The Pathophysiology of Gestational Diabetes Mellitus

 ,
, 
Abstract
1. Introduction
1.1. Glucose Regulation during Healthy Pregnancy
1.2. Classification and Prevalence of Gestational Diabetes
1.3. Forms of Gestational Diabetes
1.4. Risk Factors for Gestational Diabetes
1.5. Consequences of Gestational Diabetes
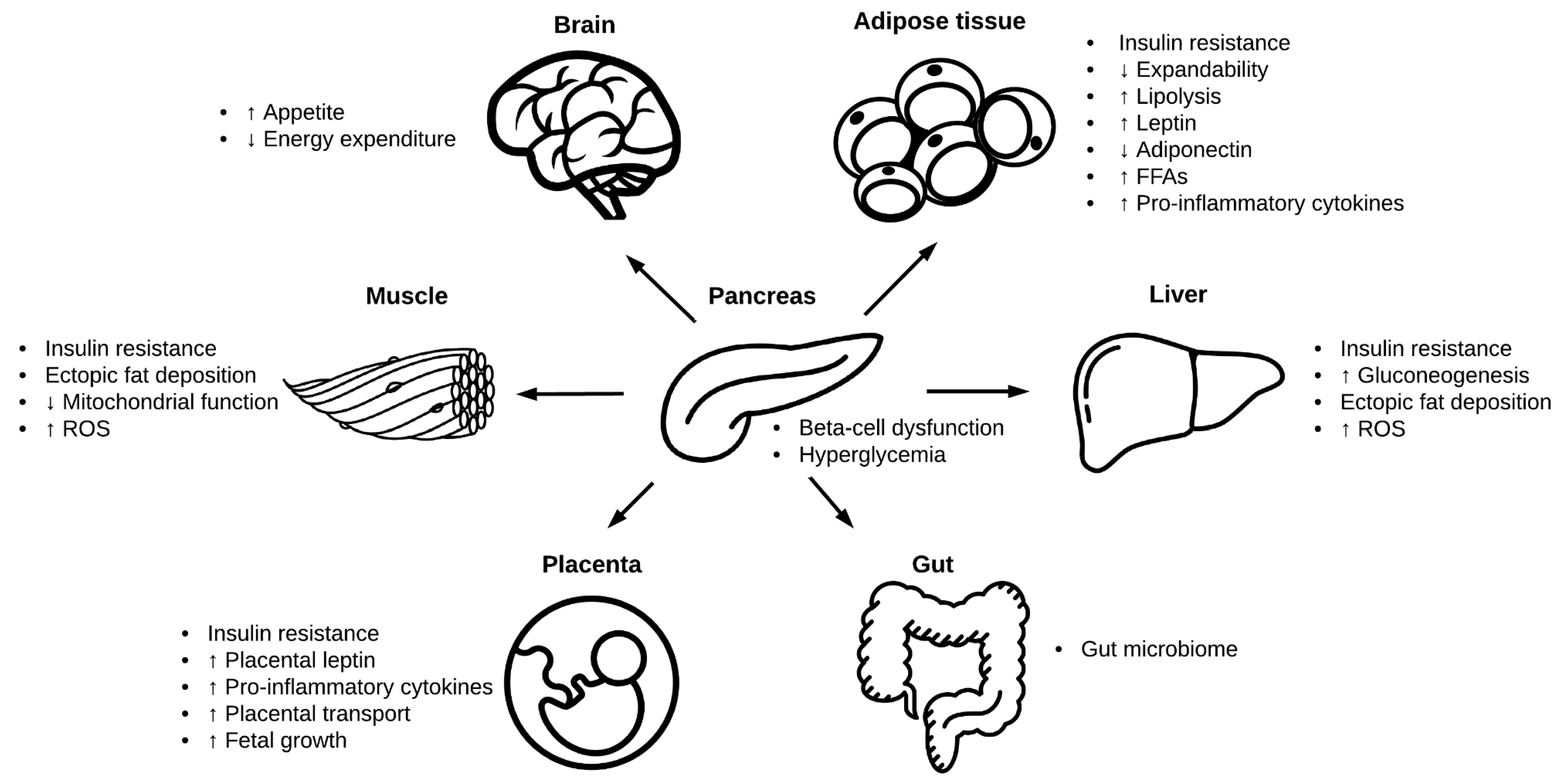
2. Pathophysiology of Gestational Diabetes
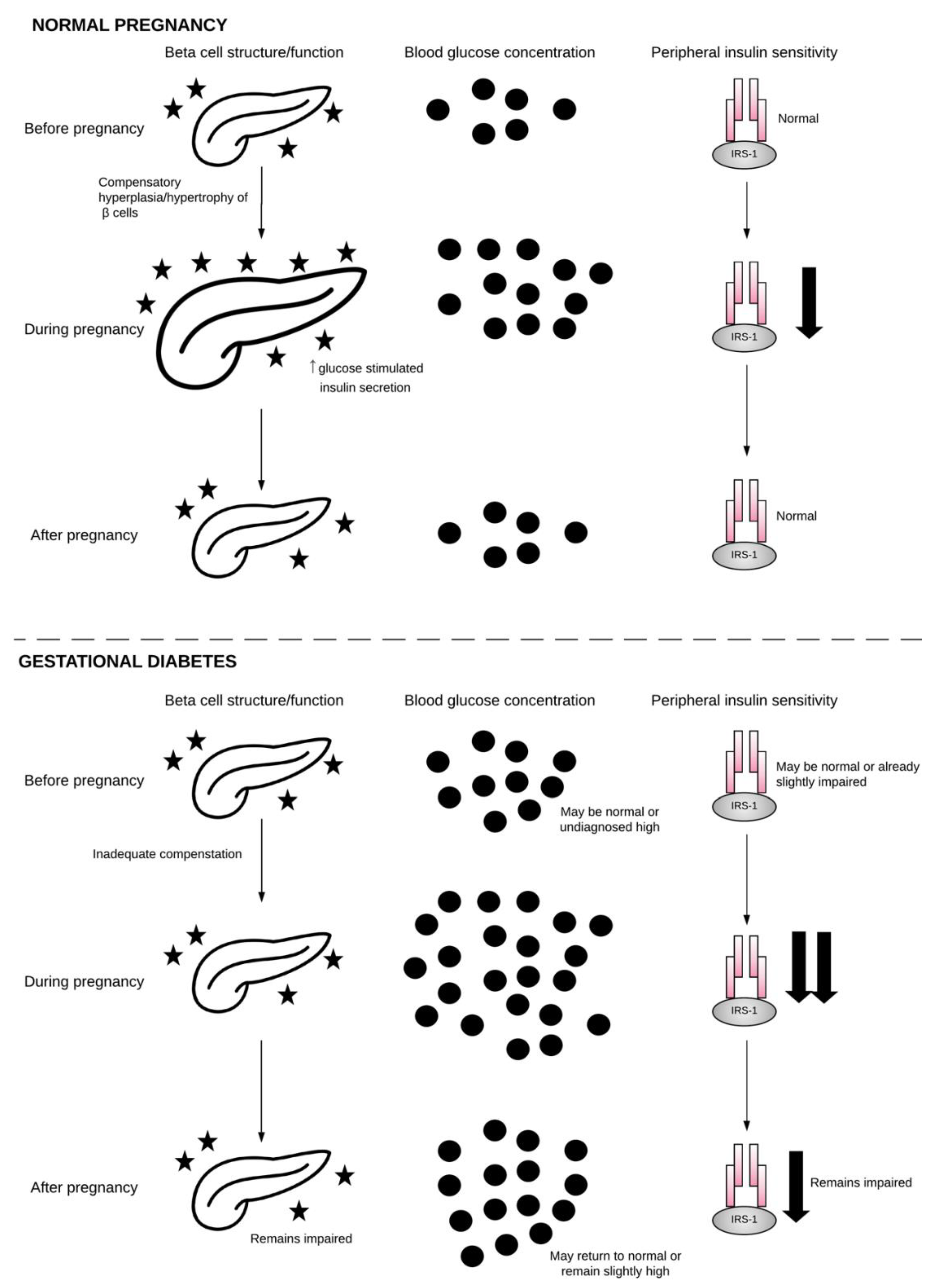
2.1. β-Cell Dysfunction
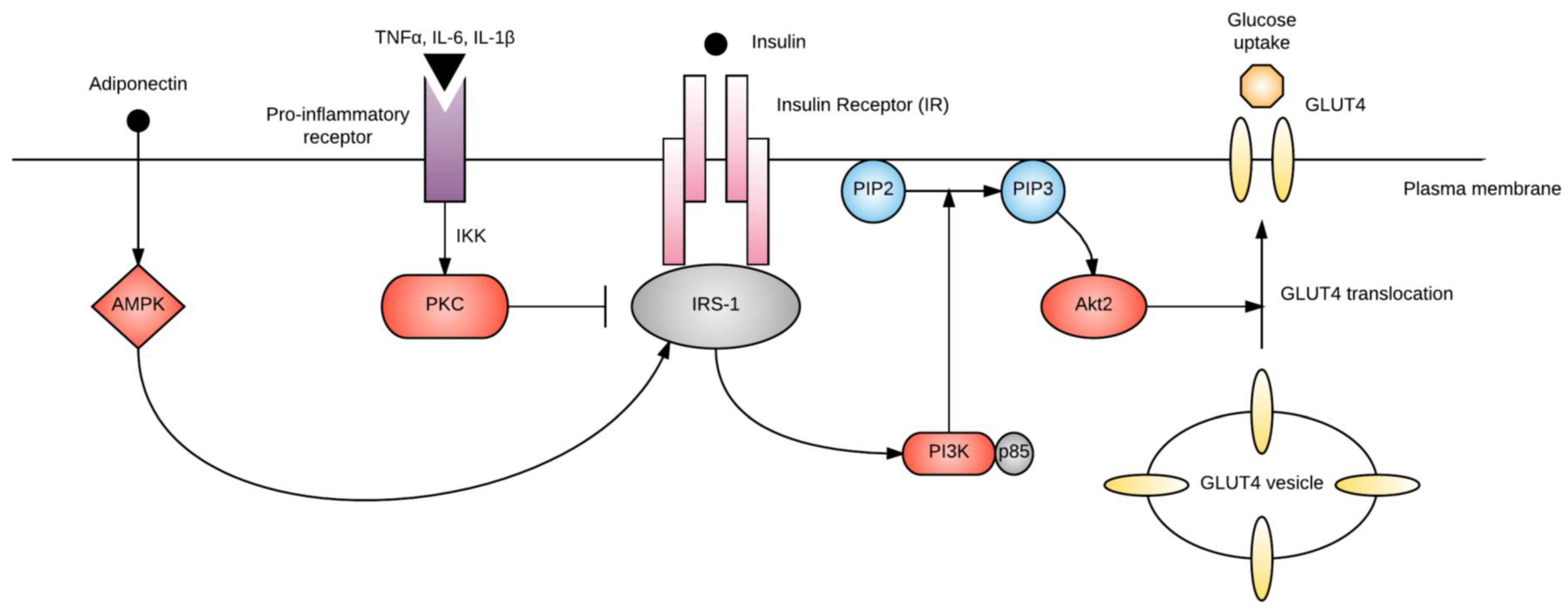
2.2. Chronic Insulin Resistance
2.3. Neurohormonal Networks
2.3.1. Leptin
2.3.2. Adiponectin
2.4. Adipose Tissue
2.4.1. Energy Storage
2.4.2. Adipose Tissue Inflammation
2.5. Liver
2.6. Skeletal and Cardiac Muscle
2.7. Gut Microbiome
2.8. Oxidative Stress
2.9. Placental Transport
3. Opportunities and Considerations for Future Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Diabetes Association. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; IDF: Brussels, Belgium, 2017. [Google Scholar]
- Feig, D.S.; Moses, R.G. Metformin Therapy during Pregnancy Good for the goose and good for the gosling too? Diabetes Care 2011, 34, 2329–2330. [Google Scholar] [CrossRef] [PubMed]
- Camelo Castillo, W.; Boggess, K.; Stürmer, T.; Brookhart, M.A.; Benjamin, D.K.; Jonsson Funk, M. Association of Adverse Pregnancy Outcomes with Glyburide vs Insulin in Women with Gestational Diabetes. JAMA Pediatr. 2015, 169, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Di Cianni, G.; Miccoli, R.; Volpe, L.; Lencioni, C.; Del Prato, S. Intermediate metabolism in normal pregnancy and in gestational diabetes. Diabetes Metab. Res. Rev. 2003, 19, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Tyzbir, E.D.; Roman, N.M.; Amini, S.B.; Sims, E.A. Longitudinal changes in insulin release and insulin resistance in nonobese pregnant women. Am. J. Obstet. Gynecol. 1991, 165, 1667–1672. [Google Scholar] [CrossRef]
- Phelps, R.L.; Metzger, B.E.; Freinkel, N. Carbohydrate metabolism in pregnancy: XVII. Diurnal profiles of plasma glucose, insulin, free fatty acids, triglycerides, cholesterol, and individual amino acids in late normal pregnancy. Am. J. Obstet. Gynecol. 1981, 140, 730–736. [Google Scholar] [CrossRef]
- Parsons, J.A.; Brelje, T.C.; Sorenson, R.L. Adaptation of islets of Langerhans to pregnancy: Increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992, 130, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; O’Sullivan, M.J.; Skyler, J.S. Insulin Action During Pregnancy: Studies with the Euglycemic Clamp Technique. Diabetes 1985, 34, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, E.; Arcidiacono, B.; Foti, D.; Brunetti, A. Gestational diabetes mellitus: An updated overview. J. Endocrinol. Investig. 2017, 40, 899–909. [Google Scholar] [CrossRef] [PubMed]
- HAPO Study Cooperative Research Group; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.M.; Vellinga, A.; Harreiter, J.; Simmons, D.; Desoye, G.; Corcoy, R.; Adelantado, J.M.; Devlieger, R.; Assche, A.V.; Galjaard, S.; et al. Epidemiology of gestational diabetes mellitus according to IADPSG/WHO 2013 criteria among obese pregnant women in Europe. Diabetologia 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Iqbal, S.; Zawacki, C.M.; Yu, D.; Brown, M.B.; Herman, W.H. Effect of selective screening for gestational diabetes. Diabetes Care 1999, 22, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Coffey, M.; Johnson, H.; Scanlon, P.; Foley, M.; Stronge, J.; O’Meara, N.M.; Firth, R.G. Universal vs. risk factor-based screening for gestational diabetes mellitus: Detection rates, gestation at diagnosis and outcome. Diabet. Med. J. Br. Diabet. Assoc. 2000, 17, 26–32. [Google Scholar] [CrossRef]
- Capula, C.; Chiefari, E.; Vero, A.; Arcidiacono, B.; Iiritano, S.; Puccio, L.; Pullano, V.; Foti, D.P.; Brunetti, A.; Vero, R. Gestational Diabetes Mellitus: Screening and Outcomes in Southern Italian Pregnant Women. Available online: https://www.hindawi.com/journals/isrn/2013/387495/ (accessed on 9 October 2018).
- Zhu, Y.; Zhang, C. Prevalence of Gestational Diabetes and Risk of Progression to Type 2 Diabetes: A Global Perspective. Curr. Diabetes Rep. 2016, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Yuen, L.; Wong, V.W. Gestational diabetes mellitus: Challenges for different ethnic groups. World J. Diabetes 2015, 6, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Moses, R.G.; Wong, V.C.K.; Lambert, K.; Morris, G.J.; Gil, F.S. Seasonal Changes in the Prevalence of Gestational Diabetes Mellitus. Diabetes Care 2016, 39, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Haneda, M.; Noda, M.; Origasa, H.; Noto, H.; Yabe, D.; Fujita, Y.; Goto, A.; Kondo, T.; Araki, E. Japanese Clinical Practice Guideline for Diabetes 2016. J. Diabetes Investig. 2018, 9, 657–697. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Go, R.C.; Aoki, M.; Riggs, A.C.; Tanizawa, Y.; Acton, R.T.; Bell, D.S.; Goldenberg, R.L.; Roseman, J.M.; Permutt, M.A. Glucokinase gene in gestational diabetes mellitus: Population association study and molecular scanning. Diabetologia 1994, 37, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Damm, P.; Kühl, C.; Buschard, K.; Jakobsen, B.K.; Svejgaard, A.; Sodoyez-Goffaux, F.; Shattock, M.; Bottazzo, G.F.; Mølsted-Pedersen, L. Prevalence and predictive value of islet cell antibodies and insulin autoantibodies in women with gestational diabetes. Diabet. Med. J. Br. Diabet. Assoc. 1994, 11, 558–563. [Google Scholar] [CrossRef]
- Buchanan, T.A.; Xiang, A.H. Gestational diabetes mellitus. J. Clin. Investig. 2005, 115, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am. J. Obstet. Gynecol. 1999, 180, 903–916. [Google Scholar] [CrossRef]
- Pendergrass, M.; Fazioni, E.; DeFronzo, R.A. Non-insulin-dependent diabetes mellitus and gestational diabetes mellitus: Same disease, another name? Diabetes Rev. 1995, 3, 566–583. [Google Scholar]
- Zajdenverg, L.; Negrato, C.A. Gestational diabetes mellitus and type 2 diabetes: Same disease in a different moment of life? Maybe not. Arch. Endocrinol. MeTable 2017, 61, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haroush, A.; Yogev, Y.; Hod, M. Epidemiology of gestational diabetes mellitus and its association with Type 2 diabetes. Diabet. Med. 2004, 21, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Metzger, B.E.; Buchanan, T.A.; Coustan, D.R.; de Leiva, A.; Dunger, D.B.; Hadden, D.R.; Hod, M.; Kitzmiller, J.L.; Kjos, S.L.; Oats, J.N.; et al. Summary and Recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care 2007, 30, S251–S260. [Google Scholar] [CrossRef] [PubMed]
- Okosun, I.S.; Chandra, K.M.D.; Boev, A.; Boltri, J.M.; Choi, S.T.; Parish, D.C.; Dever, G.E.A. Abdominal adiposity in U.S. adults: Prevalence and trends, 1960-2000. Prev. Med. 2004, 39, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Durnwald, C. Gestational diabetes: Linking epidemiology, excessive gestational weight gain, adverse pregnancy outcomes, and future metabolic syndrome. Semin. Perinatol. 2015, 39, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tobias, D.K.; Chavarro, J.E.; Bao, W.; Wang, D.; Ley, S.H.; Hu, F.B. Adherence to healthy lifestyle and risk of gestational diabetes mellitus: prospective cohort study. BMJ 2014, 349, g5450. [Google Scholar] [CrossRef] [PubMed]
- Jenum, A.K.; Mørkrid, K.; Sletner, L.; Vange, S.; Torper, J.L.; Nakstad, B.; Voldner, N.; Rognerud-Jensen, O.H.; Berntsen, S.; Mosdøl, A.; et al. Impact of ethnicity on gestational diabetes identified with the WHO and the modified International Association of Diabetes and Pregnancy Study Groups criteria: A population-based cohort study. Eur. J. Endocrinol. 2012, 166, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Anghebem-Oliveira, M.I.; Martins, B.R.; Alberton, D.; de Ramos, E.A.S.; Picheth, G.; de Rego, F.G.M. Type 2 diabetes-associated genetic variants of FTO, LEPR, PPARg, and TCF7L2 in gestational diabetes in a Brazilian population. Arch. Endocrinol. MeTable 2017, 61, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Lao, T.T.; Ho, L.-F.; Chan, B.C.P.; Leung, W.-C. Maternal Age and Prevalence of Gestational Diabetes Mellitus. Diabetes Care 2006, 29, 948–949. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, D.J.; Jovanovic, L. Low Birth Weight as a Risk Factor for Gestational Diabetes, Diabetes, and Impaired Glucose Tolerance During Pregnancy. Diabetes Care 2007, 30, S147–S149. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Wiznitzer, A.; Holcberg, G.; Mazor, M.; Sheiner, E. Family history of diabetes mellitus as an independent risk factor for macrosomia and cesarean delivery. J. Matern. Fetal Neonatal Med. 2010, 23, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Tobias, D.K.; Yeung, E.; Hu, F.B.; Zhang, C. A prospective study of prepregnancy dietary fat intake and risk of gestational diabetes. Am. J. Clin. Nutr. 2012, 95, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Schulze, M.B.; Solomon, C.G.; Hu, F.B. A prospective study of dietary patterns, meat intake and the risk of gestational diabetes mellitus. Diabetologia 2006, 49, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Taschereau-Charron, A.; Da Silva, M.S.; Bilodeau, J.-F.; Morisset, A.-S.; Julien, P.; Rudkowska, I. Alterations of fatty acid profiles in gestational diabetes and influence of the diet. Maturitas 2017, 99, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Solomon, C.G.; Hu, F.B. Dietary Fiber Intake, Dietary Glycemic Load, and the Risk for Gestational Diabetes Mellitus. Diabetes Care 2006, 29, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Bowers, K.; Tobias, D.K.; Olsen, S.F.; Chavarro, J.; Vaag, A.; Kiely, M.; Zhang, C. Prepregnancy low-carbohydrate dietary pattern and risk of gestational diabetes mellitus: A prospective cohort study. Am. J. Clin. Nutr. 2014, 99, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Sivan, E.; Boden, G. Free fatty acids, insulin resistance, and pregnancy. Curr. Diabetes Rep. 2003, 3, 319–322. [Google Scholar] [CrossRef]
- Fung, T.T.; McCullough, M.L.; Newby, P.K.; Manson, J.E.; Meigs, J.B.; Rifai, N.; Willett, W.C.; Hu, F.B. Diet-quality scores and plasma concentrations of markers of inflammation and endothelial dysfunction. Am. J. Clin. Nutr. 2005, 82, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Risk Factors for Gestational Diabetes: From an Epidemiological Standpoint. In Gestational Diabetes during and after Pregnancy; Springer: London, UK, 2010; pp. 71–81. ISBN 978-1-84882-119-4. [Google Scholar]
- Dahlquist, G. The aetiology of type 1 diabetes: An epidemiological perspective. Acta Paediatr. Oslo Nor. 1992 Suppl. 1998, 425, 5–10. [Google Scholar] [CrossRef]
- Lijinsky, W. N-Nitroso compounds in the diet. Mutat. Res. 1999, 443, 129–138. [Google Scholar] [CrossRef]
- Bao, W.; Bowers, K.; Tobias, D.K.; Hu, F.B.; Zhang, C. Prepregnancy Dietary Protein Intake, Major Dietary Protein Sources, and the Risk of Gestational Diabetes Mellitus. Diabetes Care 2013, 36, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Maslova, E.; Hansen, S.; Grunnet, L.G.; Strøm, M.; Bjerregaard, A.A.; Hjort, L.; Kampmann, F.B.; Madsen, C.M.; Thuesen, A.B.; Bech, B.H.; et al. Maternal protein intake in pregnancy and offspring metabolic health at age 9–16 y: Results from a Danish cohort of gestational diabetes mellitus pregnancies and controls. Am. J. Clin. Nutr. 2017, ajcn128637. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Colega, M.; Cai, S.; Chan, Y.H.; Padmapriya, N.; Chen, L.-W.; Soh, S.-E.; Han, W.M.; Tan, K.H.; Lee, Y.S.; et al. Higher Maternal Dietary Protein Intake Is Associated with a Higher Risk of Gestational Diabetes Mellitus in a Multiethnic Asian Cohort. J. Nutr. 2017, 147, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Lavigne, C.; Jacques, H.; Marette, A. Role of dietary proteins and amino acids in the pathogenesis of insulin resistance. Annu. Rev. Nutr. 2007, 27, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched Chain Amino Acids Cause Liver Injury in Obese/Diabetic Mice by Promoting Adipocyte Lipolysis and Inhibiting Hepatic Autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Garofano, A.; Czernichow, P.; Bréant, B. In utero undernutrition impairs rat beta-cell development. Diabetologia 1997, 40, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Ikenasio-Thorpe, B.A.; Breier, B.H.; Vickers, M.H.; Fraser, M. Prenatal influences on susceptibility to diet-induced obesity are mediated by altered neuroendocrine gene expression. J. Endocrinol. 2007, 193, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Breier, B.H.; Cutfield, W.S.; Hofman, P.L.; Gluckman, P.D. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol. Endocrinol. MeTable 2000, 279, E83-87. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Jellyman, J.K.; Han, G.; Beall, M.; Lane, R.H.; Ross, M.G. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am. J. Obstet. Gynecol. 2014, 211, 237.e1–237.e13. [Google Scholar] [CrossRef] [PubMed]
- Portha, B.; Chavey, A.; Movassat, J. Early-Life Origins of Type 2 Diabetes: Fetal Programming of the Beta-Cell Mass. Exp. Diabetes Res. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Byrn, M.; Penckofer, S. The relationship between gestational diabetes and antenatal depression. J. Obstet. Gynecol. Neonatal Nurs. 2015, 44, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.C.; Ling, L.P.; Omar, S.Z. The 50-g glucose challenge test and pregnancy outcome in a multiethnic Asian population at high risk for gestational diabetes. Int. J. Gynecol. Obstet. 2009, 105, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.K.; Kjos, S.L.; Xiang, A.; Buchanan, T.A. Long-term diabetogenic effect of single pregnancy in women with previous gestational diabetes mellitus. Lancet Lond. Engl. 1996, 347, 227–230. [Google Scholar] [CrossRef]
- Shostrom, D.C.V.; Sun, Y.; Oleson, J.J.; Snetselaar, L.G.; Bao, W. History of Gestational Diabetes Mellitus in Relation to Cardiovascular Disease and Cardiovascular Risk Factors in US Women. Front. Endocrinol. 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Action Plan for the Prevention and Control of NCDs 2013–2020; WHO: Geneva, Switzerland, 2013. [Google Scholar]
- Schwartz, R.; Gruppuso, P.A.; Petzold, K.; Brambilla, D.; Hiilesmaa, V.; Teramo, K.A. Hyperinsulinemia and macrosomia in the fetus of the diabetic mother. Diabetes Care 1994, 17, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Fetita, L.-S.; Sobngwi, E.; Serradas, P.; Calvo, F.; Gautier, J.-F. Consequences of Fetal Exposure to Maternal Diabetes in Offspring. J. Clin. Endocrinol. MeTable 2006, 91, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Gascho, C.L.L.; Leandro, D.M.K.; Ribeiro, E.; Silva, T.; Silva, J.C. Predictors of cesarean delivery in pregnant women with gestational diabetes mellitus. Rev. Bras. Ginecol. Obstet. 2017, 39, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Scifres, C.M.; Feghali, M.; Dumont, T.; Althouse, A.D.; Speer, P.; Caritis, S.N.; Catov, J.M. Large-for-Gestational-Age Ultrasound Diagnosis and Risk for Cesarean Delivery in Women With Gestational Diabetes Mellitus. Obstet. Gynecol. 2015, 126, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Esakoff, T.F.; Cheng, Y.W.; Sparks, T.N.; Caughey, A.B. The association between birthweight 4000 g or greater and perinatal outcomes in patients with and without gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2009, 200, 672.e1–672.e4. [Google Scholar] [CrossRef] [PubMed]
- Langer, O.; Yogev, Y.; Most, O.; Xenakis, E.M.J. Gestational diabetes: The consequences of not treating. Am. J. Obstet. Gynecol. 2005, 192, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Vohr, B.R.; Boney, C.M. Gestational diabetes: the forerunner for the development of maternal and childhood obesity and metabolic syndrome? J. Matern. Fetal Neonatal Med. 2008, 21, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In Utero Exposure to Maternal Hyperglycemia Increases Childhood Cardiometabolic Risk in Offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Petitt, D.J.; Bennett, P.H.; Knowler, W.C.; Baird, H.R.; Aleck, K.A. Gestational diabetes mellitus and impaired glucose tolerance during pregnancy. Long-term effects on obesity and glucose tolerance in the offspring. Diabetes 1985, 34 (Suppl. 2), 119–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Pu, Y.B.; Chow, C.C.; Yeung, V.T.; Ko, G.T.; So, W.Y.; Li, J.K.; Chan, W.B.; Ma, R.C.; Critchley, J.A.; et al. Diabetes in Hong Kong Chinese: Evidence for familial clustering and parental effects. Diabetes Care 2000, 23, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Homko, C.; Sivan, E.; Chen, X.; Reece, E.A.; Boden, G. Insulin secretion during and after pregnancy in patients with gestational diabetes mellitus. J. Clin. Endocrinol. MeTable 2001, 86, 568–573. [Google Scholar] [CrossRef]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50 (Suppl. 1), S154–159. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Verchere, C.B.; Clark, A.; Potter, K.J.; Fraser, P.E.; Raleigh, D.P.; Kahn, S.E. Toxic oligomers and islet beta cell death: Guilty by association or convicted by circumstantial evidence? Diabetologia 2010, 53, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rohm, M.; Clark, A.; Brereton, M.F. Is Type 2 Diabetes a Glycogen Storage Disease of Pancreatic β Cells? Cell MeTable 2017, 26, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Delghingaro-Augusto, V.; Nolan, C.J.; Gupta, D.; Jetton, T.L.; Latour, M.G.; Peshavaria, M.; Madiraju, S.R.M.; Joly, E.; Peyot, M.-L.; Prentki, M.; et al. Islet beta cell failure in the 60% pancreatectomised obese hyperlipidaemic Zucker fatty rat: Severe dysfunction with altered glycerolipid metabolism without steatosis or a falling beta cell mass. Diabetologia 2009, 52, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A.; Templeton, L.J.; Gertz, S.J. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 2001, 50, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.E.; Simmons, R.A. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol. Metab. 2010, 21, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Auffret, J.; Freemark, M.; Carré, N.; Mathieu, Y.; Tourrel-Cuzin, C.; Lombès, M.; Movassat, J.; Binart, N. Defective prolactin signaling impairs pancreatic β-cell development during the perinatal period. Am. J. Physiol. Endocrinol. MeTable 2013, 305, E1309–E1318. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. MeTable 2008, 10 (Suppl. 4), 32–42. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, F.A.; Aerts, L.; De Prins, F. A morphological study of the endocrine pancreas in human pregnancy. Br. J. Obstet. Gynaecol. 1978, 85, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M. Trying to understand gestational diabetes. Diabet. Med. 2014, 31, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.A.; McCurdy, C.E.; Hernandez, T.L.; Kirwan, J.P.; Catalano, P.M.; Friedman, J.E. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care 2007, 30 (Suppl. 2), S112–S119. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.E.; Kirwan, J.P.; Jing, M.; Presley, L.; Catalano, P.M. Increased Skeletal Muscle Tumor Necrosis Factor-α and Impaired Insulin Signaling Persist in Obese Women with Gestational Diabetes Mellitus 1 Year Postpartum. Diabetes 2008, 57, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Glucose sensing and the pathogenesis of obesity and type 2 diabetes. Int. J. Obes. 2005 2008, 32 (Suppl. 6), S62–S71. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Tan, S.; Gluckman, P.D.; Godfrey, K.M.; Saw, S.-M.; Teoh, O.H.; Chong, Y.-S.; Meaney, M.J.; Kramer, M.S.; Gooley, J.J.; et al. Sleep Quality and Nocturnal Sleep Duration in Pregnancy and Risk of Gestational Diabetes Mellitus. Sleep 2017, 40. [Google Scholar] [CrossRef] [PubMed]
- Facco, F.L.; Grobman, W.A.; Reid, K.J.; Parker, C.B.; Hunter, S.M.; Silver, R.M.; Basner, R.C.; Saade, G.R.; Pien, G.W.; Manchanda, S.; et al. Objectively measured short sleep duration and later sleep midpoint in pregnancy are associated with a higher risk of gestational diabetes. Am. J. Obstet. Gynecol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fukami, T.; Sun, X.; Li, T.; Desai, M.; Ross, M.G. Mechanism of Programmed Obesity in Intrauterine Fetal Growth Restricted Offspring: Paradoxically Enhanced Appetite Stimulation in Fed and Fasting States. Reprod. Sci. 2012, 19, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, A.; Harder, T.; Brunn, M.; Harder, A.; Roepke, K.; Wittrock-Staar, M.; Ziska, T.; Schellong, K.; Rodekamp, E.; Melchior, K.; et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: An epigenetic model of obesity and the metabolic syndrome. J. Physiol. 2009, 587, 4963–4976. [Google Scholar] [CrossRef] [PubMed]
- Farr, O.M.; Gavrieli, A.; Mantzoros, C.S. Leptin applications in 2015: What have we learned about leptin and obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.S.; Paglia, D.; Kwan, A.Y.; Deitel, M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat. Med. 1995, 1, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.E.; Lowe, C.; Pretz, D.; Steger, J.; Williams, L.M.; Tups, A. High-fat diet induces leptin resistance in leptin-deficient mice. J. Neuroendocrinol. 2014, 26, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Honnorat, D.; Disse, E.; Millot, L.; Mathiotte, E.; Claret, M.; Charrie, A.; Drai, J.; Garnier, L.; Maurice, C.; Durand, E.; et al. Are third-trimester adipokines associated with higher metabolic risk among women with gestational diabetes? Diabetes MeTable 2015, 41, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Maple-Brown, L.; Ye, C.; Hanley, A.J.; Connelly, P.W.; Sermer, M.; Zinman, B.; Retnakaran, R. Maternal pregravid weight is the primary determinant of serum leptin and its metabolic associations in pregnancy, irrespective of gestational glucose tolerance status. J. Clin. Endocrinol. MeTable 2012, 97, 4148–4155. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Ogawa, Y.; Sagawa, N.; Hosoda, K.; Matsumoto, T.; Mise, H.; Nishimura, H.; Yoshimasa, Y.; Tanaka, I.; Mori, T.; et al. Nonadipose tissue production of leptin: Leptin as a novel placenta-derived hormone in humans. Nat. Med. 1997, 3, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Maymó, J.L.; Gambino, Y.P.; Guadix, P.; Dueñas, J.L.; Varone, C.L.; Sánchez-Margalet, V. Activated translation signaling in placenta from pregnant women with gestational diabetes mellitus: Possible role of leptin. Horm. Metab. Res. 2013, 45, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Qiu, C.; Muy-Rivera, M.; Vadachkoria, S.; Song, T.; Luthy, D.A. Plasma adiponectin concentrations in early pregnancy and subsequent risk of gestational diabetes mellitus. J. Clin. Endocrinol. MeTable 2004, 89, 2306–2311. [Google Scholar] [CrossRef] [PubMed]
- Retnakaran, R.; Hanley, A.J.G.; Raif, N.; Connelly, P.W.; Sermer, M.; Zinman, B. Reduced adiponectin concentration in women with gestational diabetes: A potential factor in progression to type 2 diabetes. Diabetes Care 2004, 27, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.; Funahashi, T.; Shimomura, I. Molecular mechanisms of diabetes and atherosclerosis: Role of adiponectin. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, B.; Karteris, E.; Zervou, S.; Digby, J.; Hillhouse, E.W.; Vatish, M.; Randeva, H.S. Secretion of adiponectin by human placenta: Differential modulation of adiponectin and its receptors by cytokines. Diabetologia 2006, 49, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M.; Stumvoll, M. Adipokines in gestational diabetes. Lancet Diabetes Endocrinol. 2014, 2, 488–499. [Google Scholar] [CrossRef]
- Bouchard, L.; Hivert, M.-F.; Guay, S.-P.; St-Pierre, J.; Perron, P.; Brisson, D. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes 2012, 61, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Succurro, E.; Marini, M.A.; Frontoni, S.; Hribal, M.L.; Andreozzi, F.; Lauro, R.; Perticone, F.; Sesti, G. Insulin secretion in metabolically obese, but normal weight, and in metabolically healthy but obese individuals. Obesity 2008, 16, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Kantartzis, K.; Machann, J.; Schick, F.; Thamer, C.; Rittig, K.; Balletshofer, B.; Machicao, F.; Fritsche, A.; Häring, H.-U. Identification and characterization of metabolically benign obesity in humans. Arch. Intern. Med. 2008, 168, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rodriguez, R.; Lifshitz, L.M.; Bellve, K.D.; Min, S.Y.; Pires, J.; Leung, K.; Boeras, C.; Sert, A.; Draper, J.T.; Corvera, S.; et al. Human adipose tissue expansion in pregnancy is impaired in gestational diabetes mellitus. Diabetologia 2015, 58, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M. Effect of pre-existing maternal obesity, gestational diabetes and adipokines on the expression of genes involved in lipid metabolism in adipose tissue. Metabolism 2014, 63, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Kautzky-Willer, A.; Krssak, M.; Winzer, C.; Pacini, G.; Tura, A.; Farhan, S.; Wagner, O.; Brabant, G.; Horn, R.; Stingl, H.; et al. Increased intramyocellular lipid concentration identifies impaired glucose metabolism in women with previous gestational diabetes. Diabetes 2003, 52, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.; Taylor-Robinson, S.D.; Patel, N.; Allan, P.; Walker, B.R.; Johnston, D.G. Increased prevalence of non-alcoholic fatty liver disease in European women with a history of gestational diabetes. Diabetologia 2011, 54, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Bachmann, R.A.; Chen, J. Interleukin-6 and insulin resistance. Vitam. Horm. 2009, 80, 613–633. [Google Scholar] [CrossRef] [PubMed]
- Atègbo, J.-M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of adipokines and cytokines in gestational diabetes and macrosomia. J. Clin. Endocrinol. MeTable 2006, 91, 4137–4143. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.-C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-de Mouzon, S. Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes 2003, 52, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Mitton, A.; Mittion, A.; Permezel, M. In response to oxidative stress, the expression of inflammatory cytokines and antioxidant enzymes are impaired in placenta, but not adipose tissue, of women with gestational diabetes. J. Endocrinol. 2010, 204, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Burks, D.J.; White, M.F. IRS proteins and beta-cell function. Diabetes 2001, 50 (Suppl. 1), S140–S145. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, F.; Laviola, L.; Eriksson, J.W. Regional differences of insulin action in adipose tissue: Insights from in vivo and in vitro studies. Acta Physiol. Scand. 2005, 183, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Damm, P.; Prentki, M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet Lond. Engl. 2011, 378, 169–181. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.H.; Storlien, L. Muscle triglyceride and insulin resistance. Annu. Rev. Nutr. 2002, 22, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Brandon, A.E.; Turner, N.; Watt, M.J.; Bruce, C.R.; Cooney, G.J.; Kraegen, E.W. Lipid and insulin infusion-induced skeletal muscle insulin resistance is likely due to metabolic feedback and not changes in IRS-1, Akt, or AS160 phosphorylation. Am. J. Physiol. Endocrinol. MeTable 2009, 297, E67–E75. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.-E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Dekker Nitert, M.; SPRING Trial Group. Connections Between the Gut Microbiome and Metabolic Hormones in Early Pregnancy in Overweight and Obese Women. Diabetes 2016, 65, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Fugmann, M.; Breier, M.; Rottenkolber, M.; Banning, F.; Ferrari, U.; Sacco, V.; Grallert, H.; Parhofer, K.G.; Seissler, J.; Clavel, T.; et al. The stool microbiota of insulin resistant women with recent gestational diabetes, a high risk group for type 2 diabetes. Sci. Rep. 2015, 5, 13212. [Google Scholar] [CrossRef] [PubMed]
- Furet, J.-P.; Kong, L.-C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.-L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology (Baltimore) 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R.; Poulsen, S.K.; Larsen, T.M.; Bahl, M.I. Microbial enterotypes, inferred by the prevotella-to-bacteroides ratio, remained stable during a 6-month randomized controlled diet intervention with the new nordic diet. Appl. Environ. Microbiol. 2014, 80, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Jayashree, B.; Bibin, Y.S.; Prabhu, D.; Shanthirani, C.S.; Gokulakrishnan, K.; Lakshmi, B.S.; Mohan, V.; Balasubramanyam, M. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol. Cell. Biochem. 2014, 388, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mokkala, K.; Tertti, K.; Rönnemaa, T.; Vahlberg, T.; Laitinen, K. Evaluation of serum zonulin for use as an early predictor for gestational diabetes. Nutr. Diabetes 2017, 7, e253. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Hiden, U.; Desoye, G.; Froehlich, J.; Hauguel-de Mouzon, S.; Jawerbaum, A. The role of oxidative stress in the pathophysiology of gestational diabetes mellitus. Antioxid. Redox Signal. 2011, 15, 3061–3100. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Zhu, C.; Yang, H.; Geng, Q.; Ma, Q.; Long, Y.; Zhou, C.; Chen, M. Association of oxidative stress biomarkers with gestational diabetes mellitus in pregnant women: A case-control study. PLoS ONE 2015, 10, e0126490. [Google Scholar] [CrossRef] [PubMed]
- Pessler, D.; Rudich, A.; Bashan, N. Oxidative stress impairs nuclear proteins binding to the insulin responsive element in the GLUT4 promoter. Diabetologia 2001, 44, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 2010, 396, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Javadian, P.; Alimohamadi, S.; Gharedaghi, M.H.; Hantoushzadeh, S. Gestational diabetes mellitus and iron supplement; effects on pregnancy outcome. Acta Med. Iran. 2014, 52, 385–389. [Google Scholar] [PubMed]
- Puntarulo, S. Iron, oxidative stress and human health. Mol. Asp. Med. 2005, 26, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Bo, S.; Lezo, A.; Menato, G.; Gallo, M.-L.; Bardelli, C.; Signorile, A.; Berutti, C.; Massobrio, M.; Pagano, G.F. Gestational hyperglycemia, zinc, selenium, and antioxidant vitamins. Nutrition 2005, 21, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Flatt, P.R.; Brennan, L.; Newsholme, P.; McClenaghan, N.H. Detrimental actions of metabolic syndrome risk factor, homocysteine, on pancreatic beta-cell glucose metabolism and insulin secretion. J. Endocrinol. 2006, 189, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Wang, J.; Yang, M.; Shao, Y.; Liu, J.; Wu, Q.; Xu, Q.; Wang, H.; He, X.; Chen, Y.; et al. Serum homocysteine level and gestational diabetes mellitus: A meta-analysis. J. Diabetes Investig. 2016, 7, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Debreceni, B.; Debreceni, L. The role of homocysteine-lowering B-vitamins in the primary prevention of cardiovascular disease. Cardiovasc. Ther. 2014, 32, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Hiden, U.; Maier, A.; Bilban, M.; Ghaffari-Tabrizi, N.; Wadsack, C.; Lang, I.; Dohr, G.; Desoye, G. Insulin control of placental gene expression shifts from mother to foetus over the course of pregnancy. Diabetologia 2006, 49, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. Lond. Engl. 1979 2007, 113, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.N.; Jansson, T.; Powell, T.L. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am. J. Physiol. Cell Physiol. 2009, 297, C1228–C1235. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, T.; Lepercq, J.; Varastehpour, A.; Basu, S.; Catalano, P.M.; Hauguel-De Mouzon, S. Differential regulation of genes for fetoplacental lipid pathways in pregnancy with gestational and type 1 diabetes mellitus. Am. J. Obstet. Gynecol. 2009, 201, 209.e1–209.e10. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; McIntyre, H.D.; Cruickshank, J.K.; McCance, D.R.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Trimble, E.R.; Coustan, D.R.; Hadden, D.R.; et al. The hyperglycemia and adverse pregnancy outcome study: Associations of GDM and obesity with pregnancy outcomes. Diabetes Care 2012, 35, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Reichetzeder, C.; Dwi Putra, S.E.; Pfab, T.; Slowinski, T.; Neuber, C.; Kleuser, B.; Hocher, B. Increased global placental DNA methylation levels are associated with gestational diabetes. Clin. Epigenetics 2016, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Roverso, M.; Brioschi, M.; Banfi, C.; Visentin, S.; Burlina, S.; Seraglia, R.; Traldi, P.; Lapolla, A. A preliminary study on human placental tissue impaired by gestational diabetes: A comparison of gel-based versus gel-free proteomics approaches. Eur. J. Mass Spectrom. 2016, 22, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Lesseur, C.; Chen, J. Adverse Maternal Metabolic Intrauterine Environment and Placental Epigenetics: Implications for Fetal Metabolic Programming. Curr. Environ. Health Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, L.; Zhou, L.; Wu, J.; Sheng, C.; Chen, H.; Liu, Y.; Gao, S.; Huang, W. A MicroRNA Signature in Gestational Diabetes Mellitus Associated with Risk of Macrosomia. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dong, J.; Jiang, T.; Shi, Z.; Yu, B.; Zhu, Y.; Chen, D.; Xu, J.; Huo, R.; Dai, J.; et al. Early Second-Trimester Serum MiRNA Profiling Predicts Gestational Diabetes Mellitus. PLoS ONE 2011, 6, e23925. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, S.; Lambers, D.; Baccarelli, A.; Khoury, J.; Macaluso, M.; Ho, S.-M. Endocrine Disruptors: A Potential Risk Factor for Gestational Diabetes Mellitus. Am. J. Perinatol. 2016, 33, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Dolinoy, D.C. The agouti mouse model: An epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 2008, 66, S7–S11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Criteria | Pregnancies | Timing of OGTT | Steps | Glucose Load (g) | Glucose Threshold (mmol/L) | |||
|---|---|---|---|---|---|---|---|---|
| Fasting | 1 h | 2 h | 3 h | |||||
| O’Sullivan, 1964 | All | 24–28 weeks | 2 | 100 | 5.0 | 9.2 | 8.1 | 6.9 |
| WHO, 1999 | All | 24–28 weeks | 1 | 75 | 7.0 | — | 7.8 | — |
| American Diabetes Association (ADA), 2004 | High and medium risk | 14–18 weeks for high risk, 28–32 weeks for medium risk | 2 | 100 | 5.3 | 10.0 | 8.6 | 7.8 |
| National Institute for Health and Care Excellence (NICE), 2015 | High risk | As early as possible | 1 | 75 | 5.6 | — | 7.8 | — |
| IADPSG, 2010 | All | 24–28 weeks | 1 | 75 | 5.1 | 10.0 | 8.5 | — |
| WHO, 2013 | ||||||||
| ADA, 2016 | ||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. https://doi.org/10.3390/ijms19113342
Plows JF, Stanley JL, Baker PN, Reynolds CM, Vickers MH. The Pathophysiology of Gestational Diabetes Mellitus. International Journal of Molecular Sciences. 2018; 19(11):3342. https://doi.org/10.3390/ijms19113342
Chicago/Turabian StylePlows, Jasmine F, Joanna L Stanley, Philip N Baker, Clare M Reynolds, and Mark H Vickers. 2018. "The Pathophysiology of Gestational Diabetes Mellitus" International Journal of Molecular Sciences 19, no. 11: 3342. https://doi.org/10.3390/ijms19113342
APA StylePlows, J. F., Stanley, J. L., Baker, P. N., Reynolds, C. M., & Vickers, M. H. (2018). The Pathophysiology of Gestational Diabetes Mellitus. International Journal of Molecular Sciences, 19(11), 3342. https://doi.org/10.3390/ijms19113342