Physical Background of the Disordered Nature of “Mutual Synergetic Folding” Proteins




Abstract
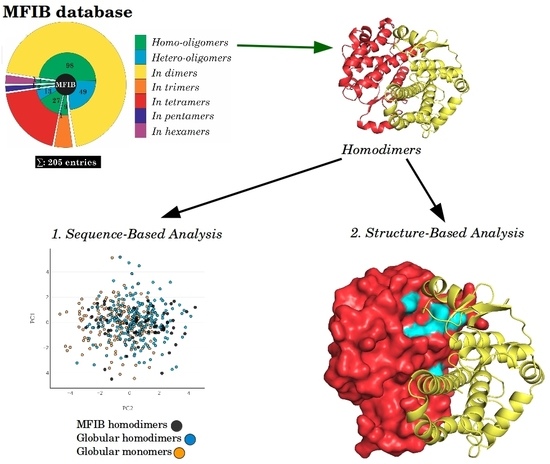
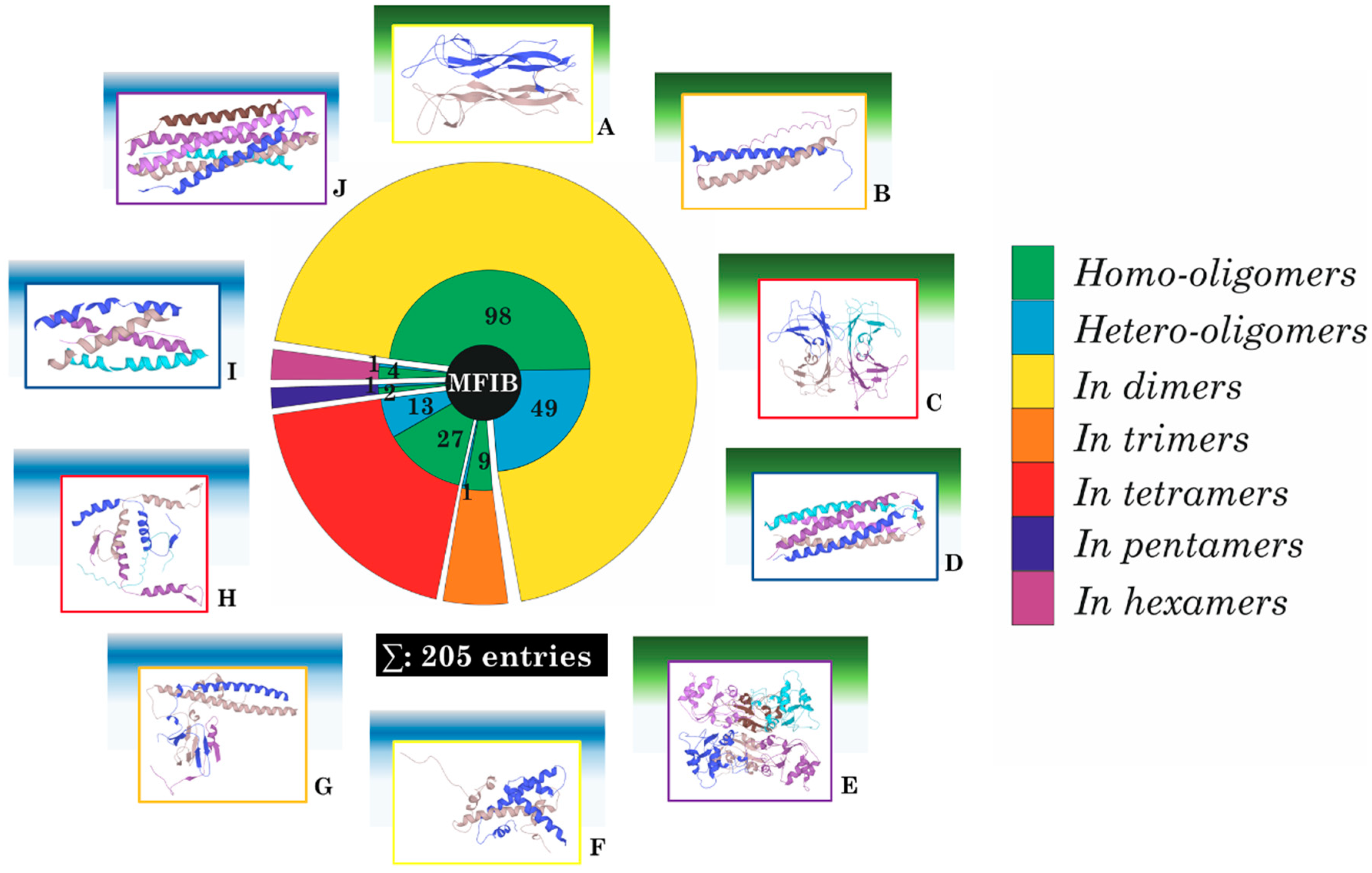
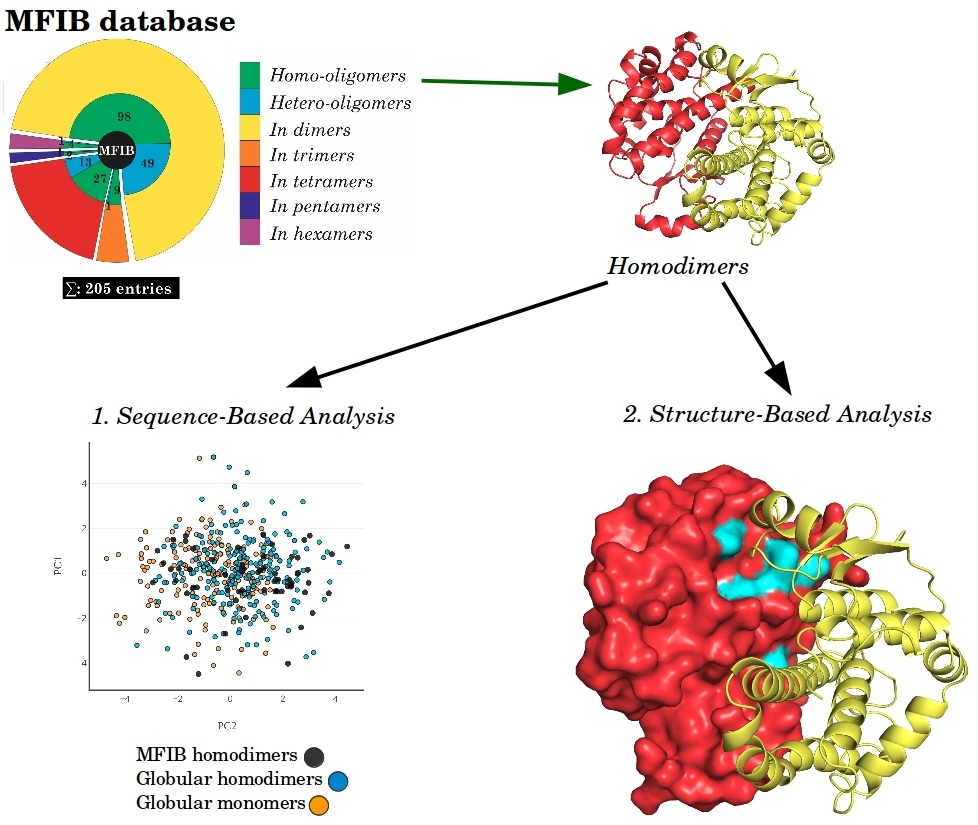{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
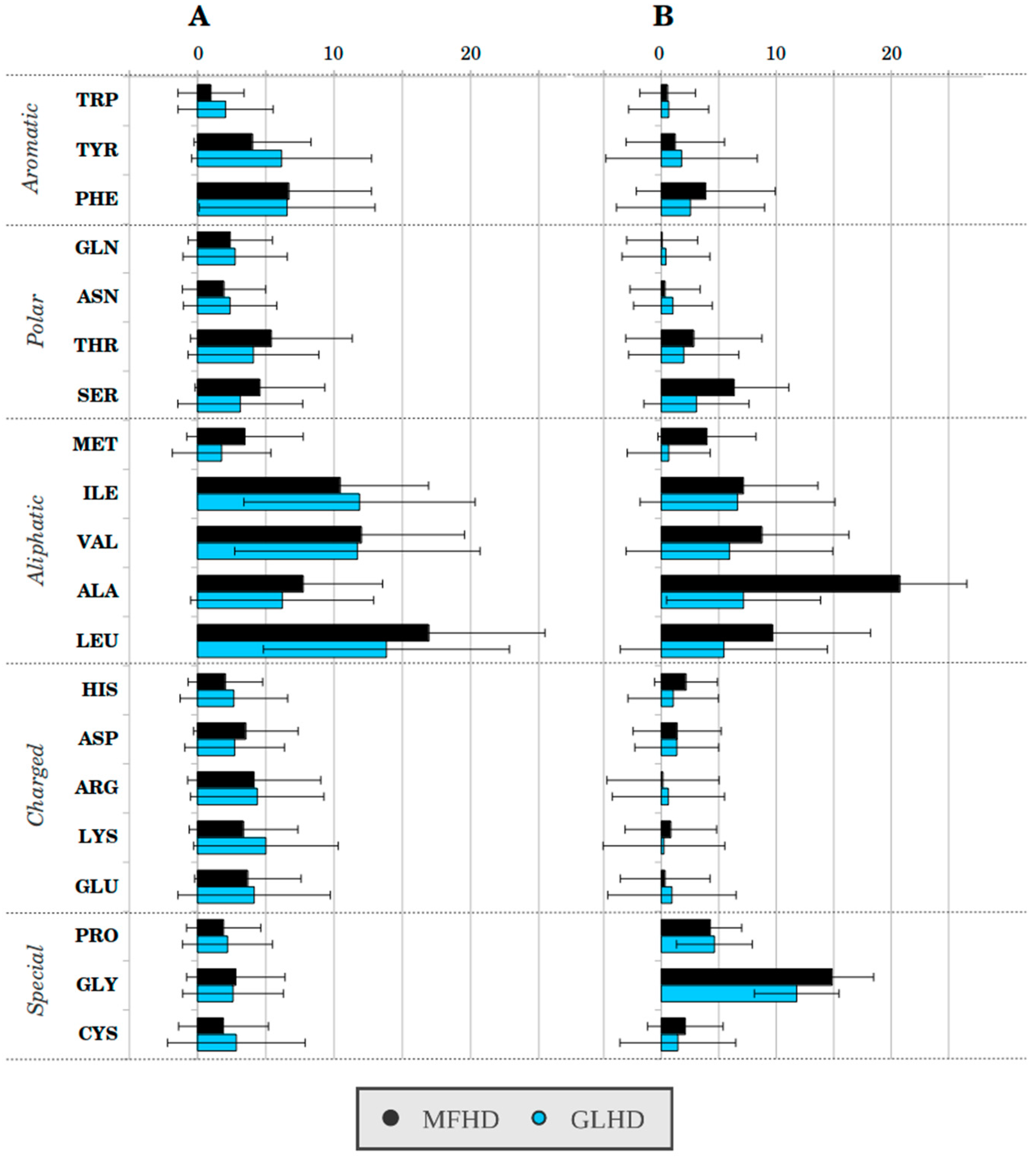{kind=link}
1. Introduction
2. Results
2.1. Sequence-Based Analysis
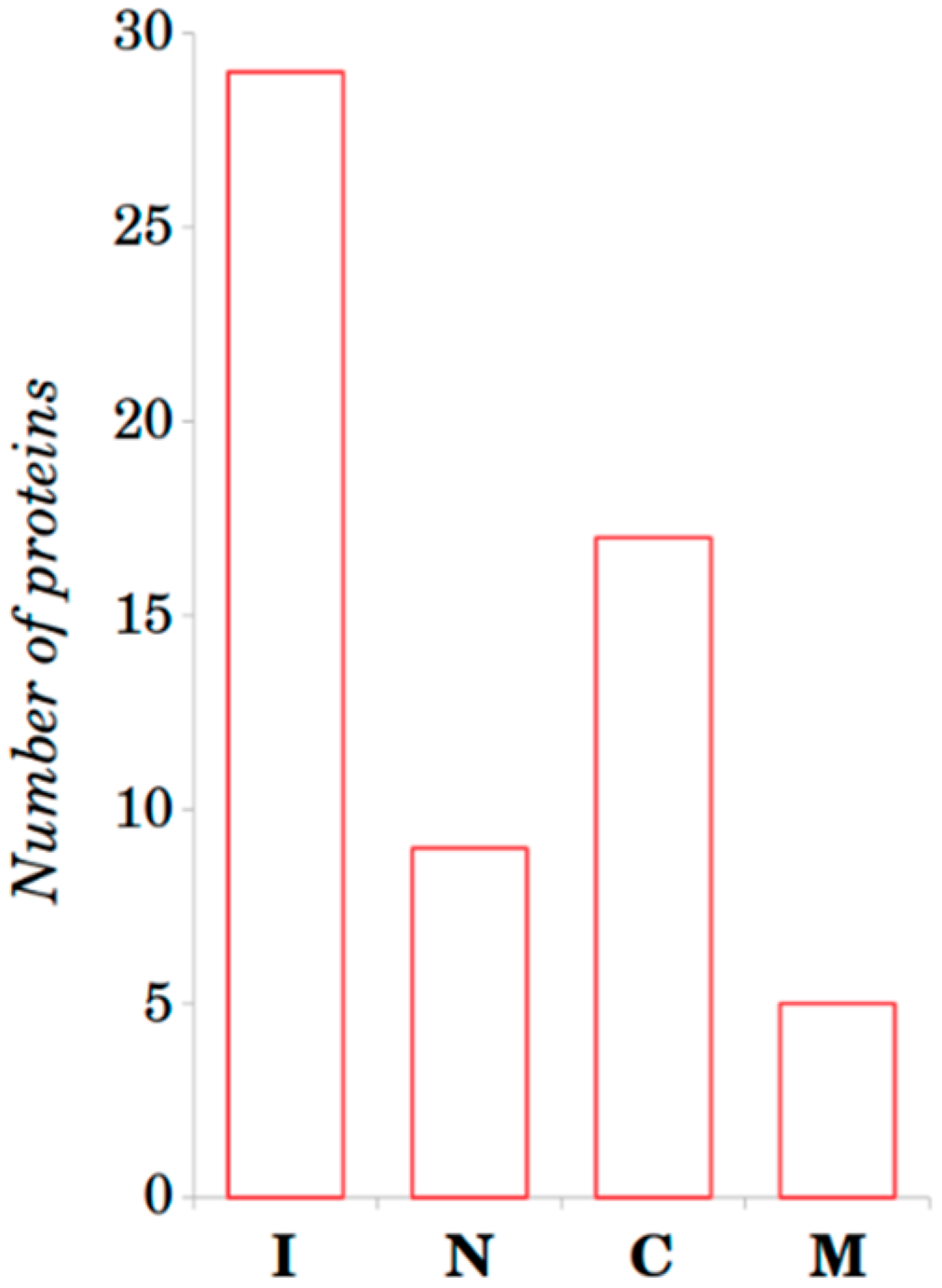
2.2. Structure-Based Analysis
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IDPs | Intrinsically disordered proteins |
| MFIB | Mutual Folding Induced by Binding database |
| DIBS | Disordered Binding Site database |
| ELMs | Short motifs of polypeptide chains |
| MFHD | MFIB homodimeric dataset |
| MSF | Mutual synergistic folding |
| GLHD | Globular homodimeric dataset |
| GLMD | Globular monomeric dataset |
| PCA | Principal component analysis |
| PDB | Protein data bank |
| RSAMPs | Residues with solvent accessible main-chain patches |
| SC/SCE | Stabilization centers/stabilization center elements |
| SASA | Solvent accessible surface area |
References
- Anfinsen, C.B.; Haber, E.; Sela, M.; White, F.H., Jr. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. USA 1961, 47, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. The formation and stabilization of protein structure. Biochem. J. 1972, 128, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Simon, I.; Dosztányi, Z. Prediction of Protein Binding Regions in Disordered Proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdos, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Tsai, C.-J.; Nussinov, R. Analysis of Ordered and Disordered Protein Complexes Reveals Structural Features Discriminating Between Stable and Unstable Monomers. J. Mol. Biol. 2004, 341, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Rumfeldt, J.A.O.; Galvagnion, C.; Vassall, K.A.; Meiering, E.M. Conformational stability and folding mechanisms of dimeric proteins. Prog. Biophys. Mol. Biol. 2008, 98, 61–84. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 2002, 415, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Garrard, S.M.; Capaldo, C.T.; Gao, L.; Rosen, M.K.; Macara, I.G.; Tomchick, D.R. Structure of Cdc42 in a complex with the GTPase-binding domain of the cell polarity protein, Par6. EMBO J. 2003, 22, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. Bioessays 2009, 31, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wu, Q.; Wang, C.; Xu, M.-Q.; Liu, Y. Mutual synergistic protein folding in split intein. Biosci. Rep. 2012, 32, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Zhang, W.; Chen, J. Synergistic folding of two intrinsically disordered proteins: Searching for conformational selection. Mol. Biosyst. 2012, 8, 198–209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fichó, E.; Reményi, I.; Simon, I.; Mészáros, B. MFIB: A repository of protein complexes with mutual folding induced by binding. Bioinformatics 2017, 33, 3682–3684. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Fichó, E.; Pancsa, R.; Simon, I.; Dosztányi, Z.; Mészáros, B. DIBS: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2017, 34, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Dobson, L.; Fichó, E.; Tusnády, G.E.; Dosztányi, Z.; Simon, I. Interplay between folding and binding modulates protein sequences, structures, functions and regulation. bioRxiv 2017, 211524. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Linding, R. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Necci, M.; Piovesan, D.; Dosztányi, Z.; Tosatto, S.C.E. MobiDB-lite: Fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics 2017, 33, 1402–1404. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Cserzö, M.; Wallin, E.; Simon, I.; von Heijne, G.; Elofsson, A. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: The dense alignment surface method. Protein Eng. 1997, 10, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Cserzö, M.; Eisenhaber, F.; Eisenhaber, B.; Simon, I. On filtering false positive transmembrane protein predictions. Protein Eng. 2002, 15, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.; Scott, R. Dehydron: A structurally encoded signal for protein interaction. Biophys. J. 2003, 85, 1914–1928. [Google Scholar] [CrossRef]
- Németh, A.; Svingor, A.; Pócsik, M.; Dobó, J.; Magyar, C.; Szilágyi, A.; Gál, P.; Závodszky, P. Mirror image mutations reveal the significance of an intersubunit ion cluster in the stability of 3-isopropylmalate dehydrogenase. FEBS Lett. 2000, 468, 48–52. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Fiser, A.; Simon, I. Stabilization centers in proteins: Identification, characterization and predictions. J. Mol. Biol. 1997, 272, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Magyar, C.; Gromiha, M.M.; Sávoly, Z.; Simon, I. The role of stabilization centers in protein thermal stability. Biochem. Biophys. Res. Commun. 2016, 471, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Simon, Á.; Dosztányi, Z.; Magyar, C.; Szirtes, G.; Rajnavölgyi, É.; Simon, I. Stabilization centers and protein stability. Theor. Chem. Acc. 2001, 106, 121–127. [Google Scholar] [CrossRef]
- Simon, A.; Dosztányi, Z.; Rajnavölgyi, E.; Simon, I. Function-related regulation of the stability of MHC proteins. Biophys. J. 2000, 79, 2305–2313. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Griep, S.; Hobohm, U. PDBselect 1992–2009 and PDBfilter-select. Nucleic Acids Res. 2010, 38, D318–D319. [Google Scholar] [CrossRef] [PubMed]
- Walshaw, J.; Woolfson, D.N. Socket: A program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol. 2001, 307, 1427–1450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xue, B.; Zhou, Y. DDOMAIN: Dividing structures into domains using a normalized domain-domain interaction profile. Protein Sci. 2007, 16, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S. Python Machine Learning; Packt Publishing Ltd.: Birmingham, UK, 2015; ISBN 9781783555147. [Google Scholar]
- The PyMOL Molecular Graphics System; Version 1.6; Schrodinger, LLC: New York, NY, USA, 2011.
- Martin, O.A. Wrappy: A Dehydron Calculator Plugin for PyMOL; IMASL-CONICET: San Luis, Argentina, 2012. [Google Scholar]
- Dosztanyi, Z. Servers for sequence-structure relationship analysis and prediction. Nucleic Acids Res. 2003, 31, 3359–3363. [Google Scholar] [CrossRef] [PubMed]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.J.; Thornton, J.M. Ion-pairs in proteins. J. Mol. Biol. 1983, 168, 867–885. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magyar, C.; Mentes, A.; Fichó, E.; Cserző, M.; Simon, I. Physical Background of the Disordered Nature of “Mutual Synergetic Folding” Proteins. Int. J. Mol. Sci. 2018, 19, 3340. https://doi.org/10.3390/ijms19113340
Magyar C, Mentes A, Fichó E, Cserző M, Simon I. Physical Background of the Disordered Nature of “Mutual Synergetic Folding” Proteins. International Journal of Molecular Sciences. 2018; 19(11):3340. https://doi.org/10.3390/ijms19113340
Chicago/Turabian StyleMagyar, Csaba, Anikó Mentes, Erzsébet Fichó, Miklós Cserző, and István Simon. 2018. "Physical Background of the Disordered Nature of “Mutual Synergetic Folding” Proteins" International Journal of Molecular Sciences 19, no. 11: 3340. https://doi.org/10.3390/ijms19113340
APA StyleMagyar, C., Mentes, A., Fichó, E., Cserző, M., & Simon, I. (2018). Physical Background of the Disordered Nature of “Mutual Synergetic Folding” Proteins. International Journal of Molecular Sciences, 19(11), 3340. https://doi.org/10.3390/ijms19113340