Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications


Abstract
1. Introduction
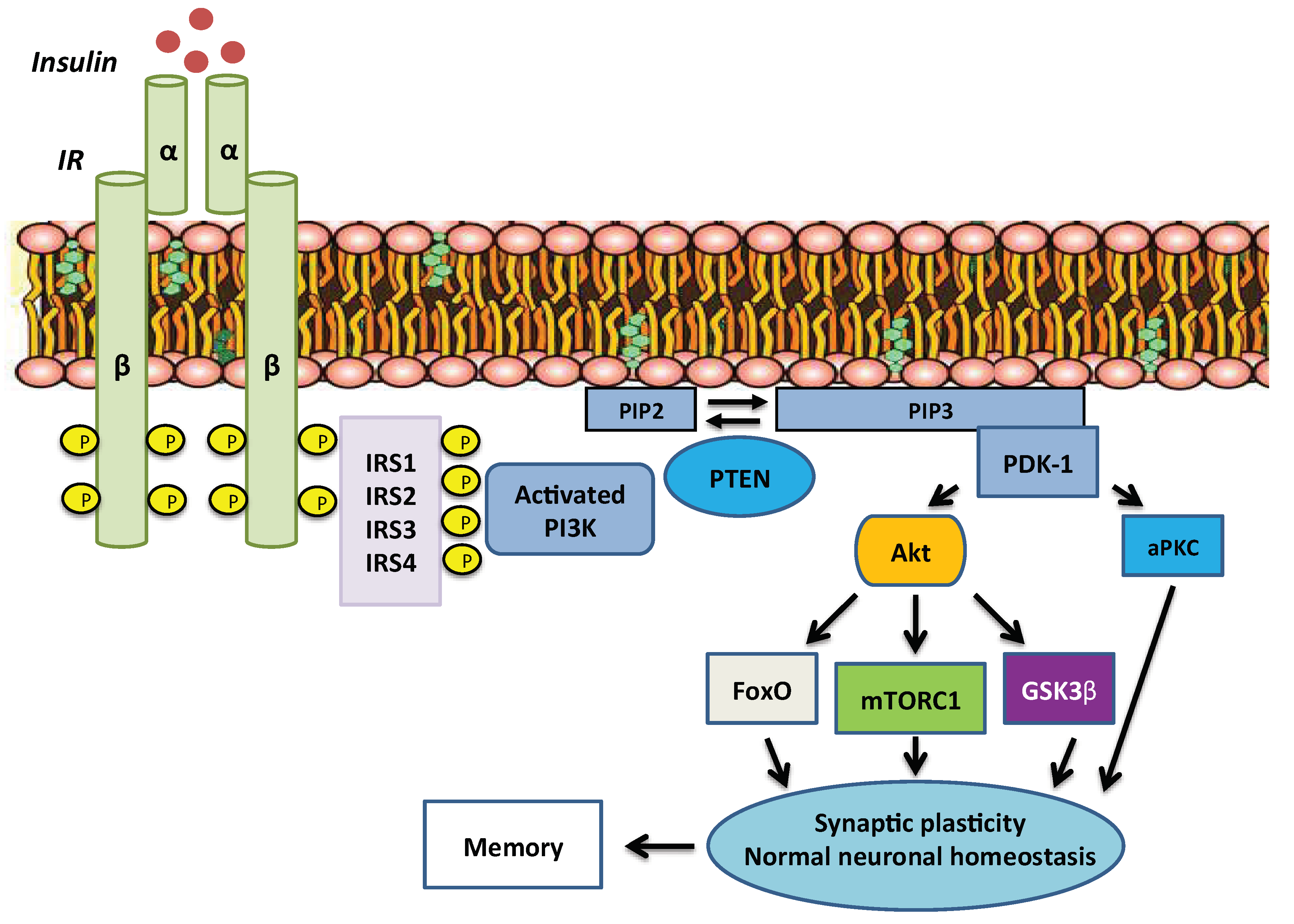
2. Overview of Insulin Signalling
3. T2DM and Neurodegeneration: The Role of Impaired Insulin Signalling, Insulin Resistance and Hyperinsulinemia
4. Insulin Signalling and Metabolism and Aβ Deposition
5. Impairment of Insulin Signalling and Tau Hyper-Phosphorylation
6. Insulin Resistance, Brain Vasculopathy and Neuroinflammation
7. Therapeutic Implications
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Apostolatos, A.; Song, S.; Acosta, S.; Peart, M.; Watson, J.E.; Bickford, P.; Cooper, D.R.; Patel, N.A. Insulin promotes neuronal survival via the alternatively spliced protein kinase CdeltaII isoform. J. Biol. Chem. 2012, 287, 9299–9310. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Strachan, M.W.; Visseren, F.L.; Kappelle, L.J.; Whitmer, R.A. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: Towards targeted interventions. Lancet Diabetes Endocrinol. 2014, 2, 246–255. [Google Scholar] [CrossRef]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, F.; Toth, C. Insulin and the brain. Curr. Diabetes Rev. 2013, 9, 102–116. [Google Scholar] [PubMed]
- Woods, S.C.; Seeley, R.J.; Baskin, D.G.; Schwartz, M.W. Insulin and the blood-brain barrier. Curr. Pharm. Des. 2003, 9, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.; Alves, C.J.; Neto, E.; Lamghari, M. Communication from the periphery to the hypothalamus through the blood-brain barrier: An in vitro platform. Int. J. Pharm. 2016, 499, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [PubMed]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.P.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Holscher, C. New drug treatments show neuroprotective effects in Alzheimer’s and Parkinson’s diseases. Neural Regen. Res. 2014, 9, 1870–1873. [Google Scholar] [CrossRef] [PubMed]
- Akintola, A.A.; van Heemst, D. Insulin, aging, and the brain: Mechanisms and implications. Front. Endocrinol. 2015, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Numan, S.; Russell, D.S. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain. Brain Res. Mol. Brain Res. 1999, 72, 97–102. [Google Scholar] [CrossRef]
- Araki, E.; Lipes, M.A.; Patti, M.E.; Bruning, J.C.; Haag, B., 3rd; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C.; et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. Off. J. Soci. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [CrossRef]
- Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 2007, 317, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Sadagurski, M.; Dong, X.C.; Myers, M.G., Jr.; White, M.F. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol. Metab. 2014, 3, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Denver, P.; English, A.; McClean, P.L. Inflammation, insulin signaling and cognitive function in aged APP/PS1 mice. Brain Behav. Immun. 2018, 70, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Anor, C.J.; O’Connor, S.; Saund, A.; Tang-Wai, D.F.; Keren, R.; Tartaglia, M.C. Neuropsychiatric Symptoms in Alzheimer Disease, Vascular Dementia, and Mixed Dementia. Neuro-Degener. Dis. 2017, 17, 127–134. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cesari, M.; Liu, F.; Dong, B.; Vellas, B. Effects of Diabetes Mellitus on Cognitive Decline in Patients with Alzheimer Disease: A Systematic Review. Can. J. Diabetes 2017, 41, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.; Escobedo, C.; Trevino, S.; Chavez, R.; Lopez-Lopez, G.; Moran, C.; Guevara, J.; Venegas, B.; Munoz-Arenas, G. Metabolic Syndrome Exacerbates the Recognition Memory Impairment and Oxidative-Inflammatory Response in Rats with an Intrahippocampal Injection of Amyloid Beta 1–42. Oxidative Med. Cell. Longev. 2018, 2018, 1358057. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.L.; Bullain, S.S.; Kawas, C.H. Late-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Rad. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Denver, P.; McClean, P.L. Distinguishing normal brain aging from the development of Alzheimer’s disease: Inflammation, insulin signaling and cognition. Neural Regen. Res. 2018, 13, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Trans. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Ratzmann, K.P.; Hampel, R. Glucose and insulin concentration patterns in cerebrospinal fluid following intravenous glucose injection in humans. Endokrinologie 1980, 76, 185–188. [Google Scholar] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. Off. J. Soci. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Zimbone, S.; Monaco, I.; Giani, F.; Pandini, G.; Copani, A.G.; Giuffrida, M.L.; Rizzarelli, E. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Martins, R.N.; Racchi, M.; Craft, S.; Helmerhorst, E. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. J. Alzheimer’s Dis. JAD 2002, 4, 369–374. [Google Scholar] [CrossRef]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. Off. J. Soci. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Kustermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Vandal, M.; White, P.J.; Tremblay, C.; St-Amour, I.; Chevrier, G.; Emond, V.; Lefrancois, D.; Virgili, J.; Planel, E.; Giguere, Y.; et al. Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes 2014, 63, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Lee, V.M. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormo, M.; Nilsson, Y.; Radesater, A.C.; Jerning, E.; Markgren, P.O.; Borgegard, T.; et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [PubMed]
- Freude, S.; Plum, L.; Schnitker, J.; Leeser, U.; Udelhoven, M.; Krone, W.; Bruning, J.C.; Schubert, M. Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes 2005, 54, 3343–3348. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.M.; Tseng, V.; Wang, J.; Wang, D.; Matyakhina, L.; Bondy, C.A. Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology 2005, 146, 5086–5091. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Chen, G.J.; Rivera, E.; Wands, J.R. Neuronal thread protein regulation and interaction with microtubule-associated proteins in SH-Sy5y neuronal cells. Cell. Mol. Life Sci. CMLS 2003, 60, 2679–2691. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Kahn, A.M.; Husid, A.; Allen, J.C.; Seidel, C.L.; Song, T. Insulin acutely inhibits cultured vascular smooth muscle cell contraction by a nitric oxide synthase-dependent pathway. Hypertension 1997, 30, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Bhamra, M.S.; Ashton, N.J. Finding a pathological diagnosis for Alzheimer’s disease: Are inflammatory molecules the answer? Electrophoresis 2012, 33, 3598–3607. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, G.; Khan, J.A.; Kumosani, T.A.; Kamal, M.A. Alzheimer’s disease and type 2 diabetes via chronic inflammatory mechanisms. Saudi J. Biol. Sci. 2015, 22, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.A.; Watson, G.S.; Montine, T.J.; Wang, Q.; Green, P.S.; Kulstad, J.J.; Cook, D.G.; Peskind, E.R.; Baker, L.D.; Goldgaber, D.; et al. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch. Neurol. 2005, 62, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Hill, M.D.; Rahimi, F.; Warden, L.A.; Halliday, G.M.; Shepherd, C.E. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2009, 19, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Watanabe, N. Ubiquitin-like protein MNSFbeta/endophilin II complex regulates Dectin-1-mediated phagocytosis and inflammatory responses in macrophages. Biochem. Biophys. Res. Commun. 2010, 401, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.; Rehman, K.; Chen, S. Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 2013, 114, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Hansen, K.; Banks, W.A. Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood-brain barrier: Protection by the antioxidant N-acetylcysteine. Brain Behav. Immun. 2012, 26, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Djelloul, M.; Taglialatela, G.; Perrone, L. Inflammatory risk factors and pathologies promoting Alzheimer’s disease progression: Is RAGE the key? Histol. Histopathol. 2015, 30, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Munch, G.; Schinzel, R.; Loske, C.; Wong, A.; Durany, N.; Li, J.J.; Vlassara, H.; Smith, M.A.; Perry, G.; Riederer, P. Alzheimer’s disease—Synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J. Neural Trans. 1998, 105, 439–461. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Cheng, Y.C.; Chen, S.J.; Yen, C.H.; Huang, R.N. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.M.; Chen, Y.L.; Pei, D.; Cheng, Y.C.; Sun, B.; Nicol, C.J.; Yen, C.H.; Chen, H.M.; Liang, Y.J.; Chiang, M.C. The neuroprotective role of metformin in advanced glycation end product treated human neural stem cells is AMPK-dependent. Biochim. Biophys. Acta 2015, 1852, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Kohler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Deng, J.; Sheng, W.; Zuo, Z. Metformin attenuates Alzheimer’s disease-like neuropathology in obese, leptin-resistant mice. Pharmacol. Biochem. Behav. 2012, 101, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, K.; Wang, R.; Liu, Y.; Kwak, Y.D.; Ma, T.; Thompson, R.C.; Zhao, Y.; Smith, L.; Gasparini, L.; et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. USA 2009, 106, 3907–3912. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-term metformin usage and cognitive function among older adults with diabetes. J. Alzheimer’s Dis. JAD 2014, 41, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Mi, J.; Jiang, Q.M.; Xu, J.M.; Tang, Y.Y.; Tian, G.; Wang, B. Metformin may produce antidepressant effects through improvement of cognitive function among depressed patients with diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 2014, 41, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Herath, P.M.; Cherbuin, N.; Eramudugolla, R.; Anstey, K.J. The Effect of Diabetes Medication on Cognitive Function: Evidence from the PATH Through Life Study. BioMed Res. Int. 2016, 2016, 7208429. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.M.; Mander, A.G.; Ames, D.; Kotowicz, M.A.; Carne, R.P.; Brodaty, H.; Woodward, M.; Boundy, K.; Ellis, K.A.; Bush, A.I.; et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 2013, 36, 2981–2987. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.; West, E.; Nolan, W.; McHale-Owen, H.; Williams, A.; Bate, C. Glimepiride protects neurons against amyloid-beta-induced synapse damage. Neuropharmacology 2016, 101, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Alp, H.; Varol, S.; Celik, M.M.; Altas, M.; Evliyaoglu, O.; Tokgoz, O.; Tanriverdi, M.H.; Uzar, E. Protective effects of beta glucan and gliclazide on brain tissue and sciatic nerve of diabetic rats induced by streptozosin. Exp. Diabetes Res. 2012, 2012, 230342. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.H.; Bahari, B.; Salari, A.A. ATP-sensitive potassium-channel inhibitor glibenclamide attenuates HPA axis hyperactivity, depression- and anxiety-related symptoms in a rat model of Alzheimer’s disease. Brain Res. Bull. 2018, 137, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Wahlqvist, M.L.; Lee, M.S.; Tsai, H.N. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J. Alzheimer’s Dis. JAD 2011, 24, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Landreth, G. Therapeutic use of agonists of the nuclear receptor PPARgamma in Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain J. Neurol. 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, X.; Blanchard, J.; Li, Y.; Iqbal, K.; Liu, F.; Gong, C.X. Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice. J. Neural Trans. 2015, 122, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martos, C.M.; Atkinson, R.A.K.; Chuah, M.I.; King, A.E.; Vickers, J.C. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimer’s Dement. 2017, 3, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shang, Y.; Jiang, L.; Shi, T.L.; Wang, L. The peroxisome proliferators activated receptor-gamma agonists as therapeutics for the treatment of Alzheimer’s disease and mild-to-moderate Alzheimer’s disease: A meta-analysis. Int. J. Neurosci. 2016, 126, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Holscher, C. The role of GLP-1 in neuronal activity and neurodegeneration. Vitam. Horm. 2010, 84, 331–354. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.; Holscher, C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Holscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- Hansen, H.H.; Barkholt, P.; Fabricius, K.; Jelsing, J.; Terwel, D.; Pyke, C.; Knudsen, L.B.; Vrang, N. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 2016, 1634, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Moller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Braendgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Kosaraju, J.; Gali, C.C.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Holsinger, R.M.; Madhunapantula, V.S.; Muthureddy Nataraj, S.K.; Basavan, D. Saxagliptin: A dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013, 72, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Kosaraju, J.; Murthy, V.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Muthureddy Nataraj, S.K.; Basavan, D. Vildagliptin: An anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.L.; Peng, C.H.; Huang, C.N. DPP-4 Inhibitor Linagliptin Attenuates Abeta-induced Cytotoxicity through Activation of AMPK in Neuronal Cells. CNS Neurosci. Ther. 2015, 21, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.R.; Barbieri, M.; Boccardi, V.; Angellotti, E.; Marfella, R.; Paolisso, G. Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 2001, 74, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Freiherr, J.; Hallschmid, M.; Frey, W.H., 2nd; Brunner, Y.F.; Chapman, C.D.; Holscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Schubert, M.; Bruning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. TEM 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. JAD 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Labuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopien, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Cheng, C.; Lin, C.H.; Tsai, Y.W.; Tsai, C.J.; Chou, P.H.; Lan, T.H. Type 2 diabetes and antidiabetic medications in relation to dementia diagnosis. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimer’s Dis. JAD 2016, 51, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Ma, Y.; Christophi, C.A.; Florez, H.; Golden, S.H.; Hazuda, H.; Crandall, J.; Venditti, E.; Watson, K.; Jeffries, S.; et al. Metformin, Lifestyle Intervention, and Cognition in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2017, 40, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Valencia, W.M.; Palacio, A.; Tamariz, L.; Florez, H. Metformin and ageing: Improving ageing outcomes beyond glycaemic control. Diabetologia 2017, 60, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Imfeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: A population-based case-control study. J. Am. Geriatr. Soc. 2012, 60, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Bencivenga, L.; Petraglia, L.; Visaggi, L.; Gioia, L.; Grieco, F.V.; de Lucia, C.; Komici, K.; Corbi, G.; Edison, P.; et al. Antidiabetic Drugs in Alzheimer’s Disease: Mechanisms of Action and Future Perspectives. J. Diabetes Res. 2017, 2017, 7420796. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A. Glucagon-like peptide 1 (GLP-1) in the treatment of diabetes. Horm. Metab. Res. 2004, 36, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Sherman, S.I.; Gorelick, F.S.; Bergenstal, R.M.; Sherwin, R.S.; Buse, J.B. Incretin-based therapies for the treatment of type 2 diabetes: Evaluation of the risks and benefits. Diabetes Care 2010, 33, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Cork, S.C.; Richards, J.E.; Holt, M.K.; Gribble, F.M.; Reimann, F.; Trapp, S. Distribution and characterisation of Glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol. Metab. 2015, 4, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, L.X.; Chen, Z.; Liu, L.B. Liraglutide prevents beta-amyloid-induced neurotoxicity in SH-SY5Y cells via a PI3K-dependent signaling pathway. Neurol. Res. 2016, 38, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.Y.; Wang, Z.J.; Holscher, C.; Yuan, L.; Zhang, J.; Sun, P.; Li, J.; Yang, W.; Wu, M.N.; Qi, J.S. Lixisenatide attenuates the detrimental effects of amyloid beta protein on spatial working memory and hippocampal neurons in rats. Behav. Brain Res. 2017, 318, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Isik, A.T.; Soysal, P.; Yay, A.; Usarel, C. The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 123, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Shingo, A.S.; Kanabayashi, T.; Kito, S.; Murase, T. Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats. Behav. Brain Res. 2013, 241, 105–111. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drugs | Effects on CNS | Ref. |
|---|---|---|
| Metformin | Restores mitochondria, attenuates AGEs effects through the activation of AMPK in human neural stem cells | [72,73] |
| Re-sensitizes impaired insulin signalling and reduces phosphorylation of tau, in neuronal cell lines | [74] | |
| Induces protein phosphatase 2A and reduces tau phosphorylation in murine neurons of Tau transgenic mouse | [75] | |
| Attenuates cognitive impairment in obese leptin resistant mice | [76] | |
| Increases the generation of amyloid beta protein, in human cell models (negative effect) | [77] | |
| Decreases the risk of cognitive decline in diabetic patients | [78] | |
| Improves depressive and cognitive performance in depressed patients | [79] | |
| Protective effect on domain of verbal learning, working memory and executive function | [80] | |
| Increases the risk of cognitive impairment in studies conducted on AD patients (negative effect) | [81] | |
| Sulfonylureas | Glimepiride protects neurons against beta amyloid induced synapse degeneration, in vitro | [82] |
| Gliclazide exerts antioxidant effect in the brain, in diabetic rats | [83] | |
| Glibenclamide decreases depression and anxiety, in a rat model of AD | [84] | |
| In combination with metformin, reduce the risk of dementia, in diabetic patients | [85] | |
| Glitazones | Neuroprotective effects in AD related to inhibition of inflammation and Aβ deposition | [86] |
| Pioglitazone reduces AD-related pathologies suppressing glial activation, in mice | [87] | |
| Pioglitazone enhances Akt signalling and tau hyperphosphorylation, in mouse model of AD | [88] | |
| In combination with leptin, pioglitazone reduces brain amyloid levels, in mouse model of AD | [89] | |
| Pioglitazone improves cognition and regional cerebral blood flow of T2DM patients | [90] | |
| Pioglitazone may provide an improvement in early stage and in mild to moderate AD in humans | [91] | |
| GLP-1 RA | Reduce apoptosis and oxidative stress; ameliorate synaptic plasticity in AD mouse model | [92] |
| Influence cellular pathways of neuronal protection and mitochondrial function | [93] | |
| Reduce tau phosphorylation, prevent synaptic loss, diminish Aβ deposition in AD mouse model | [94,95] | |
| Prevent the decline of brain glucose metabolism in AD patients with long-standing disease | [96] | |
| DPP4-i | Decrease tau phosphorylation, amyloid load and the cognitive deficits with memory improvement | [97,98] |
| Improve incretin levels, reduce Aβ deposition, tau phosphorylation, GSK-3β activation and ROS | [99] | |
| Improve glucose control and prevent worsening of cognitive function, in older patients with T2DM | [100] | |
| Insulin | Attenuates cognitive decline and enhances memory in adults with AD | [101,102] |
| In vitro inhibits apoptosis; in vivo regulates tau phosphorylation, Aβ metabolism and clearance | [103] | |
| Improves memory, mood, cerebral glucose metabolism; preserves brain volume in AD patients | [104] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 3306. https://doi.org/10.3390/ijms19113306
Tumminia A, Vinciguerra F, Parisi M, Frittitta L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. International Journal of Molecular Sciences. 2018; 19(11):3306. https://doi.org/10.3390/ijms19113306
Chicago/Turabian StyleTumminia, Andrea, Federica Vinciguerra, Miriam Parisi, and Lucia Frittitta. 2018. "Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications" International Journal of Molecular Sciences 19, no. 11: 3306. https://doi.org/10.3390/ijms19113306
APA StyleTumminia, A., Vinciguerra, F., Parisi, M., & Frittitta, L. (2018). Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. International Journal of Molecular Sciences, 19(11), 3306. https://doi.org/10.3390/ijms19113306