Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species

Abstract

1. Introduction
2. Results and Analysis
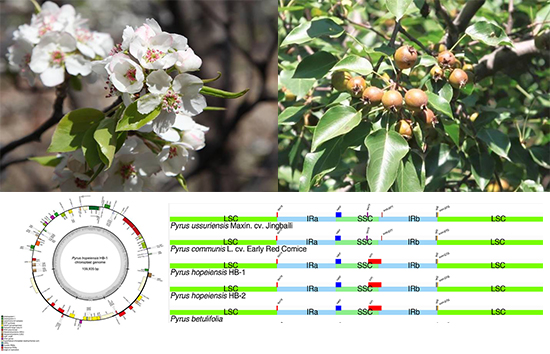
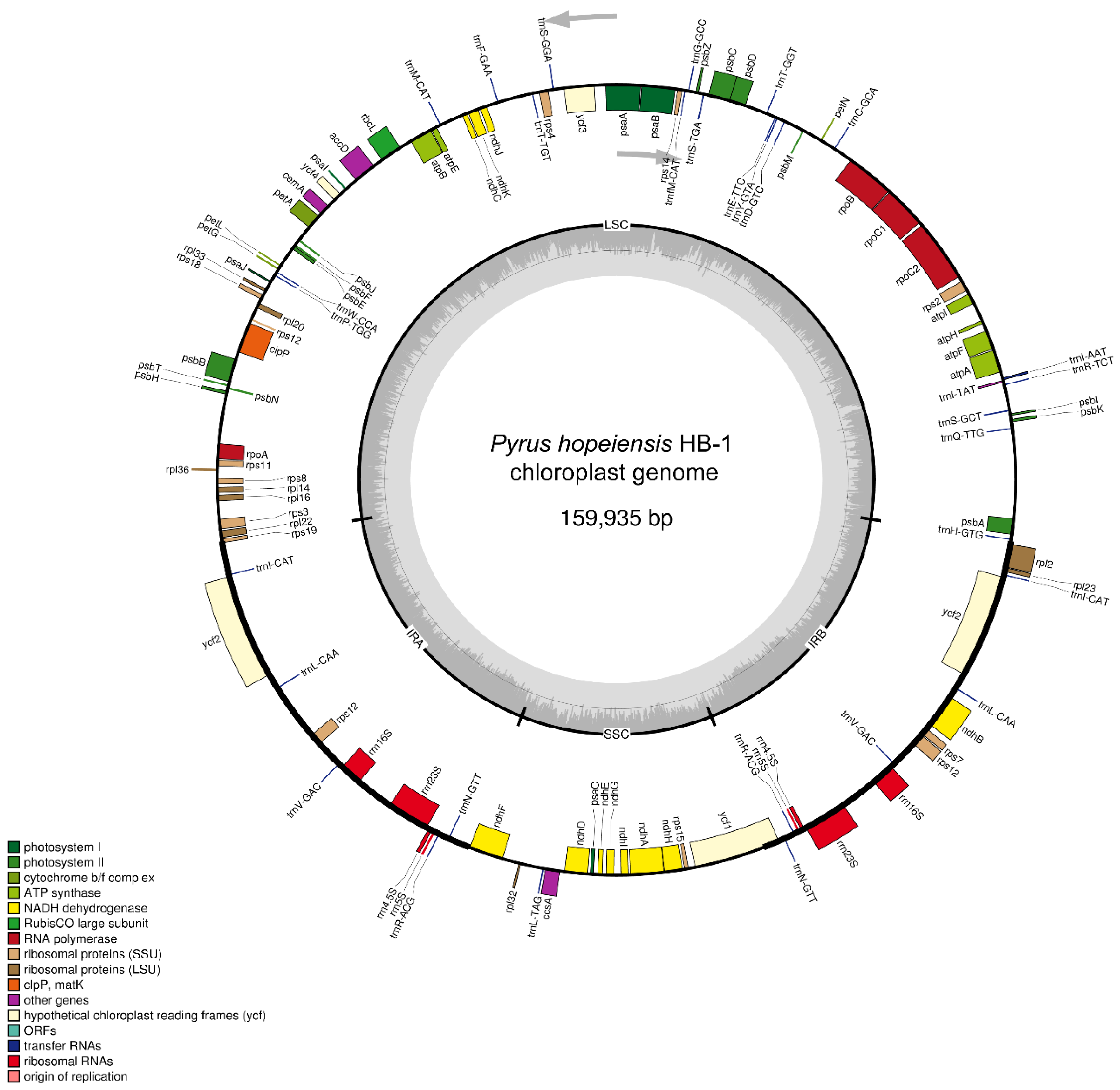
2.1. Basic Characteristics of Chloroplast Genome of P. hopeiensis
2.2. Comparison of the Basic Characteristics of the Chloroplast Genome in Five Pyrus Species
2.3. Chloroplast Gene Gain–Loss Events
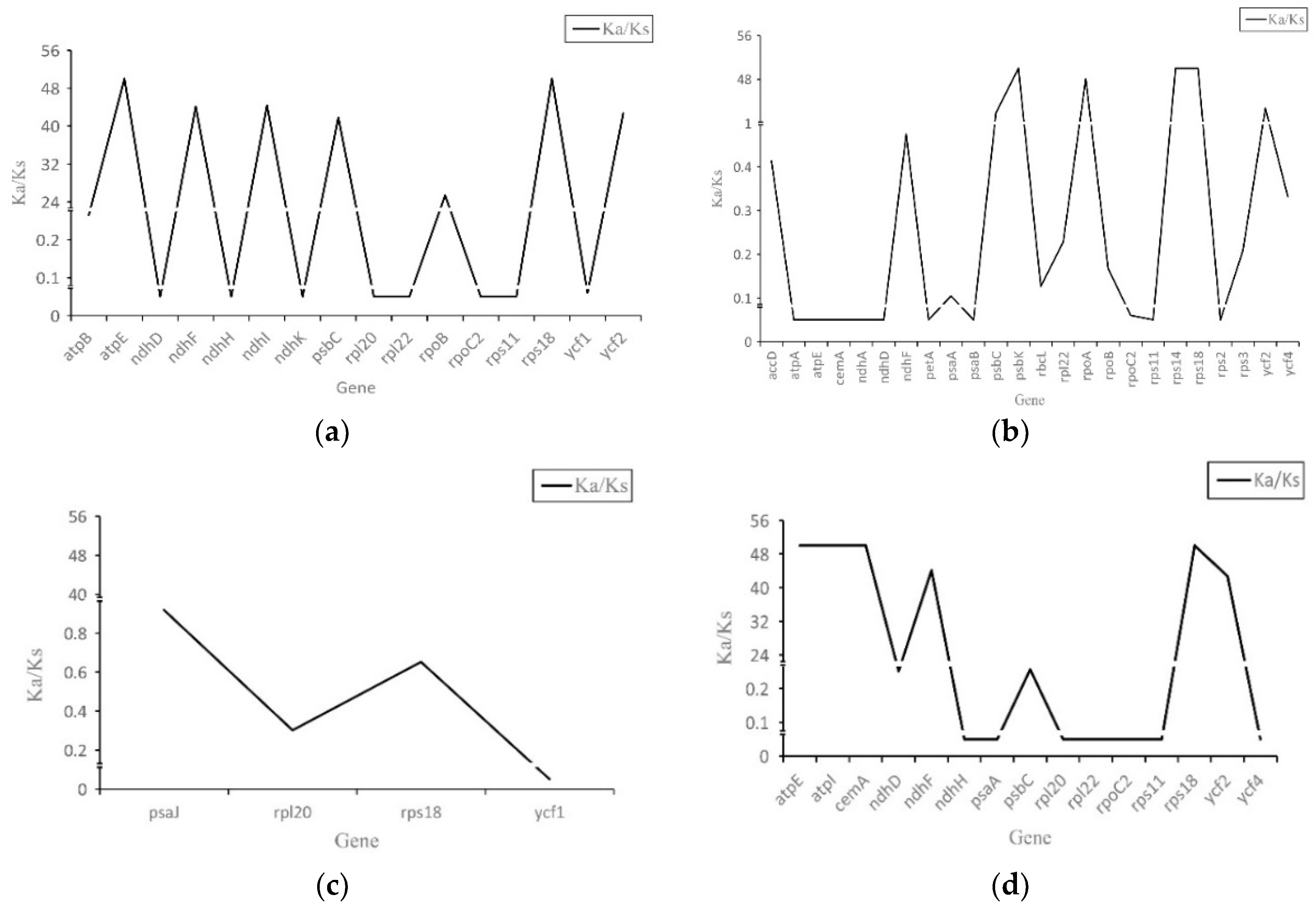
2.4. Synonymous (KS) and Nonsynonymous (KA) Substitution Rate Analysis
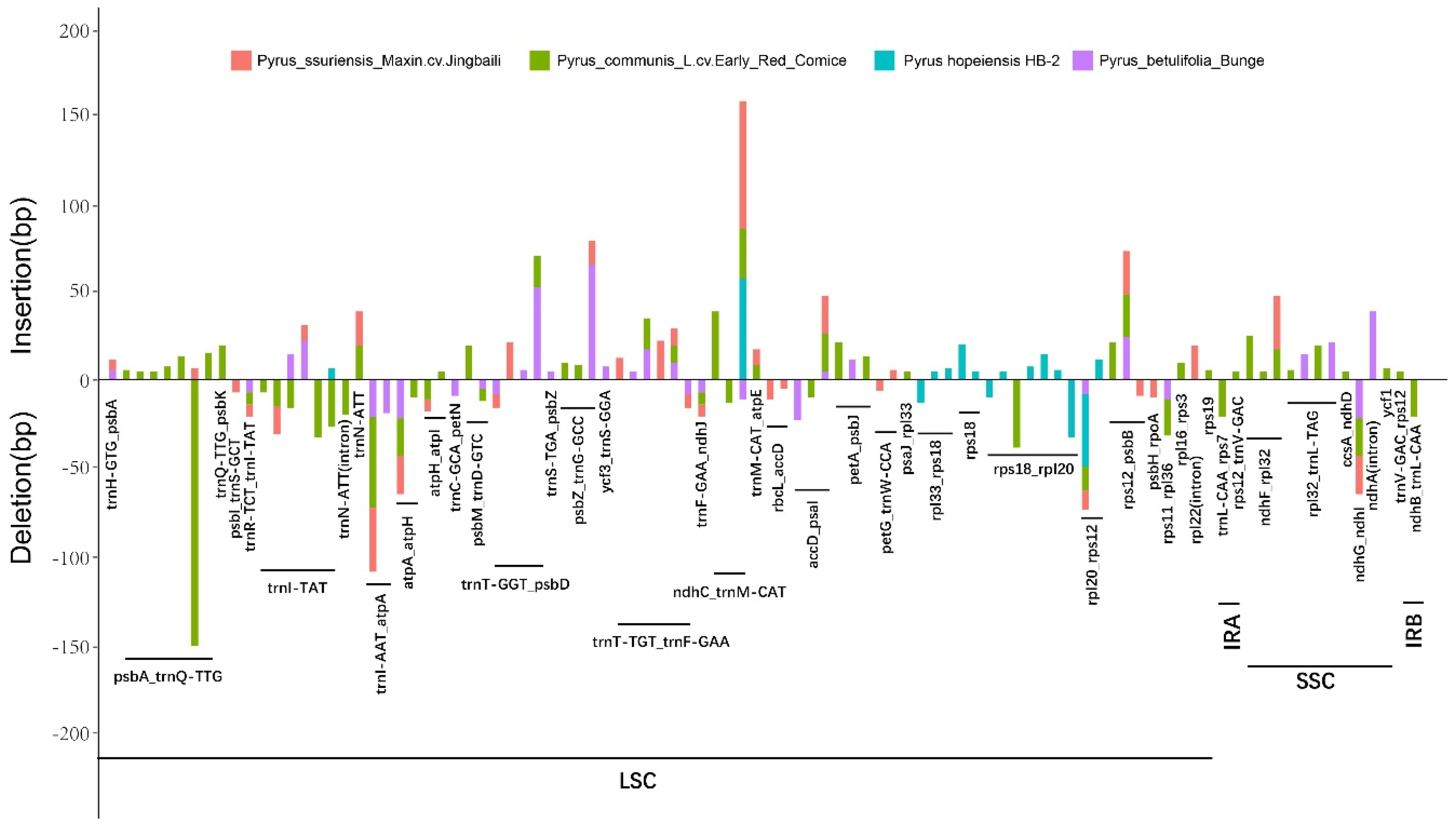
2.5. Indel Identification and Relationship of the Five Pyrus cp Genomes
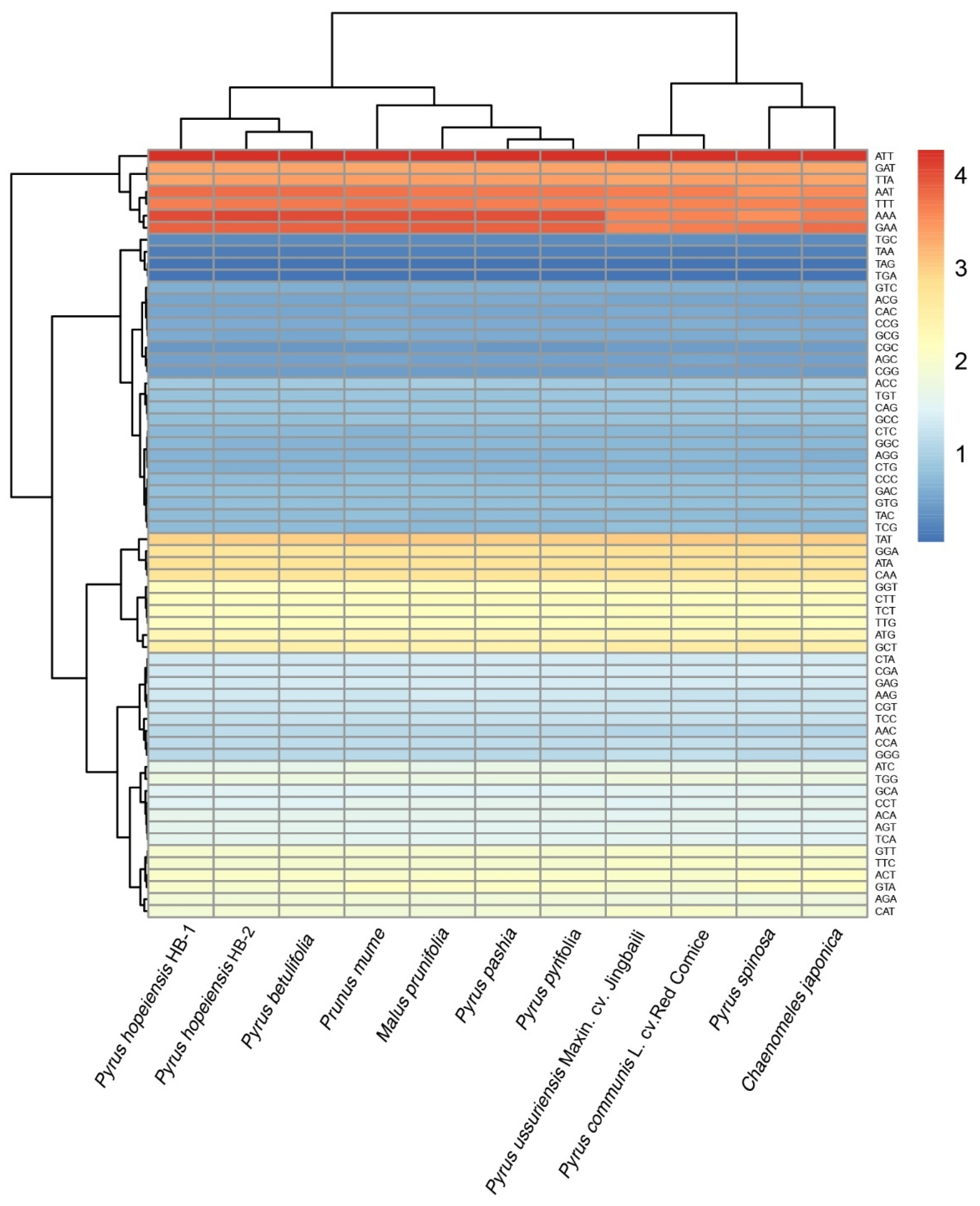
2.6. Codon Preference Analysis
2.7. Comparison of the Genome Structure in Rosaceae cp Genomes
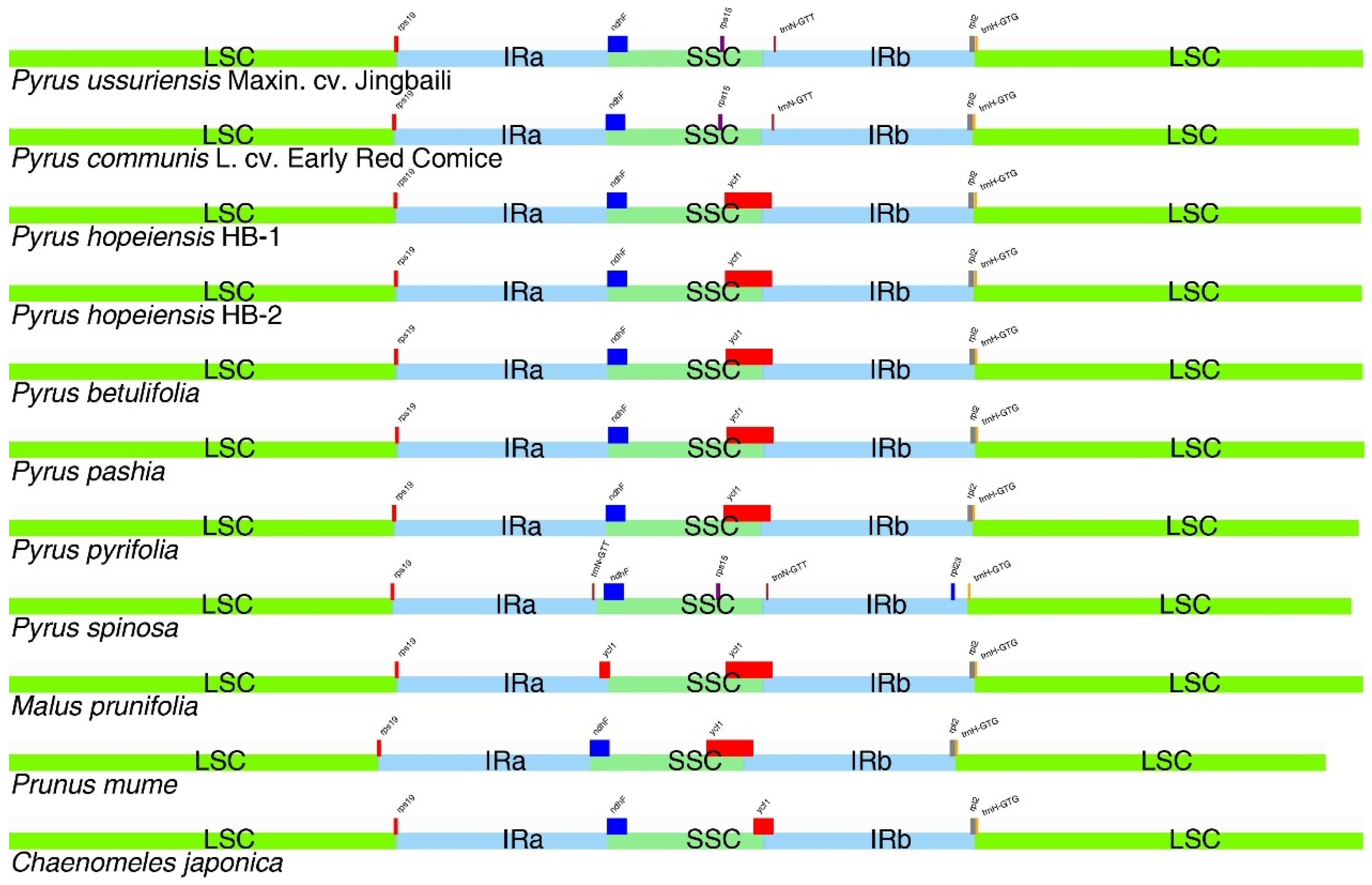
2.8. IR Contraction Analysis
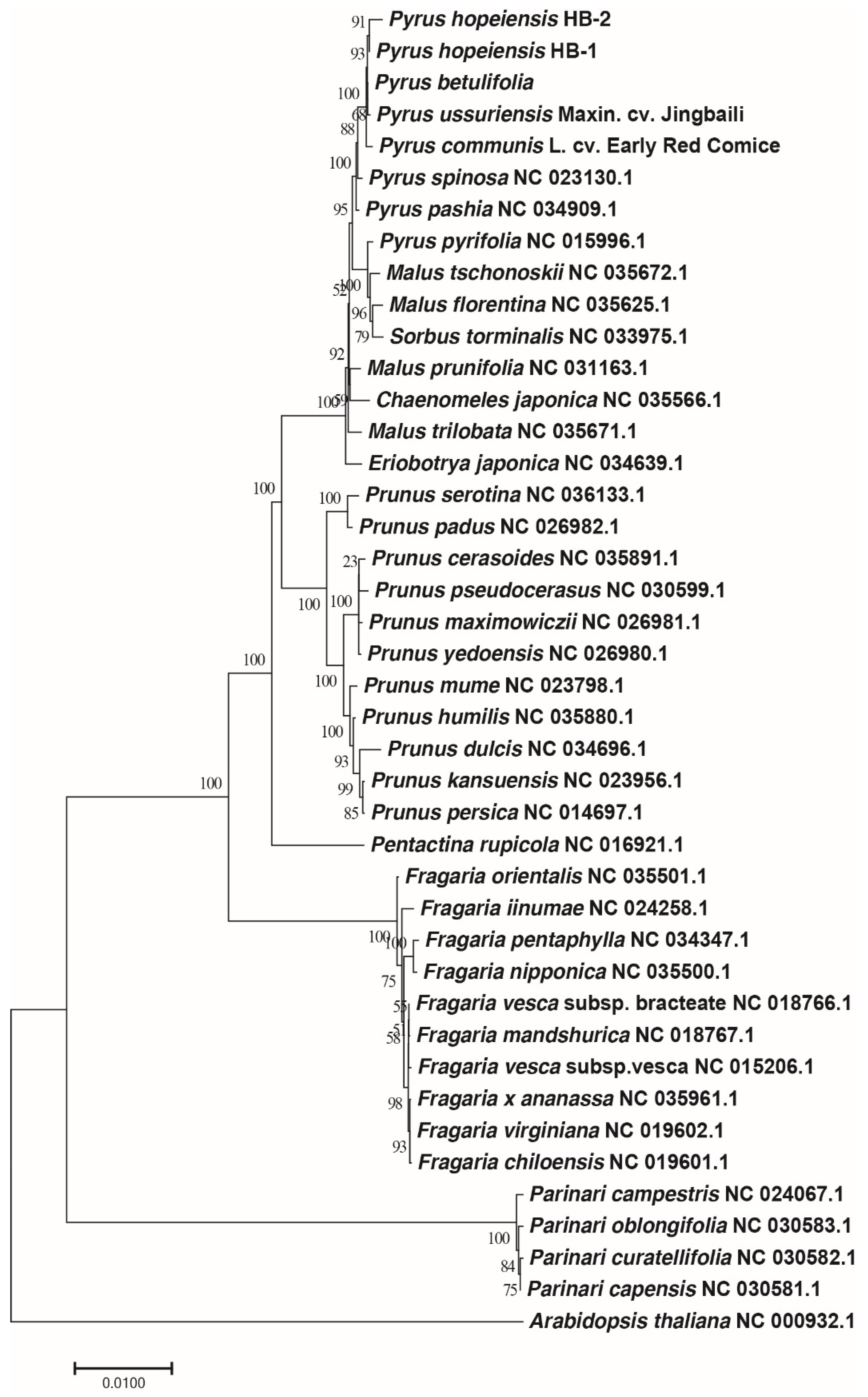
2.9. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Sequencing, Genome Assembly, and Validation
4.3. Gene Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chai, M.L.; Shen, D.X. Status and Prospects of Pear Breeding in China. J. Fruit Sci. 2003, 5, 379–383. [Google Scholar]
- Yu, D.J. Taxonomy of the Fruit Tree in China; China Agriculture Press: Beijing, China, 1979; p. 122. [Google Scholar]
- Jiang, X.F.; Chu, Q.G.; Zhang, C.S. Studies on the classification and evolution of the genus pyrus in China. J. Laiyang Agric. Coll. 1992, 9, 18–21. [Google Scholar]
- Pu, F.S.; Huang, L.S.; Sun, B.J.; Li, S.L. Study on the chromosome number of wild and cultivated pears (Pyrus sp.) in China. Acta Hortic. Sin. 1985, 12, 155–158. [Google Scholar]
- Neuhaus, H.E.; Ernes, M.J. Nonphotosyntiietic Metabolism in Plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Dumolin, S.; Demesure, B.; Etit, R.J. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor. Appl. Genet. 1995, 91, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.C. Progress in Chloroplast Genome Analysis. Prog. Biochem. Biophys. 2008, 35, 21–28. [Google Scholar]
- Bausher, M.G.; Singh, N.D.; Lee, S.B.; Jansen, R.K.; Daniell, H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: Organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Guisinger, M.M.; Chumley, T.W.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J. Mol. Evol. 2010, 70, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.L.; Ma, L.C.; Nie, B.; Wang, Y.R.; Liu, Z.P. The draft and characterization of the complete chloroplast genome of Vicia sativa cv. Lanjian No. 3. Pratacult. Sci. 2017, 34, 321–330. [Google Scholar]
- Jansen, R.K.; Saski, C.; Lee, S.B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011, 28, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Fujimoto, M.; Arimura, S.I.; Murata, J.; Tsutsumi, N.; Kadowaki, K.I. Loss of the rp132 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in populus. Gene 2007, 402, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Ge, C.F.; Zhang, H.; Qiao, Y. Advances on Chloroplast Genome Sequencing and Phylogenetic Analysis in Fruit Trees. J. Nucl. Agric. Sci. 2018, 32, 58–69. [Google Scholar]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Lyu, B.B.; He, J.H.; Wang, J.B.; Wu, X.; Wu, G.G.; Bao, D.P.; Chen, M.J.; Zhang, J.S.; Tan, Q.; et al. Codon usage bias in the straw mushroom Volvariella volvacea. Chin. J. Biotechnol. 2014, 30, 1424–1435. (In Chinese) [Google Scholar]
- Li, Y.; Kuang, X.J.; Zhu, X.X.; Zhu, Y.J.; Sun, C. Codon usage bias of Catharanthus roseus. China J. Chin. Mater. Med. 2016, 41, 4165–4168. [Google Scholar]
- Zuo, L.H.; Shang, A.Q.; Zhang, S.; Yu, X.Y.; Ren, Y.C.; Yang, M.S.; Wang, J.M. The first complete chloroplast genome sequences of Ulmus species by de novo sequencing: Genome comparative and taxonomic position analysis. PLoS ONE 2017, 12, e0171264. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, el2762. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophord). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mung bean (Vigna radiatd) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2010, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Kim, K.J. Complete chloroplast genome sequences of important oil seed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Zhou, S.L. Molecular identification of species in Prunus sect. Persica (Rosaceae), with emphasis barcodes for plants. J. Syst. Evol. 2011, 49, 138–145. [Google Scholar] [CrossRef]
- Wand, L.; Dong, W.P.; Zhou, S.L. Structural Mutations and Reorganizations in Chloroplast Genomes of Flowering Plants. Acta Bot. Boreal. Occident. Sin. 2012, 32, 1282–1288. [Google Scholar]
- Yu, D.J.; Gu, C.Z. Flora Reipublicae Popularis Sinicae; Bejing Science Press: Beijing, China, 1974; Volume 36. (In Chinese) [Google Scholar]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Wu, Y.; Zhou, H. Research progress of sugarcane chloroplast genome. J. South. Agric. 2013, 44, 17–22. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-ecient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Szmidt, A.E.; Aldén, T.; Hällgren, J.E. Paternal inheritance of chloroplast DNA in larix. Plant Mol. Biol. 1987, 9, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Nock, C.J.; Waters, D.L.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2010, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Pu, F.S.; Lin, S.H.; Chen, R.Y.; Song, W.Q.; Li, X.L. Studies on karyotype of pyrus in China(II). Acta Horticult. Sin. 1986, 13, 87–90. [Google Scholar]
- Pu, F.S.; Lin, S.H.; Song, W.Q.; Chen, R.Y.; Li, X.L. Studies on karyotype of pyrus in China(I). J. Wuhan Bot. Res. 1985, 3, 381–387. [Google Scholar]
- Lin, B.N.; Shen, D.X. Studies on the germplasmic characteristics of Pyrus by use of isozymic patterns. Acta Agric. Univ. Zhejiang Gensis 1983, 9, 235–242. [Google Scholar]
- Teng, Y.W.; Tanabe, K.; Tamura, F.; Itai, A. Genetic relationships of pear cultivars in Xin Jiang, China, as measured by RAPD markers. J. Horticult. Sci. Biotechnol. 2001, 76, 771–779. [Google Scholar] [CrossRef]
- Teng, Y.; Tanabe, K.; Tamura, F.; Itai, A. Genetic relationships of Pyrus species and cultivars native to East Asia revealed by randomly amplified polymorphic DNA markers. J. Am. Soc. Hortic. Sci. Biotech. 2002, 127, 262–270. [Google Scholar]
- Teng, Y.W.; Tanabe, K. Reconsideration on the origin of cultivated pears native to East Asia. 4thInternational Symposium of Taxonomy and Nomenclature of Cultivated Plants. Acta Hortic. 2004, 634, 175–182. [Google Scholar] [CrossRef]
- Teng, Y.W. Advances in the research on phylogeny of the genus Pyrus and the origin of pear cultivars native to East Asia. J. Fruit Sci. 2017, 34, 370–378. [Google Scholar]
- Zheng, X.Y.; Cai, D.Y.; Yao, L.H.; Teng, Y.W. Non-concerted ITS evolution, early origin and phylogenetic utility of ITS pseudogenes in Pyrus. Mol. Phylogenet. Evol. 2008, 48, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.Y.; Zheng, X.Y.; Teng, Y.W. Characterization and Phylogenetic Utility of Non-coding Chloroplast Regions trnL-trnF and accD-psaI in Pyrus. Acta Hortic. Sin. 2011, 38, 2261–2272. [Google Scholar]
- Liu, Y. Studies on Chloroplast DNA Diversity of Chinese Pear; Capital Normal University: Beijing, China, 2006. [Google Scholar]
- Raghvendra, A.S. Photosynhcsis: A Comprehensive Treatise; Cambridge University Press: Cambridge, UK, 1998; pp. 72–86. [Google Scholar]
- Kent, W.J. BLAT-the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Mott, R. EST_GENOME: A program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 1997, 13, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Abouelhoda, M.I.; Kurtz, S.; Ohlebusch, E. Replacing suffix trees with enhanced suffix arrays. J. Disc. Algo. 2004, 2, 53–86. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.P.; Zhang, Y.B.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions | Family Name | Code | List of Genes |
|---|---|---|---|
| Self-replication | Small subunit of ribosome | rps | rps2, rps3, rps4, rps7a, rps8, rps11, rps12abe, rps14, rps15, rps18, rps19 |
| rRNA Genes | rrn | rrn4.5Sa, rrn5Sa, rrn16Sa, rrn23Sa | |
| Large subunit of ribosome | rpl | rpl2ab, rpl14, rpl16, rpl20, rpl22b, rpl23a, rpl32, rpl33, rpl36 | |
| DNA dependent RNA polymerase | rpo | rpoA, rpoB, rpoC1b, rpoC2 | |
| tRNA Genes | trn | trnC-GCA, trnD-GTC, trnE-TTC, trnF-GAA, trnfM-CAT, trnG-GCC, trnH-GTG, trnI-TATb, trnI-CATa, trnI-AATb, trnL-CAAa, trnL-TAG, trnM-CAT, trnN-GTTa, trnQ-TTG, trnP-TGG, trnR-TCT, trnR-ACGa, trnS-GCT, trnS-TGA, trnS-GGA, trnT-GGT, trnT-TGT, trnV-GACa, trnW-CCA, trnY-GTA | |
| Genes for photosynthesis | Subunits of ATP synthase | atp | atpA, atpB, atpE, atpFd, atpH, atpI |
| Subunits of protochlorophyllide reductase | chl | ||
| Subunits of NADH-dehydrogenase | ndh | ndhAb, ndhBb, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | pet | petA, petG, petL, petN | |
| Subunits of photosystem Ⅰ | psa | psaA, psaB, psaC, psaJ, psaI | |
| Subunits of photosystem Ⅱ | psb | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbM, psbN, psbT, psbZ | |
| Subunit of rubisco | rbc | rbcL | |
| Other genes | Subunit of Acetyl-CoA-carboxylase | acc | accD |
| Envelop membrane protein | cem | cemA | |
| c-type cytochrome synthesis gene | ccs | ccsA | |
| Protease | clp | clpPd | |
| Translational initiation factor | inf | ||
| Maturase | mat | matK | |
| Elongation factor | tuf | ||
| Genes of unknown function | Conserved open reading frames | ycf | ycf1, ycf2a, ycf3c, ycf4 |
| Pyrus hopeiensis HB-1 | Pyrus hopeiensis HB-2 | Pyrus ussuriensis Maxin. cv. Jingbaili | Pyrus communis L. cv. Early Red Comice | Pyrus betulifolia | |
|---|---|---|---|---|---|
| Length (bp) | 159,935 | 159,981 | 160,059 | 159,834 | 160,058 |
| GC content (%) | 36.59 | 36.57 | 36.57 | 36.58 | 36.57 |
| AT content (%) | 63.41 | 63.43 | 63.43 | 63.42 | 63.43 |
| LSC length (bp) | 87,962 | 88,008 | 88,075 | 87,794 | 88,025 |
| SSC length (bp) | 19,201 | 19,201 | 19,212 | 19,260 | 19,261 |
| IR length (bp) | 26,386 | 26,386 | 26,386 | 26,390 | 26,386 |
| Gene number | 118 | 119 | 117 | 114 | 120 |
| Pseudogene number | 2 | 2 | 2 | 2 | 2 |
| Gene number in IR regions | 32 | 32 | 31 | 31 | 32 |
| Protein-coding gene number | 77 | 78 | 75 | 74 | 77 |
| Protein-coding gene (%) | 64.25 | 65.55 | 64.10 | 64.91 | 64.17 |
| rRNA gene number | 8 | 8 | 8 | 8 | 8 |
| rRNA (%) | 6.78 | 6.72 | 6.84 | 7.02 | 6.67 |
| tRNA gene number | 31 | 31 | 32 | 30 | 33 |
| tRNA (%) | 26.27 | 26.05 | 27.35 | 26.32 | 26.50 |
| Gene | Strand | Pyrus hopeiensis HB-1 | Pyrus hopeiensis HB-2 | Pyrus ussuriensis Maxin. cv. Jingbaili | Pyrus communis L. cv. Early Red Comice | Pyrus betulifolia |
|---|---|---|---|---|---|---|
| trnI-TAT | − | √ | × | √ | × | √ |
| trnI-TAT | + | × | × | √ | × | √ |
| trnN-ATT | + | × | × | √ | √ | × |
| rpoC1 | − | √ | √ | √ | √ | √ |
| ycf3 | − | √ | √ | √ | √ | √ |
| rpl22 | − | √ | √ | √ | √ | √ |
| rpl2 | − | √ | √ | √ | √ | √ |
| ndhA | − | √ | √ | √ | √ | √ |
| ndhB | + | √ | √ | √ | √ | √ |
| rpl2 | + | √ | √ | √ | √ | √ |
| rps12 | − | √ | √ | √ | √ | √ |
| rps12 | + | √ | √ | √ | √ | √ |
| trnI-AAT | + | √ | √ | × | × | × |
| trnL-TAG | − | × | × | × | × | √ |
| trnY-ATA | + | × | × | × | × | √ |
| Total | 15 | 11 | 10 | 12 | 10 | 13 |
| Specie | atpB | matK | petB | petD | psaC | psbI | psbL | psbl | Rpl20 | Rpl36 | rps16 | ycf1 | trnI-GAU | trnA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pyrus ussuriensis Maxin. cv. Jingbaili | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Pyrus communis L. cv. Early Red Comice | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Pyrus hopeiensis HB-1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 |
| Pyrus hopeiensis HB-2 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 |
| Pyrus betulifolia | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 |
| Pyrus pyrifolia | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| Pyrus spinosa | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 |
| Pyrus pashia | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 2 | 0 | 0 |
| Total number of missing genes | 2 | 1 | 5 | 5 | 1 | 1 | 7 | 7 | 1 | 2 | 5 | 2 | 7 | 7 |
| Specie | trnA-UGC | trnG-GCC | trnG-UCC | trnI-AAU | trnI-GAU | trnI-UAU | trnK-UUU | trnL | trnL-UAA | trnN-AUU | trnV-UAC | trnY-AUA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pyrus ussuriensis Maxin. cv. Jingbaili | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 |
| Pyrus communis L. cv. Early Red Comice | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Pyrus hopeiensis HB-1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pyrus hopeiensis HB-2 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pyrus betulifolia | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 |
| Pyrus pyrifolia | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 1 | 0 |
| Pyrus spinosa | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Pyrus pashia | 2 | 1 | 1 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| Total number of missing genes | 6 | 1 | 6 | 6 | 6 | 4 | 6 | 7 | 5 | 6 | 6 | 7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, J.; Li, L.; Gao, L.; Xu, J.; Yang, M. Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species. Int. J. Mol. Sci. 2018, 19, 3262. https://doi.org/10.3390/ijms19103262
Li Y, Zhang J, Li L, Gao L, Xu J, Yang M. Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species. International Journal of Molecular Sciences. 2018; 19(10):3262. https://doi.org/10.3390/ijms19103262
Chicago/Turabian StyleLi, Yongtan, Jun Zhang, Longfei Li, Lijuan Gao, Jintao Xu, and Minsheng Yang. 2018. "Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species" International Journal of Molecular Sciences 19, no. 10: 3262. https://doi.org/10.3390/ijms19103262
APA StyleLi, Y., Zhang, J., Li, L., Gao, L., Xu, J., & Yang, M. (2018). Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species. International Journal of Molecular Sciences, 19(10), 3262. https://doi.org/10.3390/ijms19103262