Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function

 ,
,  and
and
Abstract

1. Introduction
2. TMA and TMAO
2.1. Metabolism of TMA and TMAO
2.2. Genetic Determinants of TMAO
2.3. TMAO as a Plausible Contributor to Cardiovascular, Peripheral and Cerebrovascular Diseases
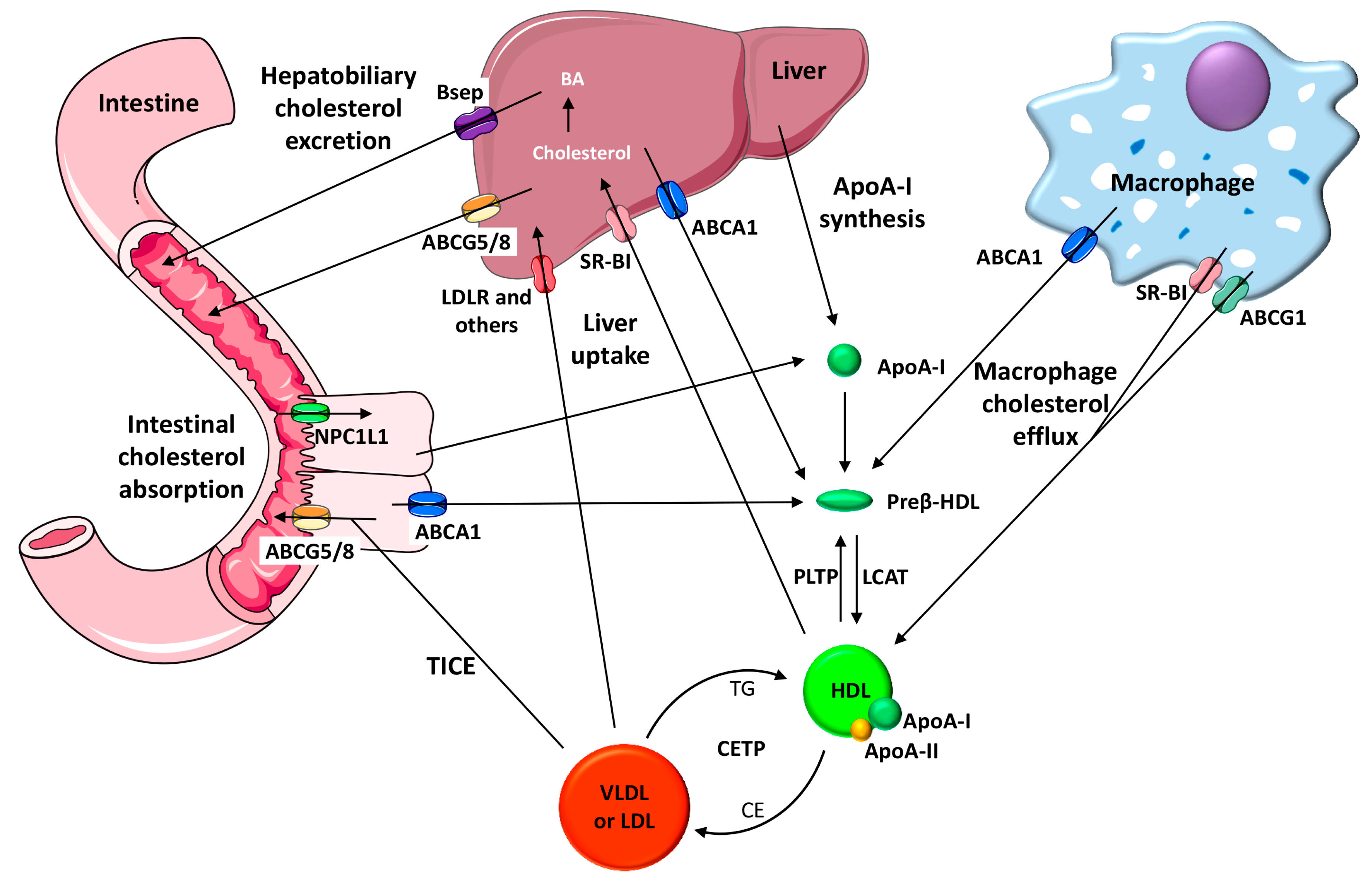
3. HDL Function and CVD
4. The Physiological Interaction between TMAO and HDL in the Context of Cardiometabolic Diseases
4.1. The Role of the Gut Microbiota in Lipid Metabolism and the Pathogenesis of CVD
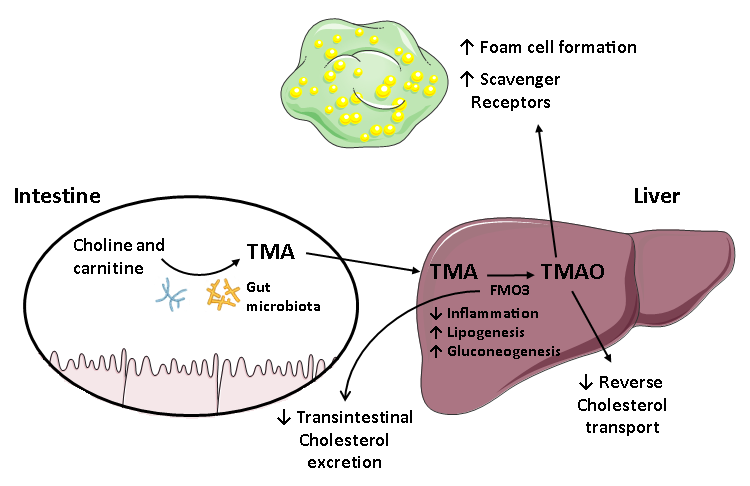
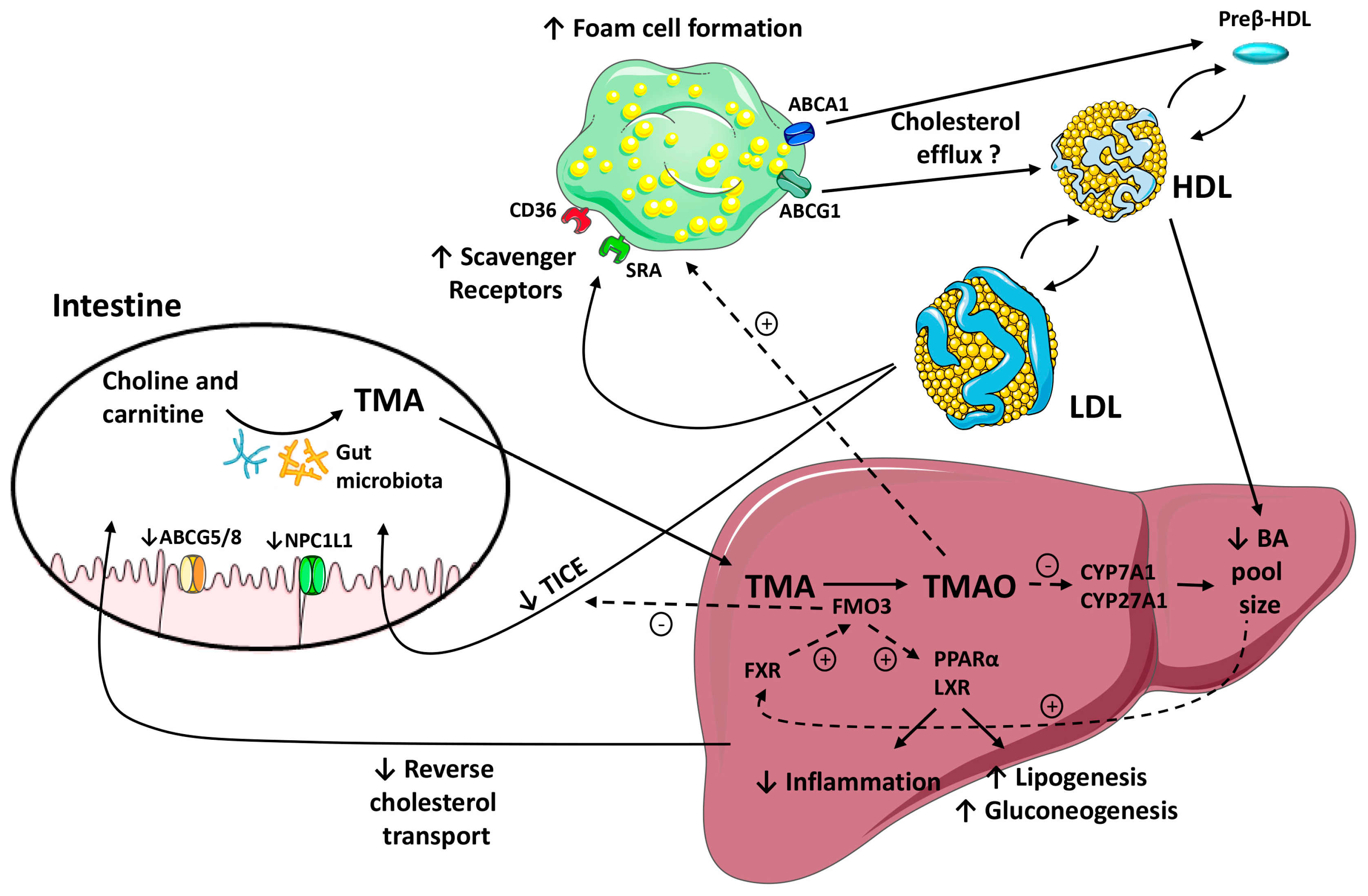
4.2. The Role of FMO3/TMAO and Nuclear Receptors on Enterohepatic Cholesterol and Bile Acid Metabolism
4.3. TMAO/FMO3 and HDL Atheroprotective Functions
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ASO | Antisense oligonucleotide |
| CD36 | Cluster of differentiation 36 |
| CETP | Cholesteryl ester transfer protein |
| CKD | Chronic kidney disease |
| CVD | Cardiovascular disease |
| FMO | Flavin monooxygenase |
| FXR | Farnesoid X receptor |
| GWAS | Genome-wide association studies |
| HDL | High-density lipoprotein |
| LCAT | Lecithin cholesterol acyltransferase |
| LDL | Low-density lipoprotein |
| LXR | Liver X receptor |
| MACE | Major adverse cardiovascular events |
| mRNA | Messenger ribonucleic acid |
| Npc1L1 | Niemann-Pick C1-Like 1 |
| OCT2 | Organic cation transporter 2 |
| PPAR | Peroxisome proliferator-activated receptor |
| SR-BI | Scavenger receptor class B type I |
| TICE | Transintestinal cholesterol excretion |
| TMA | Trimethylamine |
| TMAO | Trimethylamine-N-oxide |
| VLDL | Very low-density lipoprotein |
| γBB | γ-Butyrobetaine |
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics—2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Charach, G.; Grosskopf, I.; Rabinovich, A.; Shochat, M.; Weintraub, M.; Rabinovich, P. The association of bile acid excretion and atherosclerotic coronary artery disease. Therap. Adv. Gastroenterol. 2011, 4, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.; Tremaroli, V.; Nielsen, J.; Backhed, F. Assessing the human gut microbiota in metabolic diseases. Diabetes 2013, 62, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Kinross, J.M.; Darzi, A.W.; Nicholson, J.K. Gut microbiome-host interactions in health and disease. Genome Med. 2011, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef] [PubMed]
- Vinje, S.; Stroes, E.; Nieuwdorp, M.; Hazen, S.L. The gut microbiome as novel cardio-metabolic target: The time has come! Eur. Heart J. 2014, 35, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Levison, B.S.; Hazen, J.E.; Donahue, L.; Li, X.M.; Hazen, S.L. Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal. Biochem. 2014, 455, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, S.; Linseisen, J.; Allenspach, M.; von Eckardstein, A.; Muller, D. Plasma Concentrations of Trimethylamine-N-oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J. Nutr. 2016, 146, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.; Rohrmann, S.; Sookthai, D.; Johnson, T.; Katzke, V.; Kaaks, R.; von Eckardstein, A.; Muller, D. Intra-individual variation of plasma trimethylamine-N-oxide (TMAO), betaine and choline over 1 year. Clin. Chem. Lab. Med. 2017, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D.; et al. Gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of l-carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, C.T.; Janmohamed, A.; Smith, R.L.; Shephard, E.A.; Phillips, I.R. Missense mutation in flavin-containing mono-oxygenase 3 gene, FMO3, underlies fish-odour syndrome. Nat. Genet. 1997, 17, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Esposito, T.; Varriale, B.; D′Angelo, R.; Amato, A.; Sidoti, A. Regulation of flavin-containing mono-oxygenase (FMO3) gene expression by steroids in mice and humans. Horm. Mol. Biol. Clin. Investig. 2014, 20, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Brown, J.M. Emerging roles of flavin monooxygenase 3 in cholesterol metabolism and atherosclerosis. Curr. Opin. Lipidol. 2015, 26, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.; Merz, B.; Rist, M.J.; Ferrario, P.G.; Bub, A.; Kulling, S.E.; Watzl, B. Associations of current diet with plasma and urine TMAO in the KarMeN study: Direct and indirect contributions. Mol. Nutr. Food Res. 2017, 61, 1700363. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Awwad, H.M.; Kirsch, S.H.; Waldura, C.; Herrmann, W.; Graeber, S.; Geisel, J. Plasma trimethylamine-N-oxide following supplementation with vitamin D or D plus B vitamins. Mol. Nutr Food Res. 2017, 61, 1600358. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; von Eckardstein, A. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Zubair, N.; Conomos, M.P.; Xu, X.; Rohwer, J.E.; Krafft, C.E.; Lovejoy, J.C.; Magis, A.T. A Multi-omic Association Study of Trimethylamine N-Oxide. Cell Rep. 2018, 24, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Hai, X.; Landeras, V.; Dobre, M.A.; DeOreo, P.; Meyer, T.W.; Hostetter, T.H. Mechanism of Prominent Trimethylamine Oxide (TMAO) Accumulation in Hemodialysis Patients. PLoS ONE 2015, 10, e0143731. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, C.; Hallqvist, J.; Qureshi, A.R.; Barany, P.; Heimburger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-oxide Uptake and Efflux Transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio 2015, 6, e02481. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Craciun, S.; Balskus, E.P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Jameson, E.; Crosatti, M.; Schafer, H.; Rajakumar, K.; Bugg, T.D.; Chen, Y. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 4268–4273. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Mamer, O.A.; Akerman, B.R.; Choiniere, L.; Gaudet, D.; Hamet, P.; Treacy, E.P. In vivo variability of TMA oxidation is partially mediated by polymorphisms of the FMO3 gene. Mol. Genet. Metab. 2001, 73, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Turkanoglu Ozcelik, A.; Can Demirdogen, B.; Demirkaya, S.; Adali, O. Flavin containing monooxygenase 3 genetic polymorphisms Glu158Lys and Glu308Gly and their relation to ischemic stroke. Gene 2013, 521, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Bushueva, O.; Solodilova, M.; Churnosov, M.; Ivanov, V.; Polonikov, A. The Flavin-Containing Monooxygenase 3 Gene and Essential Hypertension: The Joint Effect of Polymorphism E158K and Cigarette Smoking on Disease Susceptibility. Int. J. Hypertens. 2014, 2014, 712169. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Cohen, C.; Newitt, R.; Shen, D.D.; Rettie, A.E.; Kestenbaum, B.R.; Himmelfarb, J.; Yeung, C.K. Association of FMO3 Variants and Trimethylamine N-Oxide Concentration, Disease Progression, and Mortality in CKD Patients. PLoS ONE 2016, 11, e0161074. [Google Scholar] [CrossRef] [PubMed]
- Hartiala, J.; Bennett, B.J.; Tang, W.H.; Wang, Z.; Stewart, A.F.; Roberts, R.; McPherson, R.; Lusis, A.J.; Hazen, S.L.; Allayee, H. Comparative genome-wide association studies in mice and humans for trimethylamine N-oxide, a proatherogenic metabolite of choline and l-carnitine. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Ho, J.E.; Chen, M.H.; Shen, D.; Cheng, S.; Larson, M.G.; Ghorbani, A.; Shi, X.; Helenius, I.T.; O′Donnell, C.J.; et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013, 18, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Awwad, H.M.; Rabagny, Y.; Graeber, S.; Herrmann, W.; Geisel, J. Plasma trimethylamine N-oxide concentration is associated with choline, phospholipids, and methyl metabolism. Am. J. Clin. Nutr. 2016, 103, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Aslibekyan, S.; Irvin, M.R.; Hidalgo, B.A.; Perry, R.T.; Jeyarajah, E.J.; Garcia, E.; Shalaurova, I.; Hopkins, P.N.; Province, M.A.; Tiwari, H.K.; et al. Genome- and CD4+ T-cell methylome-wide association study of circulating trimethylamine-N-oxide in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN). J. Nutr. Intermed. Metab. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Song, Y.; Daviglus, M.L.; Liu, K.; Van Horn, L.; Dyer, A.R.; Greenland, P. Accumulated evidence on fish consumption and coronary heart disease mortality: A meta-analysis of cohort studies. Circulation 2004, 109, 2705–2711. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; Lavie, C.J.; Fares, H.; Menezes, A.R.; O′Keefe, J.H. l-Carnitine in the secondary prevention of cardiovascular disease: Systematic review and meta-analysis. Mayo Clin. Proc. 2013, 88, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, W.H.; Buffa, J.A.; Fu, X.; Britt, E.B.; Koeth, R.A.; Levison, B.S.; Fan, Y.; Wu, Y.; Hazen, S.L. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 2014, 35, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Li, X.S.; Obeid, S.; Klingenberg, R.; Gencer, B.; Mach, F.; Raber, L.; Windecker, S.; Rodondi, N.; Nanchen, D.; Muller, O.; et al. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: A prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur. Heart J. 2017, 38, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Heaney, L.M.; Jones, D.J.; Ng, L.L. Trimethylamine N-oxide and Risk Stratification after Acute Myocardial Infarction. Clin. Chem. 2017, 63, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Wang, Z.; Fan, Y.; Wu, Y.; Hazen, S.L.; Tang, W.H. Trimethylamine N-Oxide and Mortality Risk in Patients With Peripheral Artery Disease. J. Am. Heart Assoc. 2016, 5, e004237. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liao, S.X.; He, Y.; Wang, S.; Xia, G.H.; Liu, F.T.; Zhu, J.J.; You, C.; Chen, Q.; Zhou, L.; et al. Dysbiosis of Gut Microbiota With Reduced Trimethylamine-N-Oxide Level in Patients With Large-Artery Atherosclerotic Stroke or Transient Ischemic Attack. J. Am. Heart Assoc. 2015, 4, e002699. [Google Scholar] [CrossRef] [PubMed]
- Skagen, K.; Troseid, M.; Ueland, T.; Holm, S.; Abbas, A.; Gregersen, I.; Kummen, M.; Bjerkeli, V.; Reier-Nilsen, F.; Russell, D.; et al. The Carnitine-butyrobetaine-trimethylamine-N-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis 2016, 247, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Jazwiec, R.; Dadlez, M.; Drapala, A.; Sikora, M.; Skrzypecki, J. Trimethylamine-N-oxide: A carnitine-derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can. J. Cardiol. 2014, 30, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. l-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE−/− transgenic mice expressing CETP. Atherosclerosis 2016, 244, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Lindskog Jonsson, A.; Caesar, R.; Akrami, R.; Reinhardt, C.; Fak Hallenius, F.; Boren, J.; Backhed, F. Impact of Gut Microbiota and Diet on the Development of Atherosclerosis in Apoe−/− Mice. Arterioscler Thromb. Vasc. Biol. 2018, 38, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J. Molecular regulation of HDL metabolism and function: Implications for novel therapies. J. Clin. Invest. 2006, 116, 3090–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rueckert, M.; Blanco-Vaca, F.; Kovanen, P.T.; Escola-Gil, J.C. The role of the gut in reverse cholesterol transport—Focus on the enterocyte. Prog. Lipid Res. 2013, 52, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Rader, D.J. Macrophage reverse cholesterol transport: Key to the regression of atherosclerosis? Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rueckert, M.; Escola-Gil, J.C.; Kovanen, P.T. HDL functionality in reverse cholesterol transport—Challenges in translating data emerging from mouse models to human disease. Biochim. Biophys. Acta 2016, 1861, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Escola-Gil, J.C.; Rotllan, N.; Julve, J.; Blanco-Vaca, F. In vivo macrophage-specific RCT and antioxidant and antiinflammatory HDL activity measurements: New tools for predicting HDL atheroprotection. Atherosclerosis 2009, 206, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.C.; Honeyman, M.C.; Morahan, G.; Wentworth, J.M.; Elkassaby, S.; Colman, P.G.; Fourlanos, S. Type 1 diabetes: Lessons for other autoimmune diseases? J. Autoimmun. 2008, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, V.R.; Hezaveh, R.; Reigstad, C.S.; Gopalacharyulu, P.; Yetukuri, L.; Islam, S.; Felin, J.; Perkins, R.; Boren, J.; Oresic, M.; et al. The gut microbiota modulates host energy and lipid metabolism in mice. J. Lipid Res. 2010, 51, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Mistry, R.H.; Verkade, H.J.; Tietge, U.J. Reverse Cholesterol Transport Is Increased in Germ-Free Mice-Brief Report. Arterioscler Thromb. Vasc. Biol. 2017, 37, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.C.; Buffa, J.A.; Org, E.; Wang, Z.; Levison, B.S.; Zhu, W.; Wagner, M.A.; Bennett, B.J.; Li, L.; DiDonato, J.A.; et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015, 290, 5647–5660. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.E.; Caudill, M.A. Trimethylamine-N-Oxide: Friend, Foe, or Simply Caught in the Cross-Fire? Trends Endocrinol. Metab. 2017, 28, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Charach, G.; Rabinovich, A.; Argov, O.; Weintraub, M.; Rabinovich, P. The role of bile Acid excretion in atherosclerotic coronary artery disease. Int. J. Vasc. Med. 2012, 2012, 949672. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Rolo, A.P.; Palmeira, C.M. Hepatic FXR: Key regulator of whole-body energy metabolism. Trends Endocrinol. Metab. 2011, 22, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Temel, R.E.; Brown, J.M. Biliary and nonbiliary contributions to reverse cholesterol transport. Curr. Opin. Lipidol. 2012, 23, 85–90. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, J.N.; van Dijk, T.H.; Vrins, C.L.; van Meer, H.; Havinga, R.; Bijsterveld, K.; Tietge, U.J.; Groen, A.K.; Kuipers, F. Activation of the liver X receptor stimulates trans-intestinal excretion of plasma cholesterol. J. Biol. Chem. 2009, 284, 19211–19219. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.M.; Wang, Z.; Lee, R.; Meng, Y.; Che, N.; Charugundla, S.; Qi, H.; Wu, J.; Pan, C.; Brown, J.M.; et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 2015, 56, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Shih, D.M.; Warrier, M.; Helsley, R.N.; Burrows, A.; Ferguson, D.; Brown, A.L.; Gromovsky, A.D.; Heine, M.; Chatterjee, A.; et al. The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep. 2017, 19, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ling, A.V.; Manthena, P.V.; Gearing, M.E.; Graham, M.J.; Crooke, R.M.; Croce, K.J.; Esquejo, R.M.; Clish, C.B.; Vicent, D.; et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015, 6, 6498. [Google Scholar] [CrossRef] [PubMed]
- Trenteseaux, C.; Gaston, A.T.; Aguesse, A.; Poupeau, G.; de Coppet, P.; Andriantsitohaina, R.; Laschet, J.; Amarger, V.; Krempf, M.; Nobecourt-Dupuy, E.; et al. Perinatal Hypercholesterolemia Exacerbates Atherosclerosis Lesions in Offspring by Altering Metabolism of Trimethylamine-N-Oxide and Bile Acids. Arterioscler Thromb. Vasc. Biol. 2017, 37, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Gholamhoseyniannajar, A.; Yaghoobi, M.M.; Jahani, Y.; Vahabzadeh, Z. Expression levels of heat shock protein 60 and glucose-regulated protein 78 in response to trimethylamine-N-oxide treatment in murine macrophage J774A.1 cell line. Cell. Mol. Biol. (Noisy-le-Grand) 2015, 61, 94–100. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Methods and Species | Study Findings | References |
|---|---|---|
| Metabolomics in humans | TMAO, carnitine, choline and phosphatidylcholine plasma levels correlated with CVD | [14,48] |
| GWAS in mice | The Slc30a7 gene was related to TMAO metabolism | [44] |
| GWAS in humans | SLC30A7 locus did not reach the genome-wide significance | [45] |
| Epigenome-wide study of DNA methylation in humans | No evidence of significant relationship between methylation markers and circulating TMAO levels | [47] |
| A multi-omic association study of TMAO in humans | Diet- and disease-associated metabolites were significantly associated with TMAO Proteins linked to CVD and kidney disease were correlated with TMAO | [30] |
| Metagenome analysis in humans | Gene-targeted sequencing allowed the quantification and characterization TMA-producing species | [37] |
| Metagenome analysis in humans | Gut microbiome may explain part of the population variation in plasma blood lipids | [13] |
| Global microarray expression analyses in mice | Knocking down FMO3 downregulated 136 PPARα target genes | [89] |
| Global microarray expression analyses in mice | FMO3 expression was the unique downregulated gene in mouse models of enhanced TICE | [59] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canyelles, M.; Tondo, M.; Cedó, L.; Farràs, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 2018, 19, 3228. https://doi.org/10.3390/ijms19103228
Canyelles M, Tondo M, Cedó L, Farràs M, Escolà-Gil JC, Blanco-Vaca F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. International Journal of Molecular Sciences. 2018; 19(10):3228. https://doi.org/10.3390/ijms19103228
Chicago/Turabian StyleCanyelles, Marina, Mireia Tondo, Lídia Cedó, Marta Farràs, Joan Carles Escolà-Gil, and Francisco Blanco-Vaca. 2018. "Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function" International Journal of Molecular Sciences 19, no. 10: 3228. https://doi.org/10.3390/ijms19103228
APA StyleCanyelles, M., Tondo, M., Cedó, L., Farràs, M., Escolà-Gil, J. C., & Blanco-Vaca, F. (2018). Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. International Journal of Molecular Sciences, 19(10), 3228. https://doi.org/10.3390/ijms19103228