.; Zahra, M.H.; Seno, A.; et al. Exogenous Cripto-1 Suppresses Self-Renewal of Cancer Stem Cell Model. Int. J. Mol. Sci. 2018, 19, 3345. https://doi.org/10.3390/ijms19113345
Alam MJ, Takahashi R, Afify SM, Oo AKK, Kumon K, Nawara HM, Khayrani AC, Du J, Zahra MH, Seno A, et al. Exogenous Cripto-1 Suppresses Self-Renewal of Cancer Stem Cell Model. International Journal of Molecular Sciences. 2018; 19(11):3345. https://doi.org/10.3390/ijms19113345
Chicago/Turabian StyleAlam, Md Jahangir, Ryota Takahashi, Said M. Afify, Aung Ko Ko Oo, Kazuki Kumon, Hend M. Nawara, Aprilliana Cahya Khayrani, Juan Du, Maram H. Zahra, Akimasa Seno, and et al. 2018. "Exogenous Cripto-1 Suppresses Self-Renewal of Cancer Stem Cell Model" International Journal of Molecular Sciences 19, no. 11: 3345. https://doi.org/10.3390/ijms19113345
APA StyleAlam, M. J., Takahashi, R., Afify, S. M., Oo, A. K. K., Kumon, K., Nawara, H. M., Khayrani, A. C., Du, J., Zahra, M. H., Seno, A., Salomon, D. S., & Seno, M. (2018). Exogenous Cripto-1 Suppresses Self-Renewal of Cancer Stem Cell Model. International Journal of Molecular Sciences, 19(11), 3345. https://doi.org/10.3390/ijms19113345