iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line




Abstract
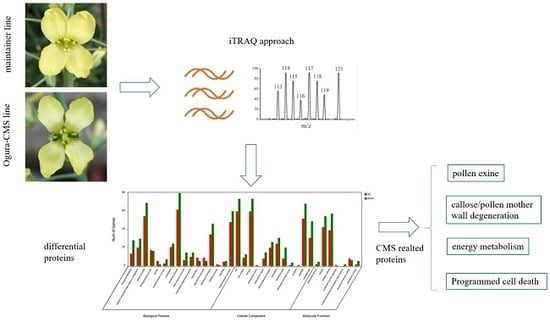
1. Introduction
2. Results
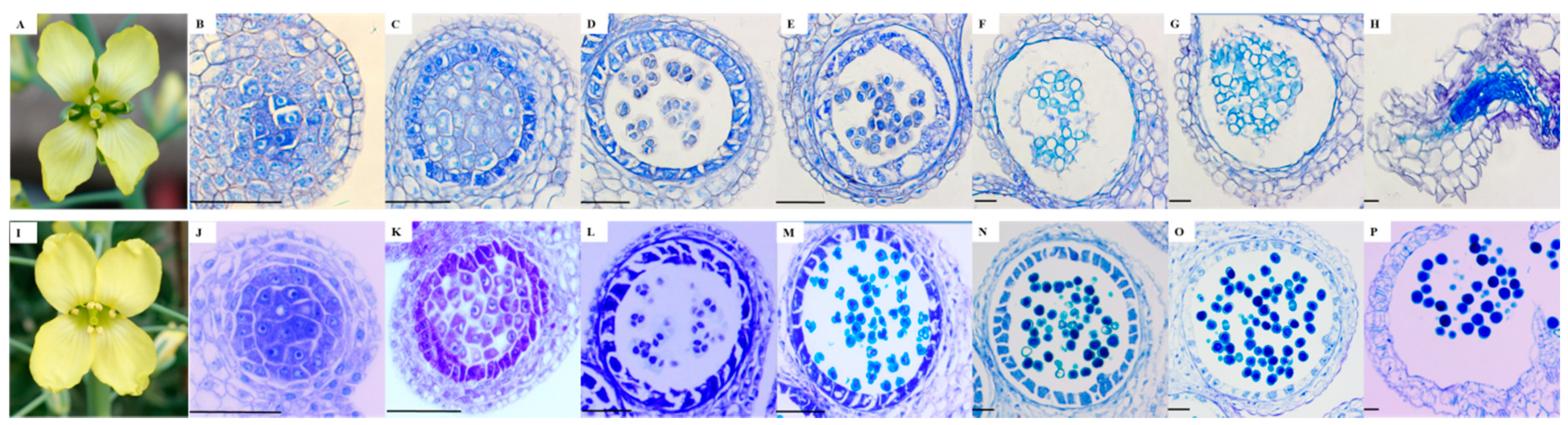
2.1. Morphological and Microscopic Examination
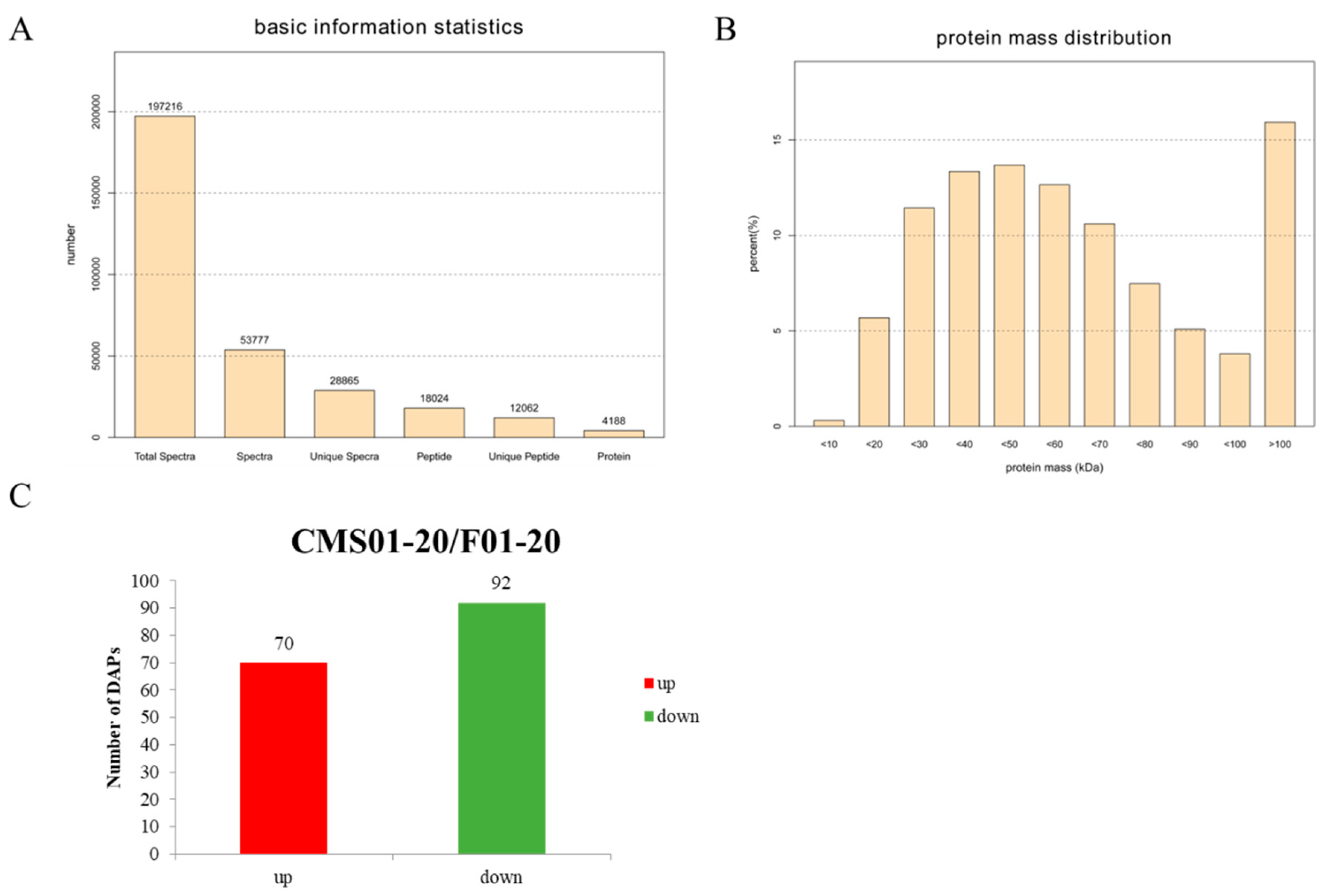
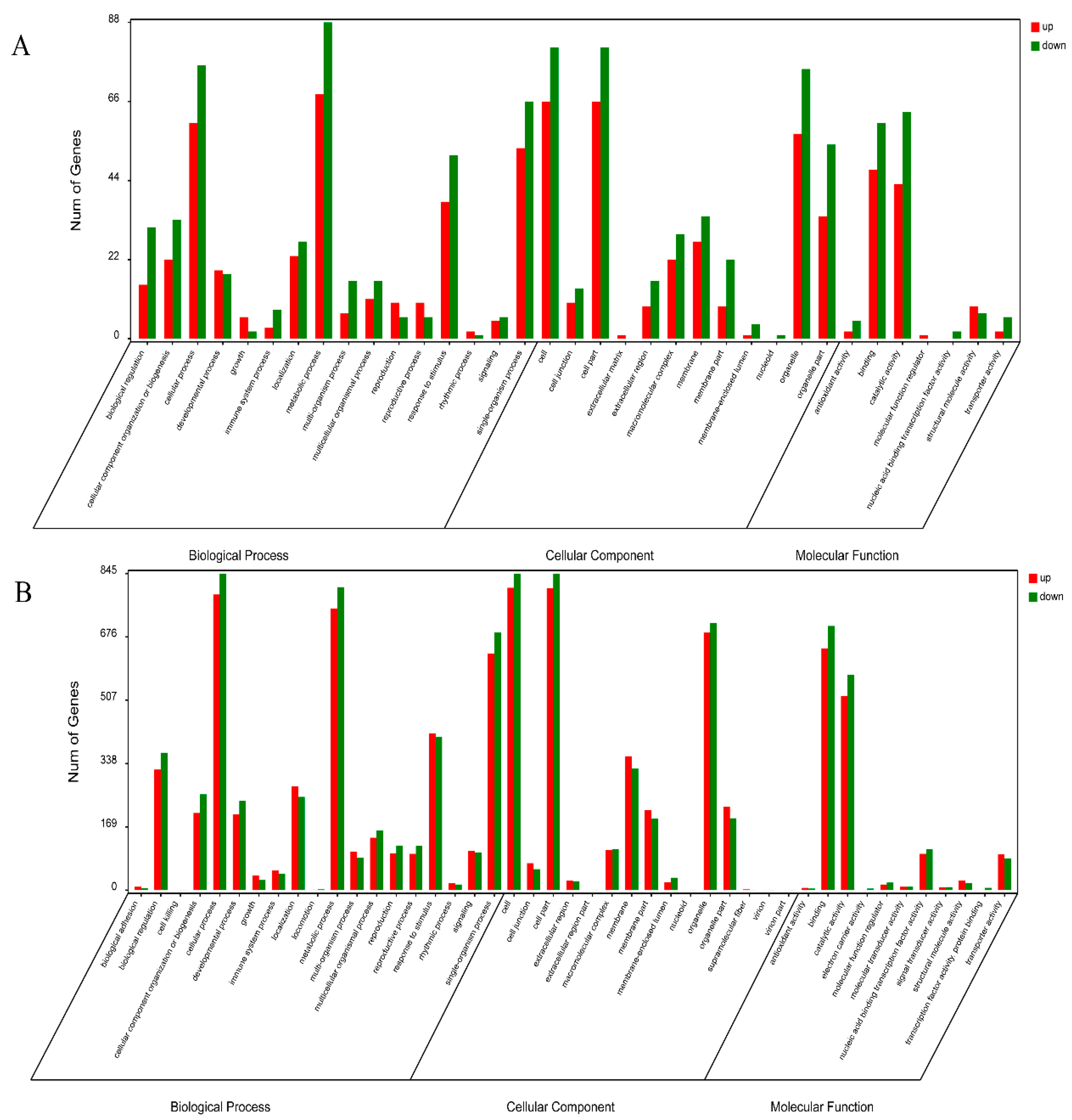
2.2. Overview of the Protein Species Identified Using iTRAQ Data
2.3. Overview of the DAPs between CMS01-20 and F01-20
2.4. DAPs Involved in Oxidative Phosphorylation and TCA Cycle
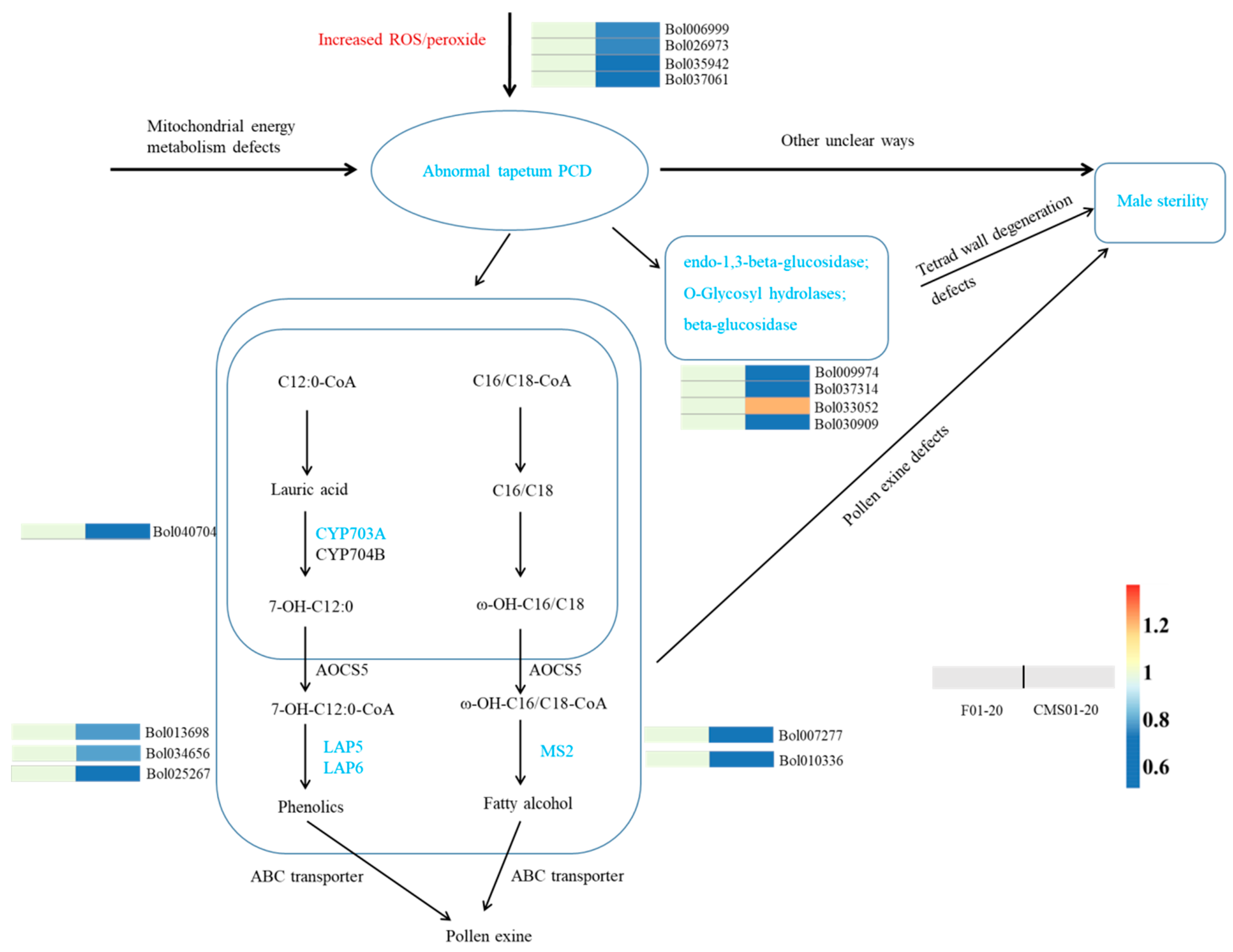
2.5. Other Ogura-CMS Related DAPs and Pathways
2.6. Joint Proteome–Transcriptome Analysis
3. Materials and Methods
3.1. Plant Materials and Sample Preparation
3.2. Microscopy
3.3. iTRAQ Analysis and Protein Species Annotation
3.4. RNA-Seq Analysis and Conjoint Analysis with Proteome Data
3.5. Quantitative RT-PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fang, Z.Y.; Liu, Y.M.; Yang, L.M.; Wang, X.W.; Zhuang, M.; Zhang, Y.Y.; Sun, P.T. A survey of research in genetic breedings of cabbage in China. Acta Hort. Sin. 2002, 29, S657–S663. [Google Scholar]
- Prakash, C.; Verma, T. Heterosis in cytoplasmic male sterile lines of cabbage. Crucif. Newslett. 2004, 25, 49–50. [Google Scholar]
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Y.; Sun, P.T.; Liu, Y.M.; Yang, L.M.; Wang, X.W.; Hou, A.F.; Bian, C.S. A male sterile line with dominant gene (MS) in cabbage (Brassica oleracea var. capitata) and its utilization for hybrid seed production. Euphytica 1997, 97, 265–268. [Google Scholar]
- Fang, Z.Y.; Sun, P.T.; Liu, Y.M. Investigation of different types of male sterility and application of dominant male sterility in cabbage. China Veg. 2001, 1, 6–10. [Google Scholar]
- Ogura, H. Studies of a new male-sterility in Japanese radish, with special reference to the utilization of this sterility towards the practical raising of hybrid seeds. Mem. Fac. Agric. Kagoshima Univ. 1968, 6, 39–78. [Google Scholar]
- Bannerrot, H.; Boulidard, L.; Couderon, Y.; Temple, J. Transfer of cytoplasmic male sterility from Raphanus sativus to Brassica oleracea. Proc. Eucarpia Meet. Crucif. 1974, 25, 52–54. [Google Scholar]
- Sigareva, M.; Earle, E. Direct transfer of a cold-tolerant Ogura male-sterile cytoplasm into cabbage (Brassica oleracea ssp. capitata) via protoplast fusion. Theor. Appl. Genet. 1997, 94, 213–220. [Google Scholar] [CrossRef]
- Walters, W.T.; Mutschler, A.M.; Earle, D.E. Protoplast fusion-derived Ogura male-sterile cauliflower with cold tolerance. Plant Cell Rep. 1992, 10, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, G.; Primard, C.; Vedel, F.; Chetrit, P.; Remy, R.; Renard, M. Intergeneric cytoplasmic hybridization in Cruciferae by protoplast fusion. Mol. Genet. Genomics 1983, 191, 244–250. [Google Scholar] [CrossRef]
- Wang, Q.B.; Zhang, Y.Y.; Fang, Z.Y.; Liu, Y.M.; Yang, L.M.; Zhuang, M. Chloroplast and mitochondrial SSR help to distinguish allo-cytoplasmic male sterile types in cabbage (Brassica oleracea L. var. capitata). Mol. Breed. 2012, 30, 709–716. [Google Scholar] [CrossRef]
- Chase, C.D. Cytoplasmic male sterility: A window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, W.; Huang, Q.; Qin, X.; Yu, C.; Wang, L.; Li, S.; Zhu, R.; Zhu, Y. Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 2014, 19, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, R.; Saxena, R.K.; Davila, J.; Shah, T.; Chen, W.; Xiao, Y.; Fan, G.; Saxena, K.B.; Alverson, A.J.; Spillane, C.; et al. Cytoplasmic male sterility-associated chimeric open reading frames identified by mitochondrial genome sequencing of four Cajanus genotypes. DNA Res. 2013, 20, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Glimelius, K. Cytoplasmic male-sterility and nuclear encoded fertility restoration. Plant Mitochond. 2011, 1, 469–491. [Google Scholar]
- Woodson, J.D.; Chory, J. Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 2008, 9, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Duroc, Y.; Hiard, S.; Vrielynck, N.; Ragu, S.; Budar, F. The Ogura sterility-inducing protein forms a large complex without interfering with the oxidative phosphorylation components in rapeseed mitochondria. Plant Mol. Biol. 2009, 70, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, S.; Budar, F.; Lancelin, D.; Small, I.; Defrance, M.C.; Pelletier, G. Sequence and transcript analysis of the Nco2.5 Ogura specific fragment correlated with cytoplasmic male sterility in Brassica cybrids. Mol. Gen. Genet. 1992, 235, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Grelon, M.; Budar, F.; Bonhomme, S.; Pelletier, G. Ogura cytoplasmic male-sterility (CMS)-associated orf138 is translated into a mitochondrial membrane polypeptide in male-sterile Brassica cybrids. Mol. Gen. Genet. 1994, 243, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, S.; Makaroff, C.A. Characterization of the radish mitochondrial orfB locus: Possible relationship with male sterility in Ogura radish. Curr. Genet. 1993, 24, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Duroc, Y.; Gaillard, C.; Hiard, S.; Defrance, M.C.; Pelletier, G.; Budar, F. Biochemical and functional characterization of ORF138, a mitochondrial protein responsible for Ogura cytoplasmic male sterility in Brassiceae. Biochimie 2005, 87, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Kim, W.K.; Lim, Y.P.; Kim, Y.K.; Hur, Y. Ogura-CMS in Chinese cabbage (Brassica rapa ssp. pekinensis) causes delayed expression of many nuclear genes. Plant Sci. 2013, 199, 7–17. [Google Scholar] [PubMed]
- Wang, S.; Wang, C.; Zhang, X.X.; Chen, X.; Liu, J.J.; Jia, X.F.; Jia, S.Q. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Zhang, L.; Liu, Y.; Li, Z.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. Normal and abortive buds transcriptomic profiling of broccoli ogu cytoplasmic male sterile line and its maintainer. Int. J. Mol. Sci. 2018, 19, 2501. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Sun, C.; Li, H.; Hu, S.; Lei, L.; Kang, J. Integrated analysis of transcriptome and proteome changes related to the Ogura cytoplasmic male sterility in cabbage. PLoS ONE 2018, 13, e0193462. [Google Scholar] [CrossRef] [PubMed]
- Mihr, C.; Baumgärtner, M.; Dieterich, J.H.; Schmitz, U.K.; Braun, H.P. Proteomic approach for investigation of cytoplasmic male sterility (CMS) in Brassica. J. Plant Physiol. 2001, 158, 787–794. [Google Scholar] [CrossRef]
- Sheoran, I.S.; Sawhney, V.K. Proteome analysis of the normal and Ogura (ogu) CMS anthers of Brassica napus to identify proteins associated with male sterility. Botany 2010, 88, 217–230. [Google Scholar] [CrossRef]
- Liu, X.; Han, F.; Kong, C.; Fang, Z.; Yang, L.; Zhang, Y.; Zhuang, M.; Liu, Y.; Li, Z.; Lv, H. Rapid introgression of the Fusarium wilt resistance gene into an elite cabbage line through the combined application of a microspore culture, genome background analysis, and disease resistance-specific marker assisted foreground selection. Front. Plant Sci. 2017, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- González-Melendi, P.; Uyttewaal, M.; Morcillo, C.N.; Mora, J.R.H.; Fajardo, S.; Budar, F.; Lucas, M.M. A light and electron microscopy analysis of the events leading to male sterility in Ogu-INRA CMS of rapeseed (Brassica napus). J. Exp. Bot. 2008, 59, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Tratt, J. Identifying the Wall-Degrading Enzymes Responsible for Microspore Release from the Pollen Tetrad. Ph.D. Thesis, University of Bath, Bath, UK, 2016. [Google Scholar]
- Balk, J.; Leaver, C.J. The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochrome c release. Plant Cell 2001, 13, 1803–1818. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wan, C.; Kong, J.; Zhang, Z.; Li, Y.; Zhu, Y. Programmed cell death during microgenesis in a Honglian CMS line of rice is correlated with oxidative stress in mitochondria. Funct. Plant Biol. 2004, 31, 369–376. [Google Scholar] [CrossRef]
- Luo, D.P.; Xu, H.; Liu, Z.L.; Guo, J.X.; Li, H.Y.; Chen, L.T.; Fang, C.; Zhang, Q.Y.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Moon, S.; Lee, Y.S.; Zhu, L.; Liang, W.; Zhang, D.; Jung, K.H.; An, G. Defective Tapetum Cell Death 1 (DTC1) regulates ROS levels by binding to metallothionein during tapetum degeneration. Plant Physiol. 2016, 170, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Zhu, L.; Tu, L.; Deng, F.; Yuan, D.; Zhang, X. Cotton GhCKI disrupts normal male reproduction by delaying tapetum programmed cell death via inactivating starch synthase. Plant J. 2013, 75, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, X.; Han, S.; He, T.; Zhang, H.; Yang, L.; Yang, S.; Gai, J. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J. Proteomics 2016, 138, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. J. Proteomics 2018, 179, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, C.; Wei, H.; Song, M.; Meng, Y.; Ma, J.; Fan, S.; Yu, S. iTRAQ-facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). J. Proteomics 2015, 126, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zeng, Y.; An, J.; Ye, J.; Xu, Q.; Deng, X. An integrative analysis of transcriptome and proteome provides new insights into carotenoid biosynthesis and regulation in sweet orange fruits. J. Proteomics 2012, 75, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- Lou, P.; Kang, J.; Zhang, G.; Bonnema, G.; Fang, Z.; Wang, X. Transcript profiling of a dominant male sterile mutant (Ms-cd1) in cabbage during flower bud development. Plant Sci. 2007, 172, 111–119. [Google Scholar] [CrossRef]
- Chu, P.; Yan, G.; Yang, Q.; Zhai, L.; Zhang, C.; Zhang, F.; Guan, R. iTRAQ-based quantitative proteomics analysis of Brassica napus, leaves reveals pathways associated with chlorophyll deficiency. J. Proteomics 2015, 113, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.B.; Fang, Y.N.; Pan, Z.Y.; Sun, L.; Deng, X.X.; Grosser, J.W.; Guo, W.W. iTRAQ-based quantitative proteomics analysis revealed alterations of carbohydrate metabolism pathways and mitochondrial proteins in a male sterile Cybrid pummelo. J. Proteome Res. 2014, 13, 2998–3015. [Google Scholar] [CrossRef] [PubMed]
- Sabar, M.; Gagliardi, D.; Balk, J.; Leaver, C.J. ORFB is a subunit of F1F(O)-ATP synthase: Insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 2003, 4, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Edqvist, J.; Farbos, I.; Glimelius, K. Male-sterile tobacco displays abnormal mitochondrial atp1 transcript accumulation and reduced floral ATP/ADP ratio. Plant Mol. Biol. 2000, 42, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Ducos, E.; Touzet, P.; Boutry, M. The male sterile G, cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001, 26, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, S.; Yi, P.; Wan, C.; Chen, Z.; Zhu, Y. A Honglian CMS line of rice displays aberrant F0 of F0 F1-ATPase. Plant Cell Rep. 2007, 26, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gao, F.; Ji, Y.; Liu, Y.; Dan, Z.; Yang, P.; Zhu, Y.; Li, S. ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, T.; Toriyama, K. Genetic regulation of sporopollenin synthesis and pollen exine development. Annu. Rev. Plant Biol. 2011, 62, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 1995, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Dobritsa, A.A.; Lei, Z.; Nishikawa, S.I.; Urbanczyk-Wochniak, E.; Huhman, D.V.; Preuss, D.; Sumner, L.W. LAP5 and LAP6 encode anther-specific proteins with similarity to chalcone synthase essential for pollen exine development in Arabidopsis thaliana. Plant Physiol. 2010, 110, 157446. [Google Scholar] [CrossRef]
- Morant, M.; Jørgensen, K.; Schaller, H.; Pinot, F.; Møller, B.L.; Werck-Reichhart, D.; Bak, S. CYP703 is an ancient cytochrome P450 in land plants catalyzing in-chain hydroxylation of lauric acid to provide building blocks for sporopollenin synthesis in pollen. Plant Cell 2007, 19, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.G.M.; Hodge, R.; Kalantidis, K.; Florack, D.; Wilson, Z.A.; Mulligan, B.J.; Stiekema, W.J.; Scott, R.; Pereira, A. The Arabidopsis MALE STERILITY 2 protein shares similarity with reductases in elongation/condensation complexes. Plant J. 1997, 12, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, X.H.; Zhang, K.; Shi, J.; De Oliveira, S.; Schreiber, L.; Shanklin, J.; Zhang, D. Male Sterile2 encodes a plastid-localized fatty acyl carrier protein reductase required for pollen exine development in Arabidopsis. Plant Physiol. 2011, 157, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.; Chater, C.C.; Kamisugi, Y.; Cuming, A.C.; Wellman, C.H.; Beerling, D.J.; Fleming, A.J. Conservation of Male Sterility 2 function during spore and pollen wall development supports an evolutionarily early recruitment of a core component in the sporopollenin biosynthetic pathway. New Phytol. 2015, 205, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Piffanelli, P.; Ross, J.H.E.; Murphy, D.J. Biogenesis and function of the lipidic structures of pollen grains. Sexual Plant Reprod. 1998, 11, 65–80. [Google Scholar] [CrossRef]
- Preuss, D.; Rhee, S.Y.; Davis, R.W. Tetrad analysis possible in Arabidopsis with mutation of the QUARTET (QRT) genes. Science 1994, 264, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Osborne, E.; Poindexter, P.D.; Somerville, C.R. Microspore separation in the quartet 3 mutants of Arabidopsis is impaired by a defect in a developmentally regulated polygalacturonase required for pollen mother cell wall degradation. Plant Physiol. 2003, 133, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zhang, X.; Zhu, Y.; Zhu, W.; Xie, H.; Wang, X. Metabolism of reactive oxygen species in cotton cytoplasmic male sterility and its restoration. Plant Cell Rep. 2007, 26, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Gechev, T.S.; Hille, J. Hydrogen peroxide as a signal controlling plant programmed cell death. J. Cell Biol. 2005, 168, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Von Malek, B.; van der Graaff, E.; Schneitz, K.; Keller, B. The Arabidopsis male-sterile mutant dde2-2 is defective in the ALLENE OXIDE SYNTHASE gene encoding one of the key enzymes of the jasmonic acid biosynthesis pathway. Planta 2002, 216, 187–192. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Description | Up/Down in CMS Line | |
|---|---|---|---|
| Oxidative phosphorylation | Bol015119 | NADH-ubiquinone oxidoreductase B18 subunit | down |
| Bol009135 | ATP synthase subunit d, mitochondrial-like | down | |
| Bol025034 | ATP synthase subunit d, mitochondrial-like | down | |
| Bol017288 | mitochondrial ATP synthase 6 KD subunit | down | |
| Bol025922 | mitochondrial ATP synthase 6 KD subunit | down | |
| Bol015469 | ATP synthase 6 kDa subunit | down | |
| Bol012326 | cytochrome c | down | |
| Bol010838 | cytochrome c oxidase subunit Vc | down | |
| TCA cycle | Bol022522 | pyruvate dehydrogenase E1 component subunit beta-2 | up |
| Bol008536 | pyruvate dehydrogenase E1 component subunit beta-2 | up | |
| Bol008657 | 2-oxoglutarate dehydrogenase | up | |
| Bol029048 | aconitate hydratase 1 | up | |
| Bol029509 | citrate synthase 4 | up | |
| pollen wall | Bol013698 | LAP5; Chalcone and stilbene synthase family protein | down |
| Bol025267 | LAP6; Chalcone and stilbene synthase family protein | down | |
| Bol007277 | MS2; fatty acyl-CoA reductase | down | |
| Bol034656 | LAP5; Chalcone and stilbene synthase family protein | down | |
| Bol040704 | cytochrome P450 703A2 | down | |
| Bol010336 | MS2; fatty acyl-CoA reductase | down | |
| tetrad wall | Bol009974 | probable glucan endo-1,3-beta-glucosidase A6 | down |
| Bol037314 | O-Glycosyl hydrolases family 17 protein; | down | |
| Bol033052 | beta-d-xylosidase 1 | up | |
| Bol030909 | beta-glucosidase 43 isoform X2 | down | |
| PCD | Bol006999 | catalase-3 | down |
| Bol026973 | catalase-3 | down | |
| Bol035942 | allene oxide synthase | down | |
| Bol037061 | peroxisomal | down | |
| Bol005496 | stromal ascorbate peroxidase | down | |
| Bol004624 | glutathione S-transferase F9 | down | |
| Bol033376 | glutathione S-transferase F9 | down |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, F.; Zhang, X.; Yang, L.; Zhuang, M.; Zhang, Y.; Li, Z.; Fang, Z.; Lv, H. iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line. Int. J. Mol. Sci. 2018, 19, 3180. https://doi.org/10.3390/ijms19103180
Han F, Zhang X, Yang L, Zhuang M, Zhang Y, Li Z, Fang Z, Lv H. iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line. International Journal of Molecular Sciences. 2018; 19(10):3180. https://doi.org/10.3390/ijms19103180
Chicago/Turabian StyleHan, Fengqing, Xiaoli Zhang, Limei Yang, Mu Zhuang, Yangyong Zhang, Zhansheng Li, Zhiyuan Fang, and Honghao Lv. 2018. "iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line" International Journal of Molecular Sciences 19, no. 10: 3180. https://doi.org/10.3390/ijms19103180
APA StyleHan, F., Zhang, X., Yang, L., Zhuang, M., Zhang, Y., Li, Z., Fang, Z., & Lv, H. (2018). iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line. International Journal of Molecular Sciences, 19(10), 3180. https://doi.org/10.3390/ijms19103180