HSCVFNT: Inference of Time-Delayed Gene Regulatory Network Based on Complex-Valued Flexible Neural Tree Model

Abstract
1. Introduction
2. Results
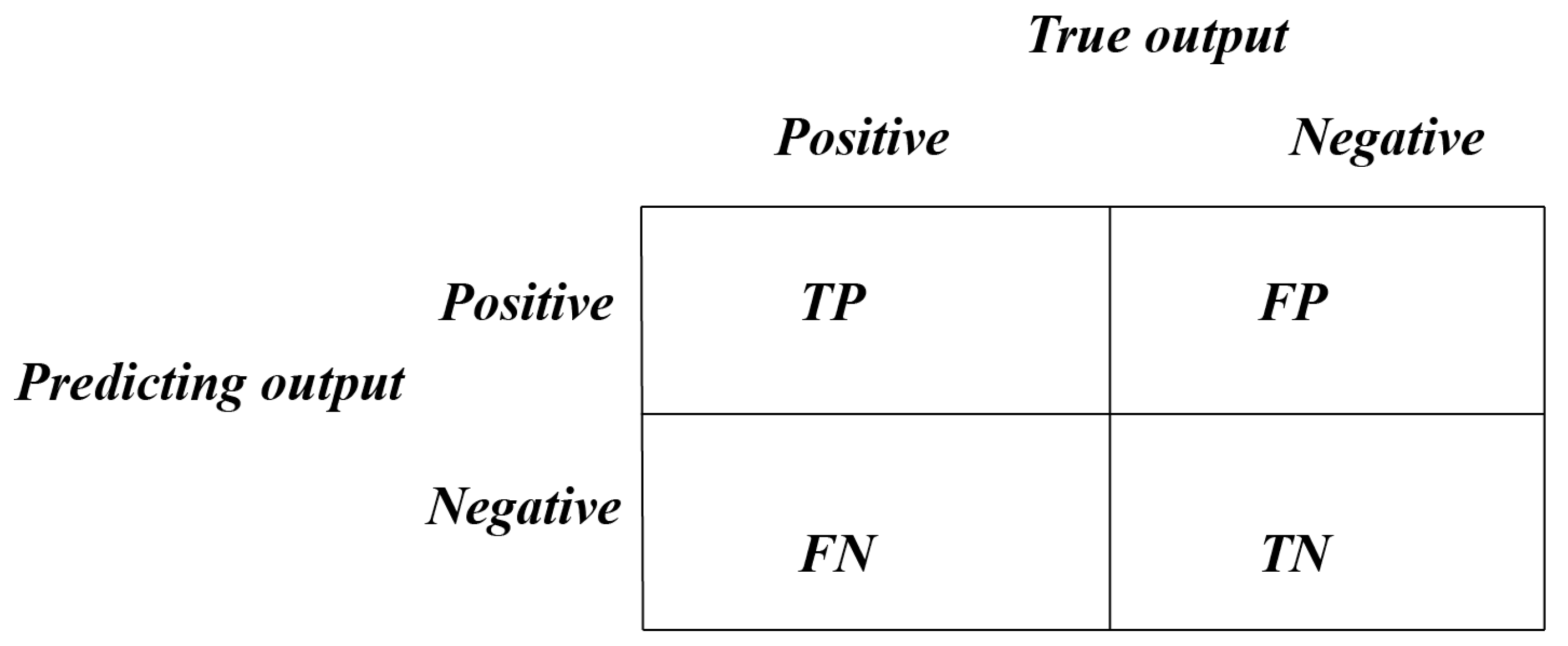
2.1. Datasets and Evaluation Metrics
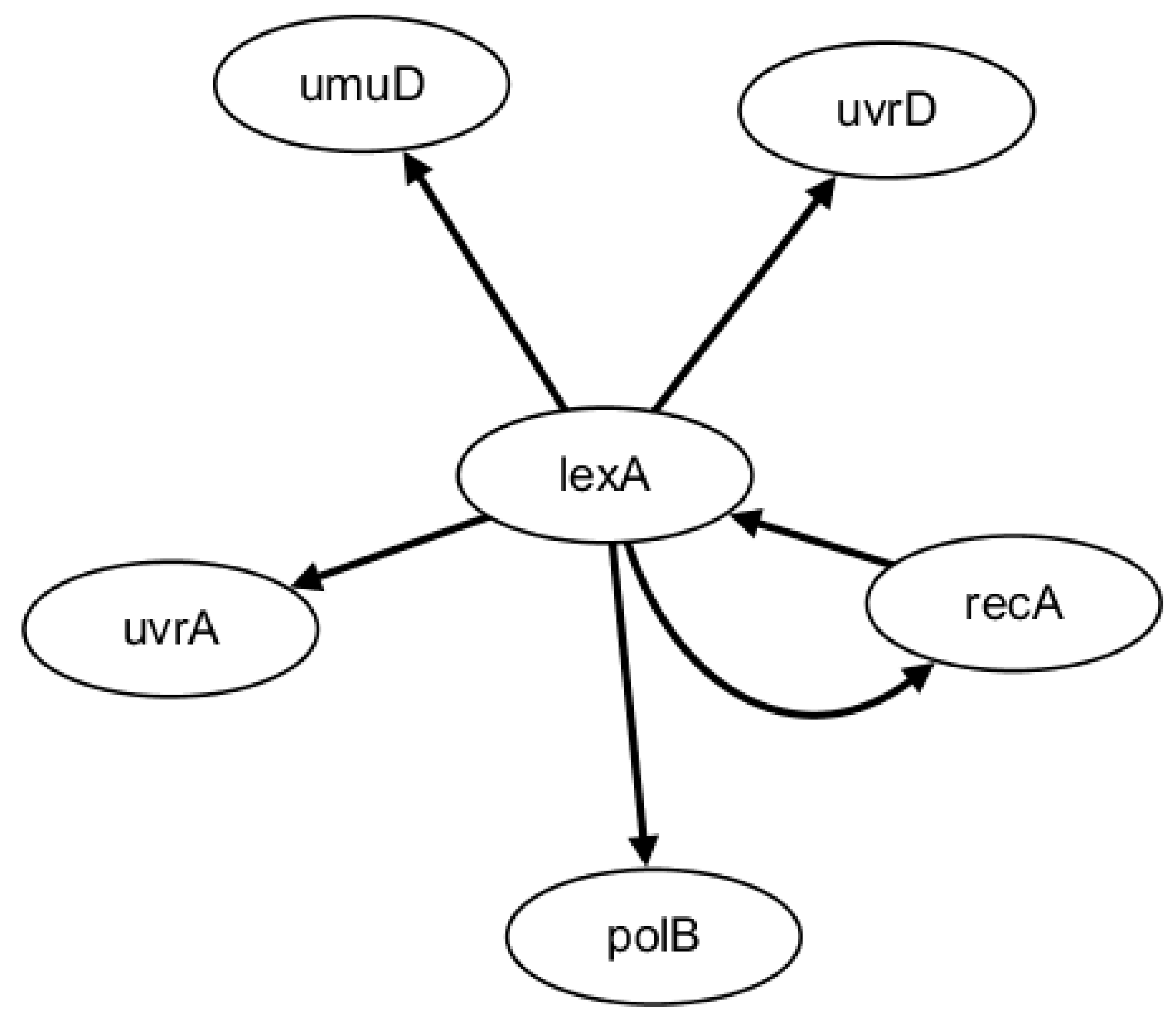
2.2. Network Construction of Real Data of E. coli SOSDNA Repair Network
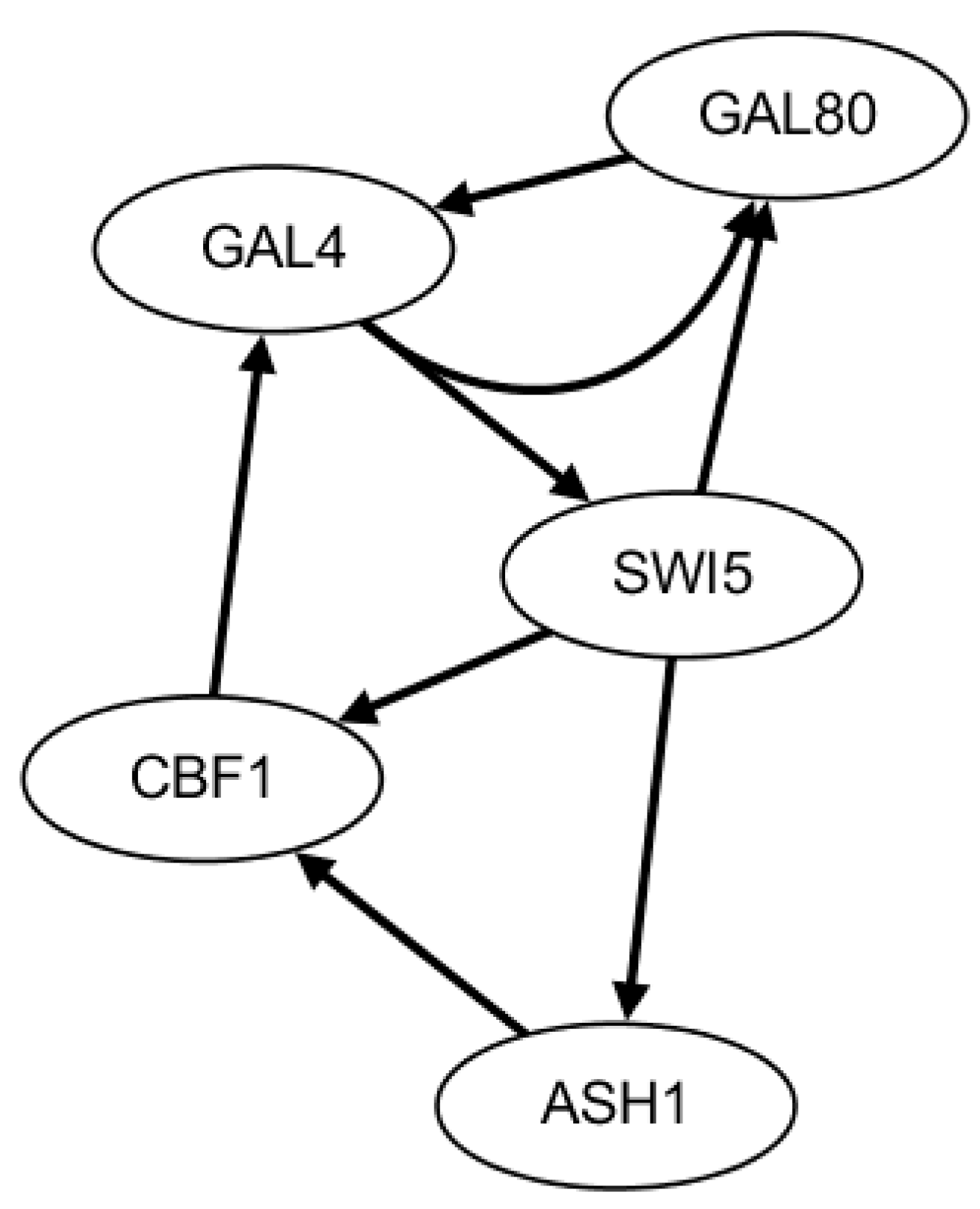
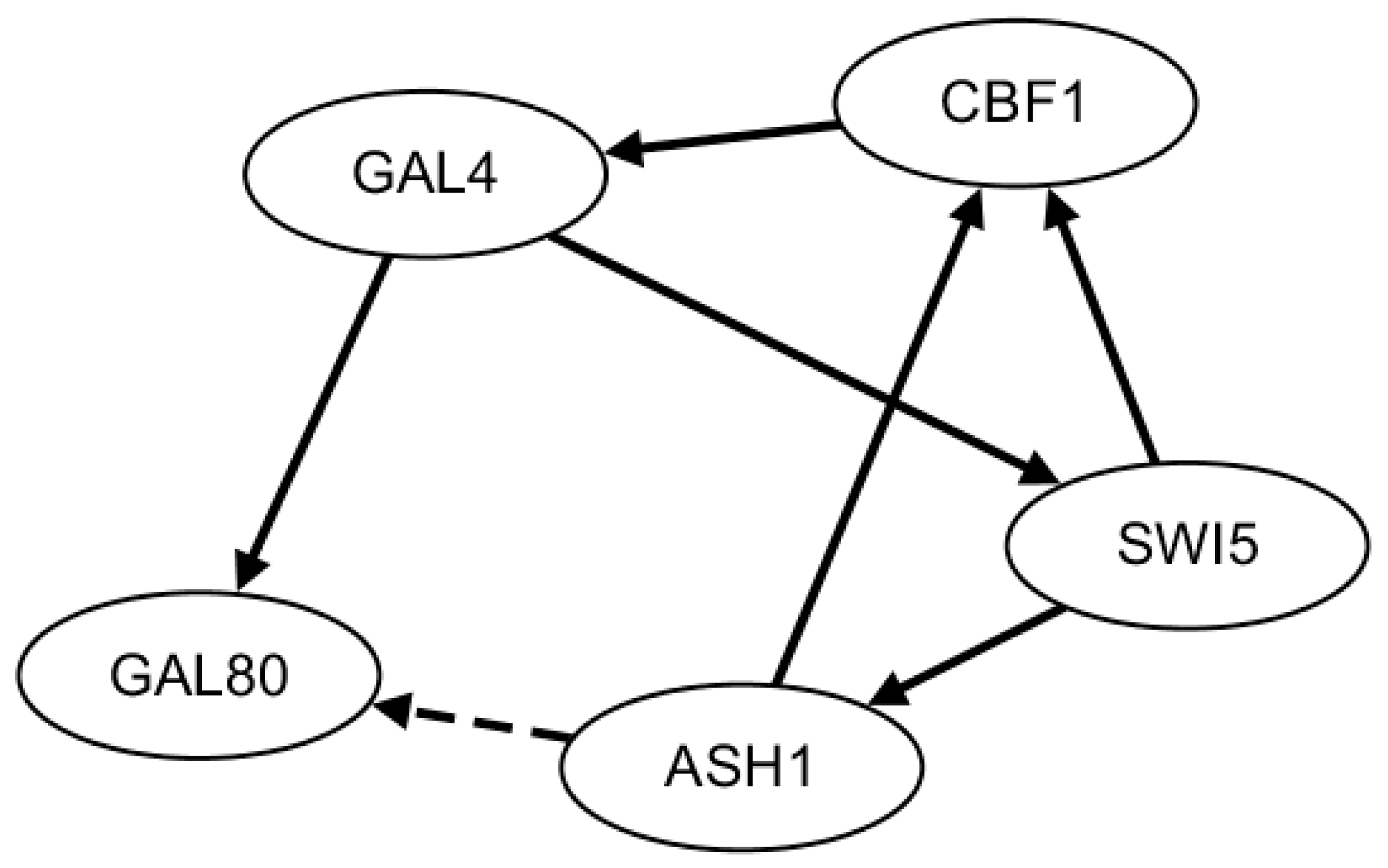
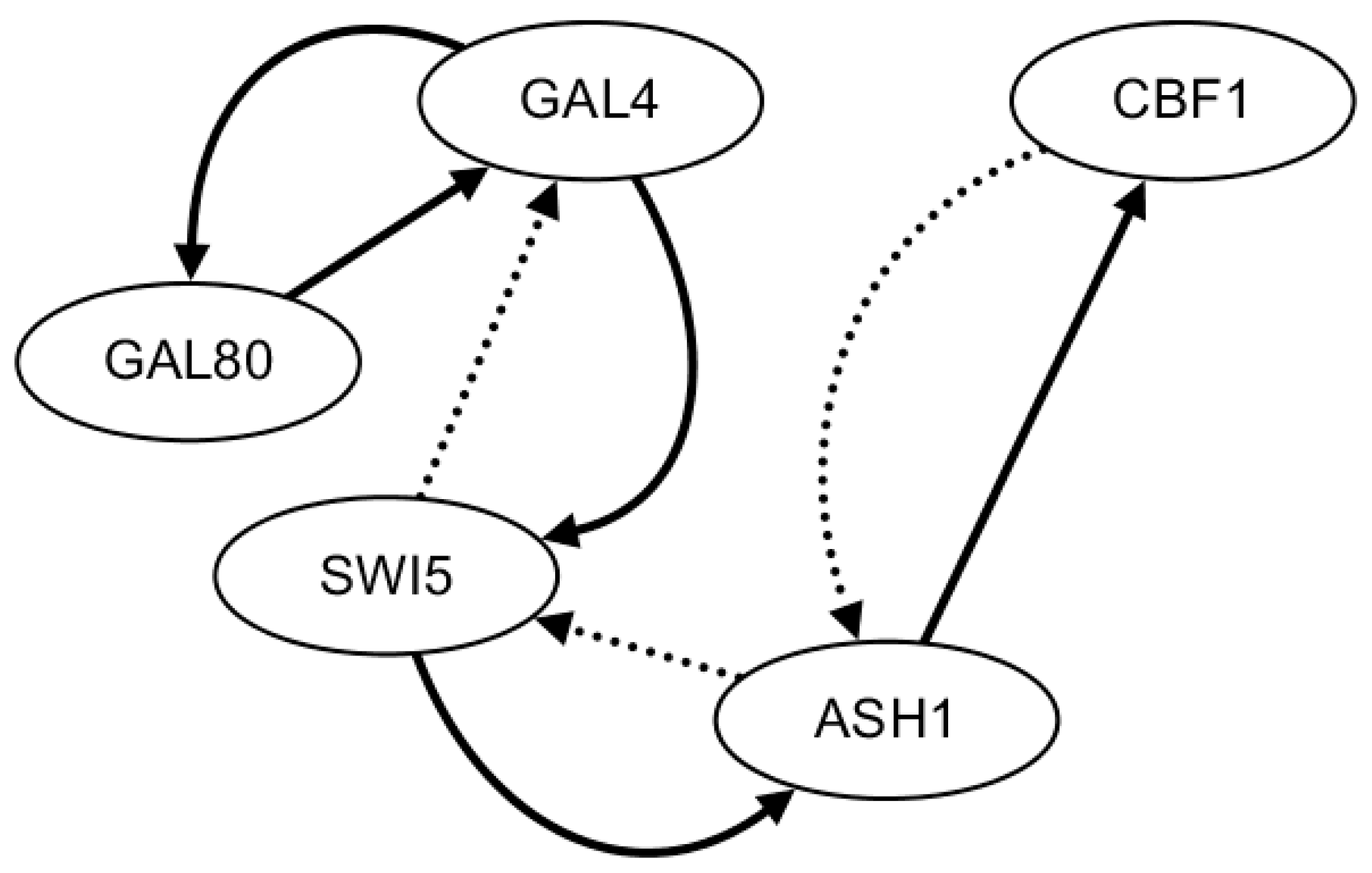
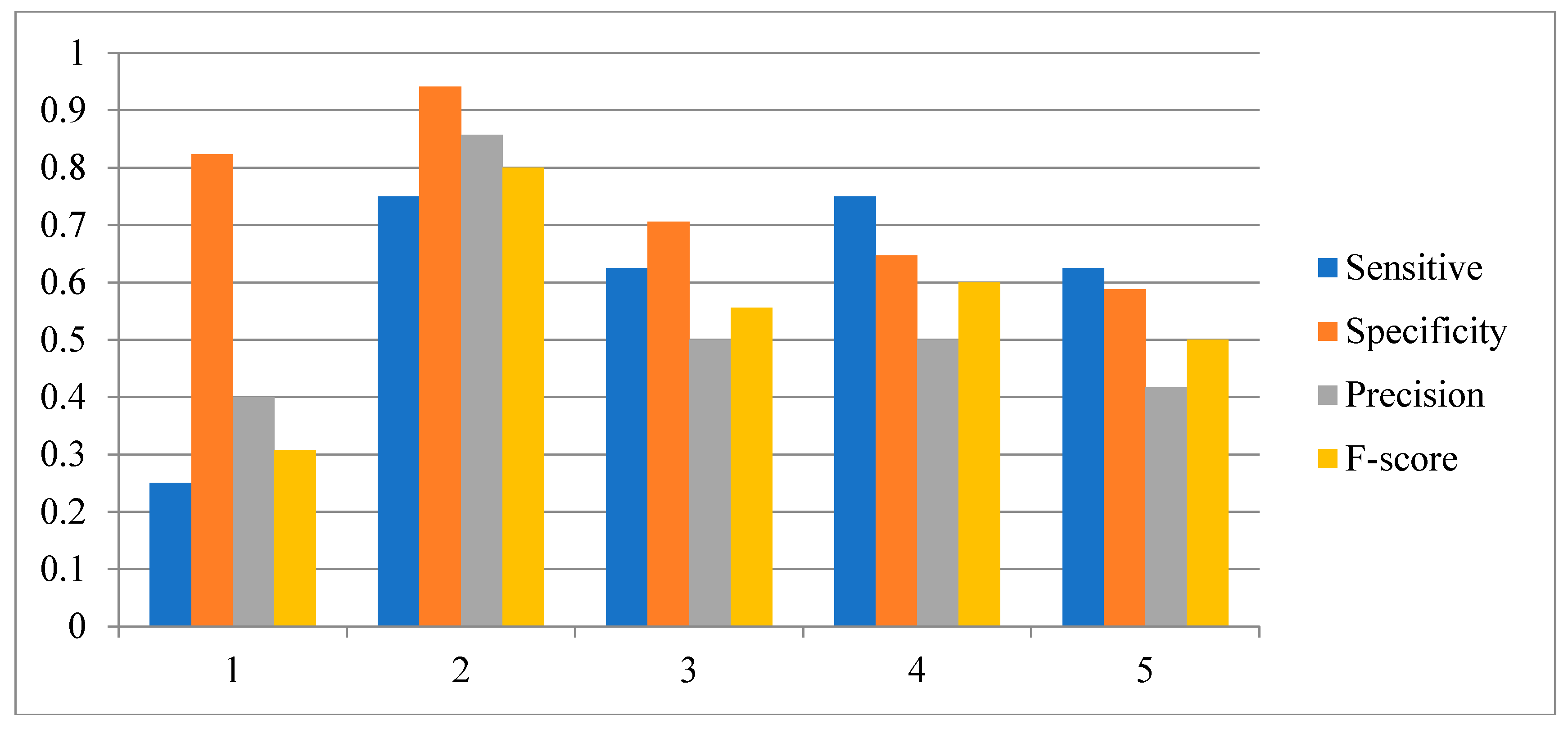
2.3. Network Construction of Real Data of the IRMA Network
3. Discussion
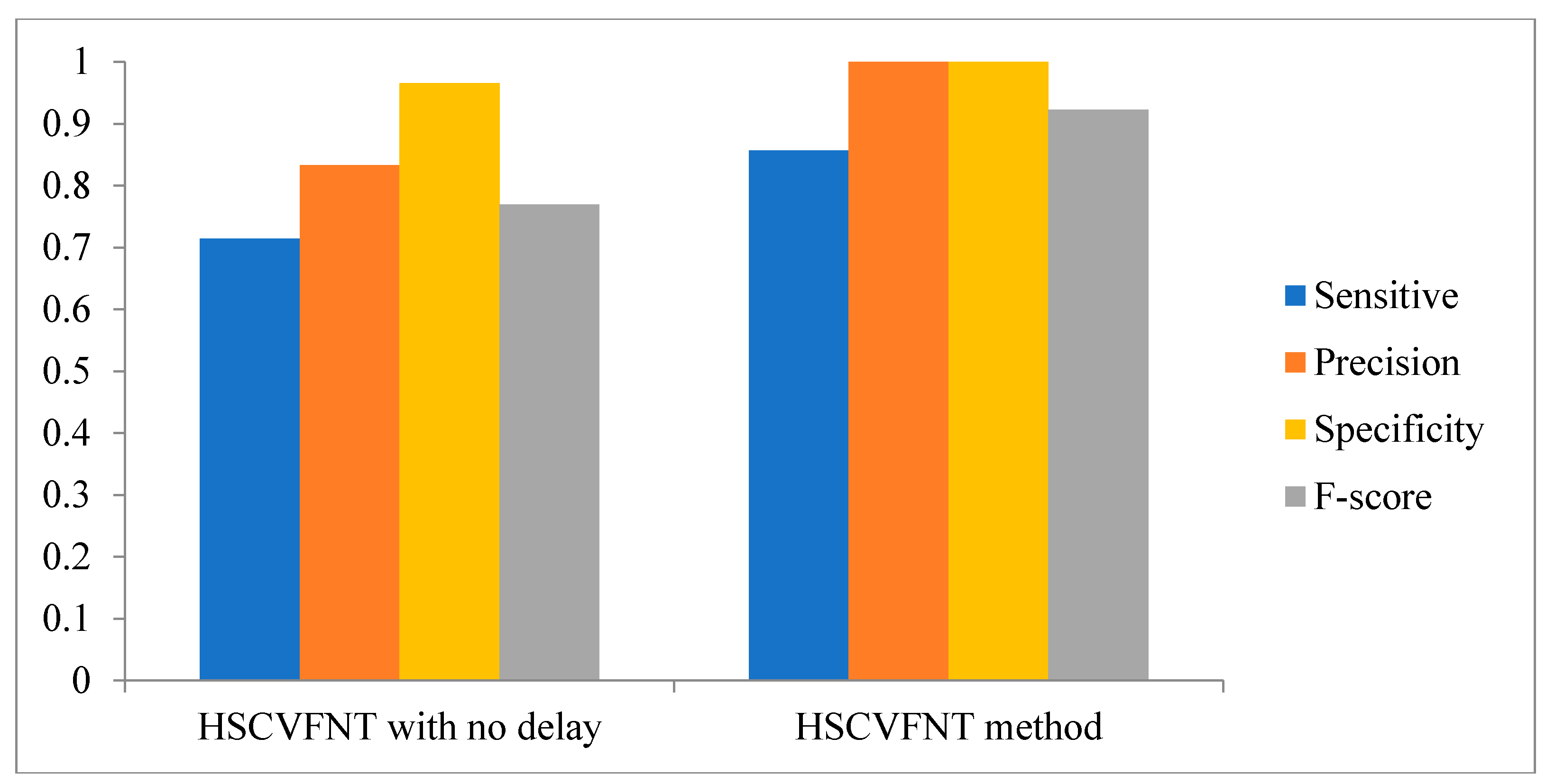
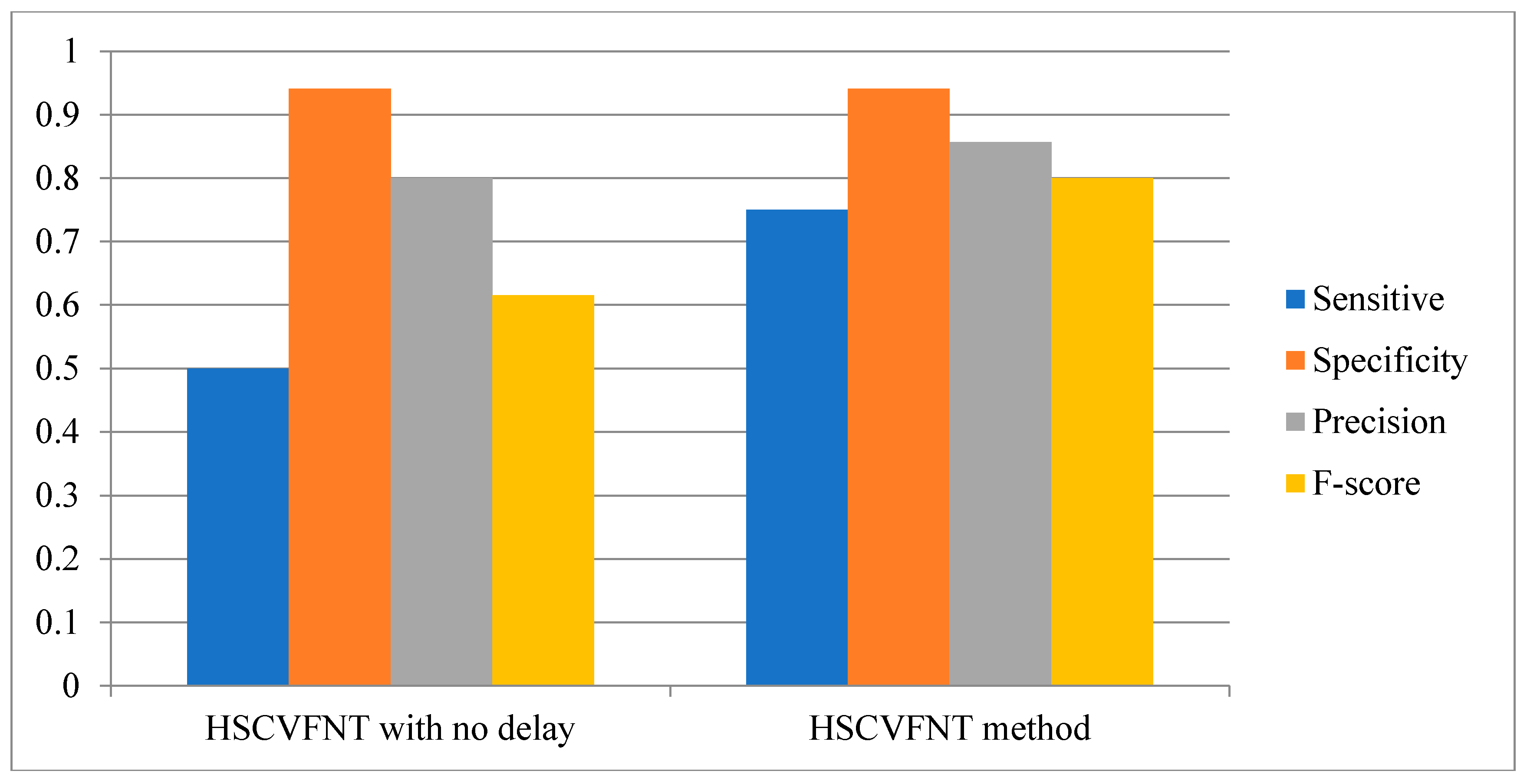
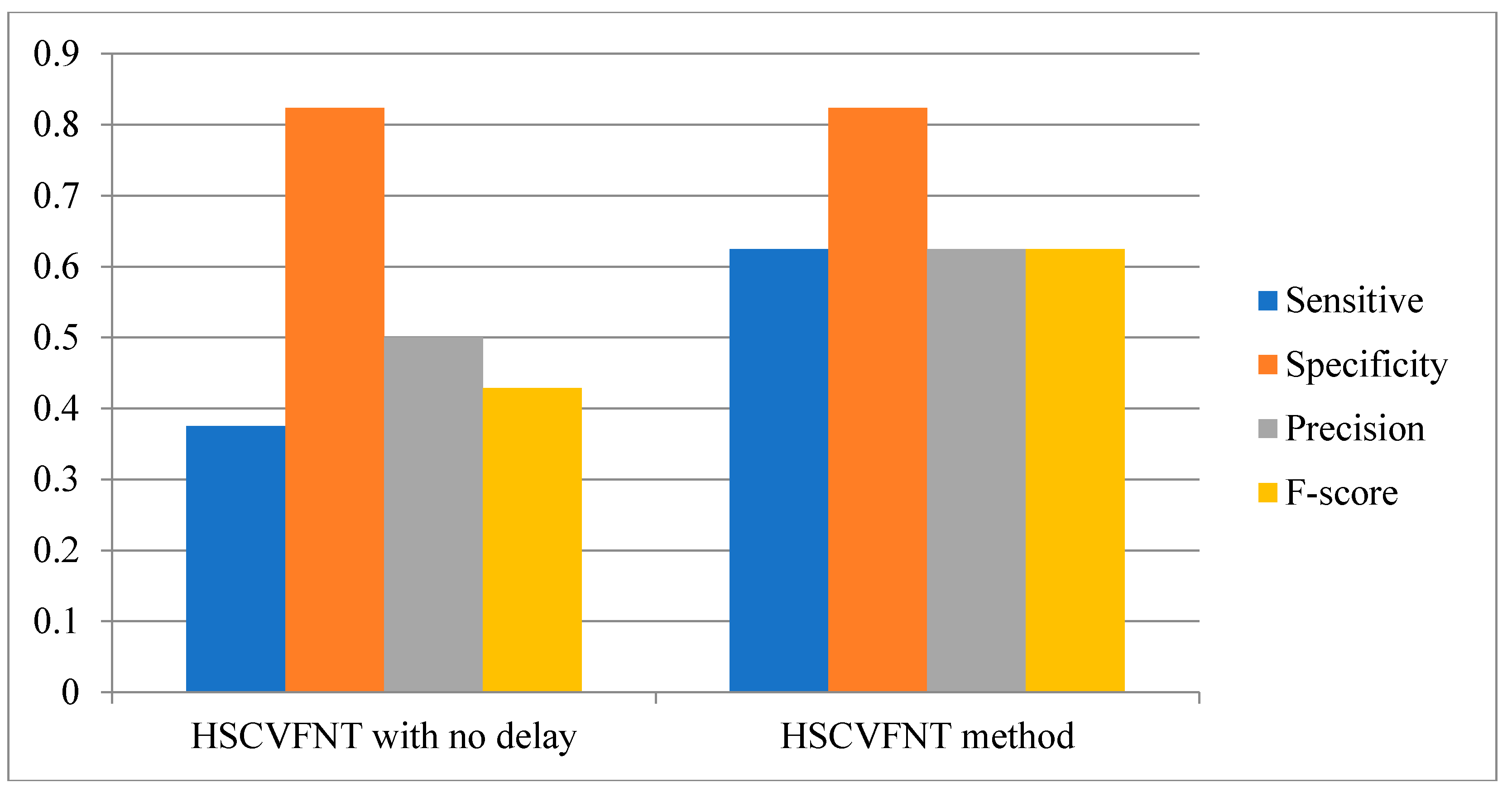
3.1. Influence of Time-Delayed Factor
3.2. Influence of the Number of Candidate Regulatory Factors
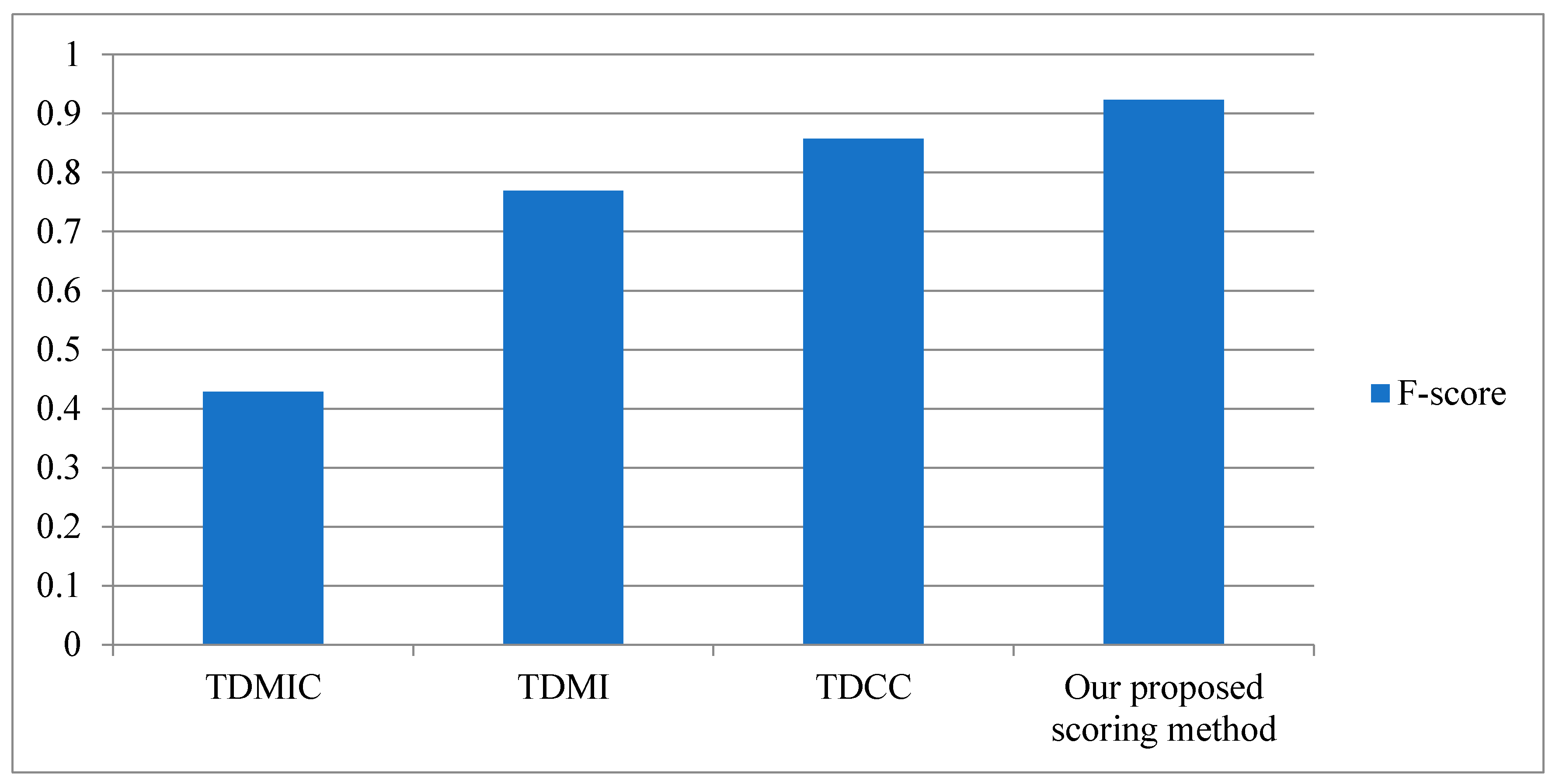
3.3. Performance of Our Proposed Scoring Method
3.4. Performance of Our Selected Stochastic Optimizer
4. Method
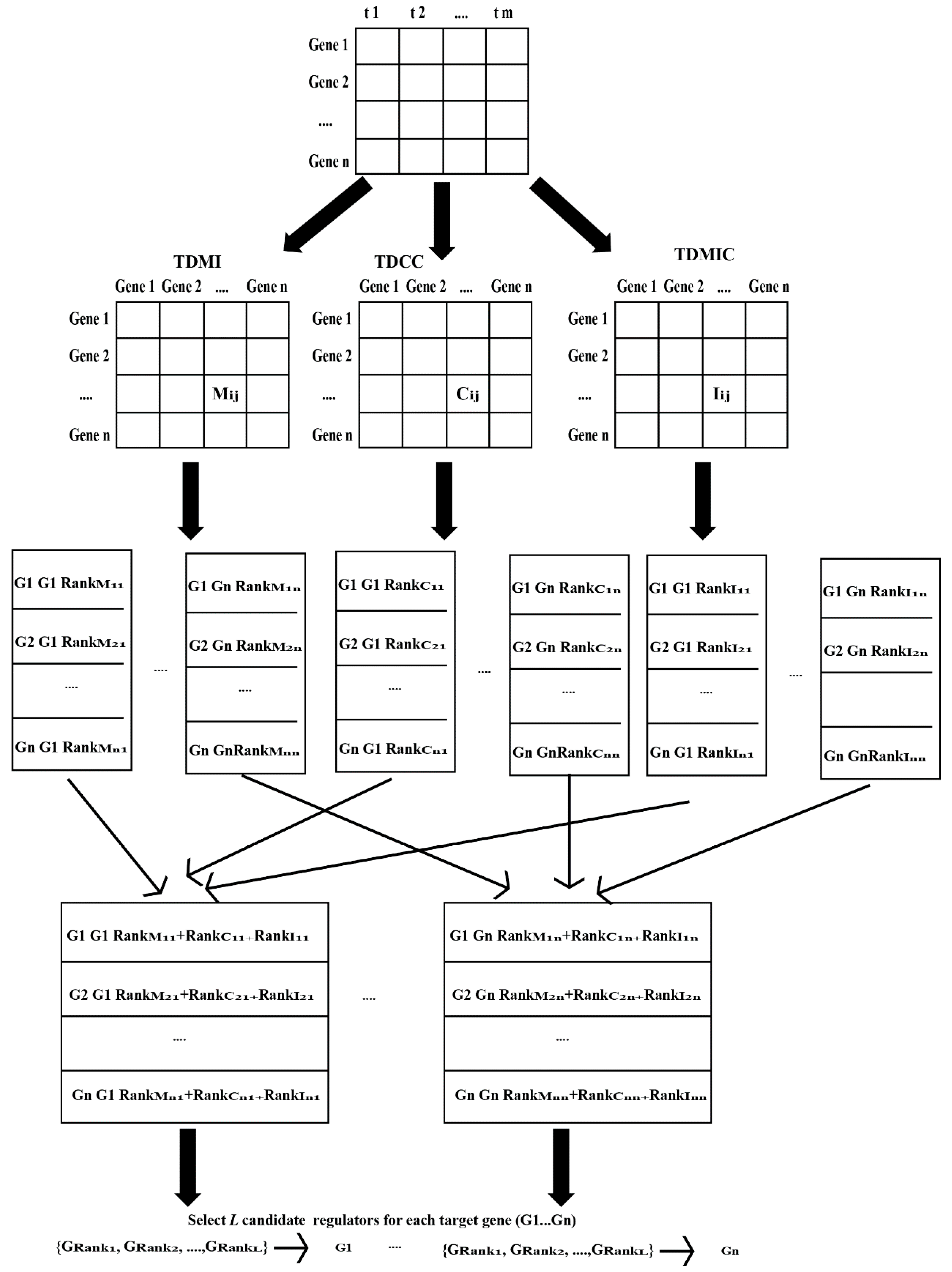
4.1. Our Proposed Scoring Method
4.1.1. Time-Delayed Mutual Information
4.1.2. Time-Delayed Maximum Information Coefficient
4.1.3. Time-Delayed Correlation Coefficient (TDCC)
4.1.4. Our Proposed Scoring Method
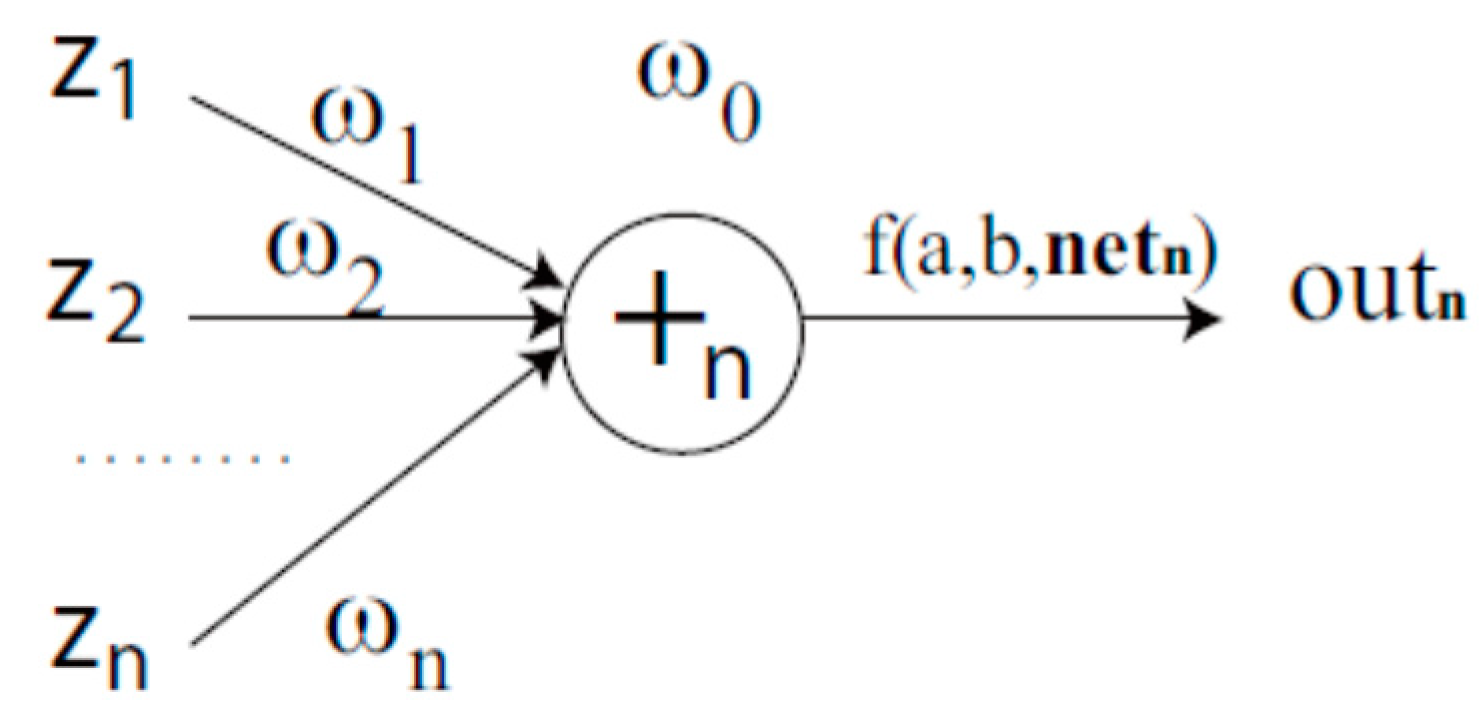
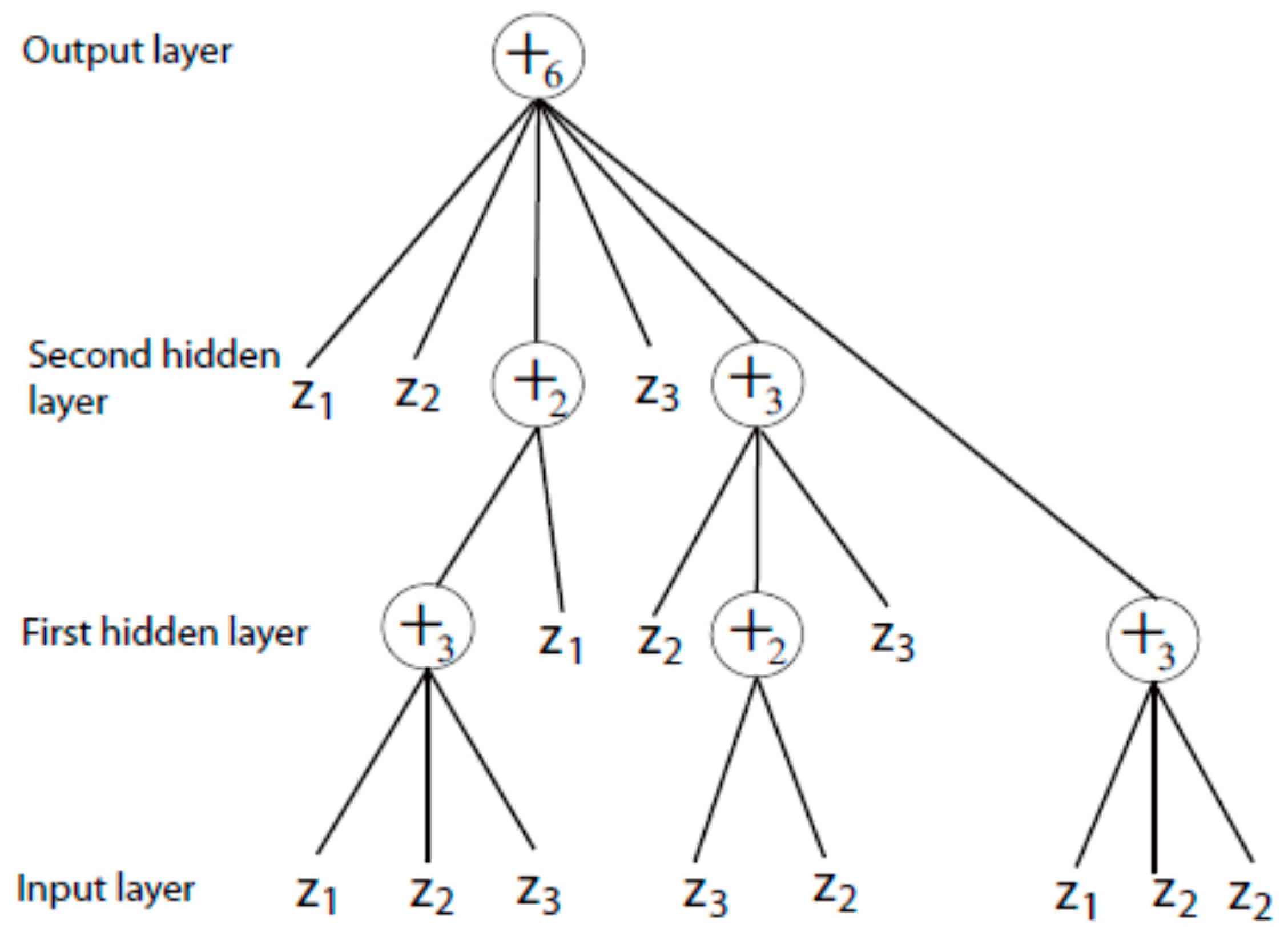
4.2. Complex-Valued Flexible Neural Tree
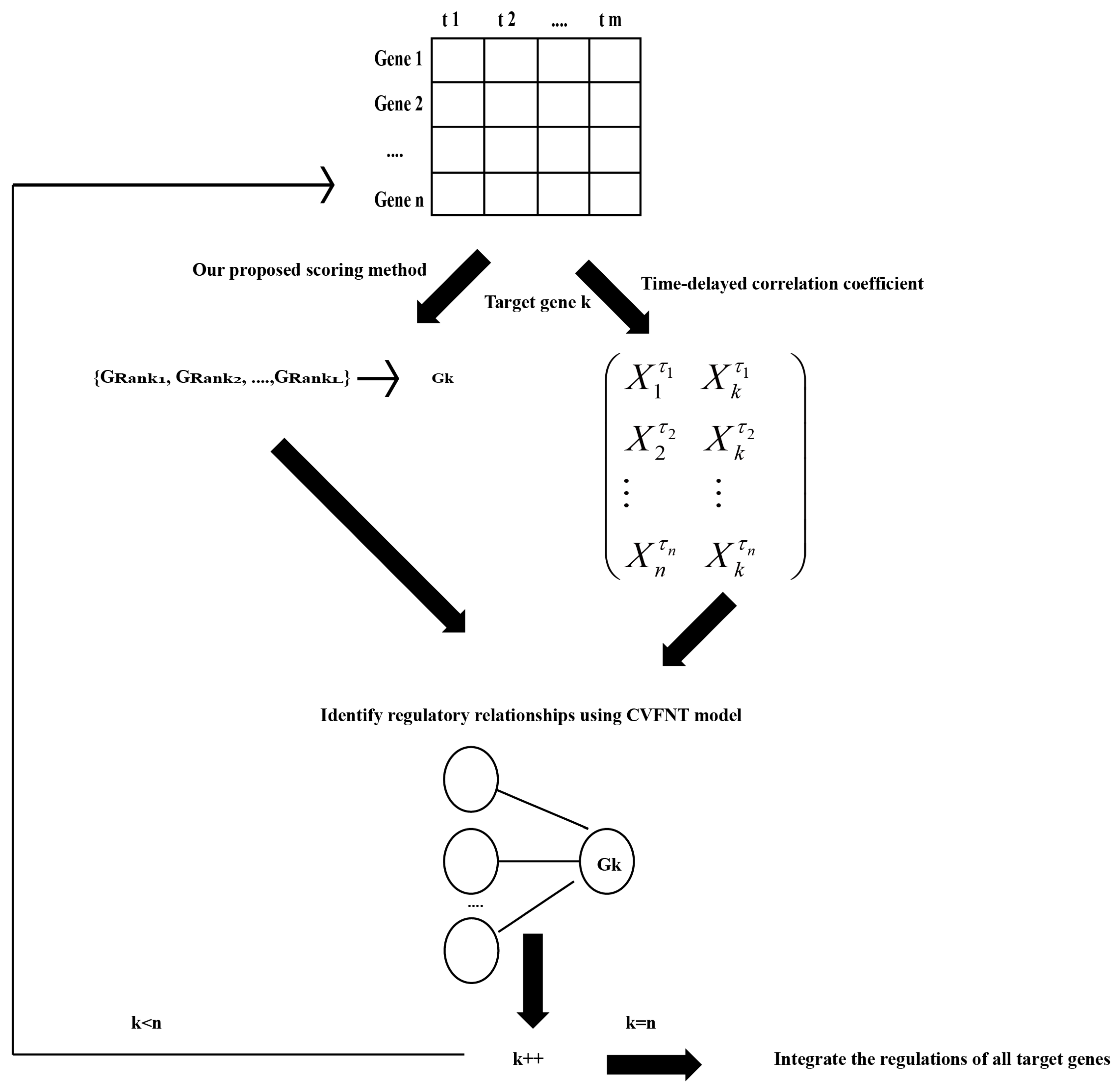
4.3. Flowchart of Our Method
- (1)
- Let gene expression data to be , where . Maximal time lag and the number of candidate regulatory factors are defined in advance.
- (2)
- For target gene , our proposed scoring method is utilized to select a candidate regulatory factor set with data , which is described in detail in Section 4.1.4.
- (3)
- For target gene , its time-delayed time series matrix is created. The algorithm is described in Figure 19. Time-delayed time series matrix between gene and its regulatory factors is listed in Equation (16):where is the optimal time lag between gene and gene , , and .
- (4)
- According to the candidate regulatory set and time-delayed time series matrix of gene k, the CVFNT model is utilized to identify the time-delayed regulatory relationships. The gene expression data of candidate regulatory factors are utilized as input data. The gene expression data of target gene k are utilized as output data. The optimization process of the CVFNT model is described as follows:(a) Initialize the population, containing the structure, real-valued and complex-valued parameters of the CVFNT model.(b) All individuals in the population are evaluated, using root mean squared error (RMSE):where is the number of data points, is the actual output of i-th time point and is the predicted output of the i-th time point.If the optimal CVFNT model is found or the maximum number of iterations is reached, the optimization process is over; otherwise, go to (c).
- (5)
- According to the structure of the optimized CVFNT model, input genes are seen as the regulatory factors. Obtain the regulations between target gene k and its regulatory factors.
- (6)
- . If , go to (2); otherwise, the regulations of all target genes are integrated in order to infer a time-delayed gene regulatory network.
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SOS | Save Our Soul |
| TDSS | time-delayed S-system |
| GRN | gene regulatory network |
| CVFNT | complex-valued flexible neural tree |
| TP | True Positive |
| FP | False Positive |
| FN | False Negative |
| TN | True Negative |
References
- Chan, T.E.; Stumpf, M.P.H.; Babtie, A.C. Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst. 2017, 5, 251. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Shao, J.; Zhao, Z.; Wang, P.; Qin, J.; Deng, Y.; Boheler, K.R.; Wang, J.; Yan, B. PTHGRN: Unraveling post-translational hierarchical gene regulatory networks using PPI, ChIP-seq and gene expression data. Nucleic Acids Res. 2014, 42, W130–W136. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Yuan, C.A.; Huang, Z.H.; Huang, D.S. Pupylation sites prediction with ensemble classification model. Int. J. Data Min. Bioin. 2017, 18, 91–104. [Google Scholar] [CrossRef]
- Elizabeth, N.E. Gene networks Network analysis gets dynamic. Nat. Rev. Genet. 2008, 9, 897. [Google Scholar]
- Teichmann, S.A.; Babu, M.M. Gene regulatory network growth by duplication. Nat. Genet. 2004, 36, 492. [Google Scholar] [CrossRef] [PubMed]
- Karlebach, G.; Shamir, R. Minimally perturbing a gene regulatory network to avoid a disease phenotype: The glioma network as a test case. BMC Syst. Biol. 2010, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Reagan, R.L.; Uratsu, S.L.; Phu, M.L.; Albrecht, U.; Zhao, W.; Davis, C.E.; Bowman, K.D.; Dandekar, A.M. Gene regulatory networks elucidating huanglongbing disease mechanisms. PLoS ONE 2013, 8, e74256. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Michoel, T.; Peer, Y.V.D. Prediction of a gene regulatory network linked to prostate cancer from gene expression, microRNA and clinical data. Bioinformatics 2010, 26, I638. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.; Noman, N.; Iba, H. Reverse engineering gene regulatory network from microarray data using linear time-variant model. BMC Bioinform. 2010, 11, S56. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.H.; Salleh, S.M.; Arif, S.; Zainudin, M.; Firdaus-Raih, M. Reconstructing gene regulatory networks from knock-out data using Gaussian Noise Model and Pearson Correlation Coefficient. Comput. Biol. Chem. 2015, 59, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.V.; Lewis, Z.V. On the Weierstrass-Mandelbrot fractal function. Proc. R. Soc. Lond. Ser. A 1980, 370, 459–484. [Google Scholar] [CrossRef]
- Guariglia, E. Harmonic Sierpinski Gasket and Applications. Entropy 2018, 20, 714. [Google Scholar] [CrossRef]
- Xenitidis, P.; Seimenis, I.; Kakolyris, S.; Adamopoulos, A. Evaluation of artificial time series microarray data for dynamic gene regulatory network inference. J. Theor. Biol. 2017, 426, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; You, Z.H.; Huang, D.S. CIPPEI: Computational identification of protein pupylation sites based on protein physicochemical properties and evolutionary information. Oncotarget 2017, 8, 108867–108879. [Google Scholar] [PubMed]
- Zhang, D.; Song, H.; Yu, L.; Wang, Q.G.; Ong, C. Set-values filtering for discrete time-delay genetic regulatory networks with time-varying parameters. Nonlinear Dyn. 2012, 69, 693–703. [Google Scholar] [CrossRef]
- Takada, M.; Hori, Y.; Hara, S. Existence of Oscillations in Cyclic Gene Regulatory Networks with Time Delay. Mathematics 2012, 47, 1203–1209. [Google Scholar]
- Parmar, K.; Blyuss, K.B.; Kyrychko, Y.N.; Hogan, S.J. Time-Delayed Models of Gene Regulatory Networks. Comput. Math. Method. Med. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Shi, M.; Shen, Q.; Zhu, J.; Shi, P. Filter Design with Adaptation to Time-Delay Parameters for Genetic Regulatory Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 15, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gong, P.; Li, H.N.; Perkins, E.J.; Wang, N.; Zhang, C.Y. Gene regulatory network inference and validation using relative change ratio analysis and time-delayed dynamic Bayesian network. EURASIP J. Bioinform. Syst. Biol. 2014, 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Chueh, T.H.; Lu, H. Inference of biological pathway from gene expression profiles by time delay boolean networks. PLoS ONE 2012, 7, e4209. [Google Scholar] [CrossRef] [PubMed]
- Zoppoli, P.; Morganella, S.; Ceccarelli, M. TimeDelayed-ARACNE: Reverse engineering of gene networks from time-course data by an information theoretic approach. BMC Bioinform. 2010, 11, 154. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.R.; Chetty, M.; Xuan Vinh, N.X. Incorporating time-delays in S-System model for reverse engineering genetic networks. BMC Bioinform. 2013, 14, 196. [Google Scholar] [CrossRef] [PubMed]
- Mundra, P.A.; Zheng, J.; Niranjan, M.; Welsch, R.E.; Rajapakse, J.C. Inferring Time-Delayed Gene Regulatory Networks Using Cross-Correlation and Sparse Regression. Lec. Notes Comput, Sci. 2013, 7875, 64–75. [Google Scholar]
- Li, Y.; Chen, H.; Zheng, J.; Ngom, A. The Max-Min High-Order Dynamic Bayesian Network for Learning Gene Regulatory Networks with Time-Delayed Regulations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Kordmahalleh, M.M.; Sefidmazgi, M.G.; Harrison, S.H. A Homaifar, Identifying time-delayed gene regulatory networks via an evolvable hierarchical recurrent neural network. Biodata Min. 2017, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Guariglia, E. Entropy and Fractal Antennas. Entropy 2016, 18, 84. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, K.; Liu, Z.P.; Duval, B.; Richer, J.M.; Zhao, X.M.; Hao, J.K.; Chen, L. NARROMI: A noise and redundancy reduction technique improves accuracy of gene regulatory network inference. Bioinformatics 2013, 29, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, S.W.; Guo, W.F.; Wei, Z.G.; Chen, L. Inference of Gene Regulatory Network Based on Local Bayesian Networks. PLoS Comput. Biol. 2016, 12, e1005024. [Google Scholar] [CrossRef] [PubMed]
- Akhand, M.A.H.; Nandi, R.N.; Amran, S.M.; Murase, K. Gene Regulatory Network inference incorporating Maximal Information Coefficient into Minimal Redundancy Network. ICEEICT 2015, 38, 723–724. [Google Scholar]
- Liu, W.; Zhu, W.; Liao, B.; Chen, H.; Ren, S.; Cai, L. Improving gene regulatory network structure using redundancy reduction in the MRNET algorithm. RSC Adv. 2017, 7, 23222–23233. [Google Scholar] [CrossRef]
- Trabelsi, C.; Bilaniuk, O.; Zhang, Y.; Serdyuk, D.; Subramanian, S. Deep Complex Networks. ICLR 2018, 2018, 1–19. [Google Scholar]
- Li, S. An Modified Error Function for the Complex-value Backpropagation Neural Networks. Neural Inf. Process. Lett. Rev. 2005, 9, 1–7. [Google Scholar]
- You, C.; Hong, D. Nonlinear blind equalization schemes using complex-valued multilayer feedforward neural networks. IEEE T. Neur. Net. Learn. Syst. 1998, 9, 1442–1455. [Google Scholar]
- Popa, C.A. Complex-Valued Deep Belief Networks. Lect. Notes Comput. Sci. 2018, 10878, 72–78. [Google Scholar]
- Xiong, T.; Bao, Y.; Hu, Z.; Chiong, R. Forecasting interval time series using a fully complex-valued RBF neural network with DPSO and PSO algorithms. Inform. Sci. 2015, 305, 77–92. [Google Scholar] [CrossRef]
- Saoud, L.S.; Rahmoune, F.; Tourtchine, V.; Baddari, K. Fully Complex Valued Wavelet Network for Forecasting the Global Solar Irradiation. Neural Process. Lett. 2017, 45, 475–505. [Google Scholar] [CrossRef]
- Savitha, R.; Suresh, S.; Sundararajan, N. Fast learning Circular Complex-valued Extreme Learning Machine (CC-ELM) for real-valued classification problems. Inform. Sci. 2012, 187, 277–290. [Google Scholar] [CrossRef]
- Ronen, M.; Rosenberg, R.; Shraiman, B.I.; Alon, U. Assigning numbers to the arrows: Parameterizing a gene regulationnetwork by using accurate expression kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 10555–10560. [Google Scholar] [CrossRef] [PubMed]
- Cantone, I.; Marucci, L.; Iorio, F.; Ricci, M.; Belcastro, V.; Bansal, M.; Santini, S.; di Bernardo, M.; di Bernardo, D.; Cosma, M. A yeastsynthetic network for in vivo assessment of reverse-engineeringand modeling approaches. Cell 2009, 137, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Perrin, B.E.; Ralaivola, L.; Mazurie, A.; Bottani, S.; Mallet, J.; D’Alche-Buc, F. Gene networks inference using dynamic Bayesian networks. Bioinformatics 2003, 138–148. [Google Scholar] [CrossRef]
- Noman, N.; Iba, H. Reverse engineering genetic networks using evolutionary computation. Genome Inform. 2005, 16, 205–214. [Google Scholar] [PubMed]
- Mundra, P.; Zheng, J.; Niranjan, M.; Welsch, R.; Rajapakse, J. Inferring time-delayed gene regulatory networks using crosscorrelation and sparse regression. In Bioinformatics Research and Applications; Springer: Cham, Switzerland, 2013; Volume LNCS7875, pp. 64–75. [Google Scholar]
- Zou, M.; Conzen, S. A new dynamic Bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data. Bioinformatics 2005, 21, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Husmeier, D. Sensitivity and specificity of inferring genetic regulatory interactions from microarray experiments with dynamic Bayesian networks. Bioinformatics 2003, 19, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Raza, K.; Alam, M. Recurrent neural network based hybrid model for reconstructing gene regulatory network. Comput. Biol. Chem. 2016, 64, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Ogundijo, O.E.; Elmas, A.; Wang, X. Reverse engineering gene regulatory networks from measurement with missing values. EURASIP J. Bioinform. Syst. Biol. 2017, 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.M.; He, K.; Lu, L.; Cao, Y.; Liu, J.D.; Chen, L. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics 2012, 28, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Kwon, Y.K. A novel mutual information-based Boolean network inference method from time-series gene expression data. PLoS ONE 2017, 12, e0171097. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.N.; Chen, L.; Wu, D.; Zhang, L.M. Maximal Information Coefficient for Feature Selection for Clinical Document Classification. Acta Phys. Chim. Sin. 2012, 28, 963–970. [Google Scholar]
- Albanese, D.; Filosi, M.; Visintainer, R.; Riccadonna, S.; Jurman, G.; Furlanello, C. Minerva and minepy: A C engine for the MINE suite and its R, Python and MATLAB wrappers. Bioinformatics 2013, 29, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Reshef, D.N.; Reshef, Y.A.; Finucane, H.K.; Grossman, S.R.; McVean, G.; Turnbaugh, P.; Lander, E.; Mitzenmacher, M.; Sabeti, P. Detecting novel associations in large data sets. Science 2011, 334, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, B.; Dong, J.; Abraham, A. Time-series forecasting using flexible neural tree model. Inform. Sci. 2005, 174, 219–235. [Google Scholar] [CrossRef]
- Chen, Y.; Abraham, A.; Yang, B. Feature selection and classification using flexible neural tree. Neurocomputing 2006, 70, 305–313. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, B.; Meng, Q. Small-time scale network traffic prediction based on flexible neural tree. Appl. Soft Comput. 2012, 12, 274–279. [Google Scholar] [CrossRef]
- Ojha, V.K.; Schiano, S.; Wu, C.Y.; Snášel, V.; Abraham, A. Predictive modeling of die filling of the pharmaceutical granules using the flexible neural tree. Neural Comput. Appl. 2018, 29, 467–481. [Google Scholar] [CrossRef]
- Bao, W.; Wang, D.; Chen, Y. Classification of Protein Structure Classes on Flexible Neutral Tree. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Yuan, C.A.; Zhang, Y.H.; Han, K.; Nandi, A.K.; Barry, H.; Huang, D.S. Mutli-features prediction of protein translational modification sites. IEEE/ACM Tran. Comput. Biol. Bioinform. 2018, 15, 1453–1460. [Google Scholar]
- Jia, L.; Zhang, W.; Yang, B. Identification of Nonlinear System Based on Complex-Valued Flexible Neural Network. In Intelligent Data Engineering and Automated Learning—IDEAL 2017; Springer: Cham, Switzerland, 2017; pp. 154–162. [Google Scholar]
- Amin, M.F.; Murase, K. Single-layered complex-valued neural network for real-valued classification problems. Neurocomputing 2009, 72, 945–955. [Google Scholar] [CrossRef]
- Rashid, S.; Saraswathi, S.; Kloczkowski, A.; Sundaram, S.; Kolinski, A. Protein secondary structure prediction using a small training set (compact model) combined with a Complex-valued neural network approach. BMC Bioinform. 2016, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, Y.H.; Jiang, M.Y. Reverse engineering of gene regulatory networks using flexible neural tree models. Neurocomputing 2013, 99, 458–466. [Google Scholar] [CrossRef]
- Yang, X.S.; He, X.S. Bat algorithm: Literature review and applications. Int. J. Bio-Inspir. Comput. 2013, 5, 141–149. [Google Scholar] [CrossRef]
- Gandomi, A.H.; Yang, X.S.; Alavi, A.H.; Talatahari, S. Bat algorithm for constrained optimization tasks. Neural Comput. Appl. 2013, 22, 1239–1255. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
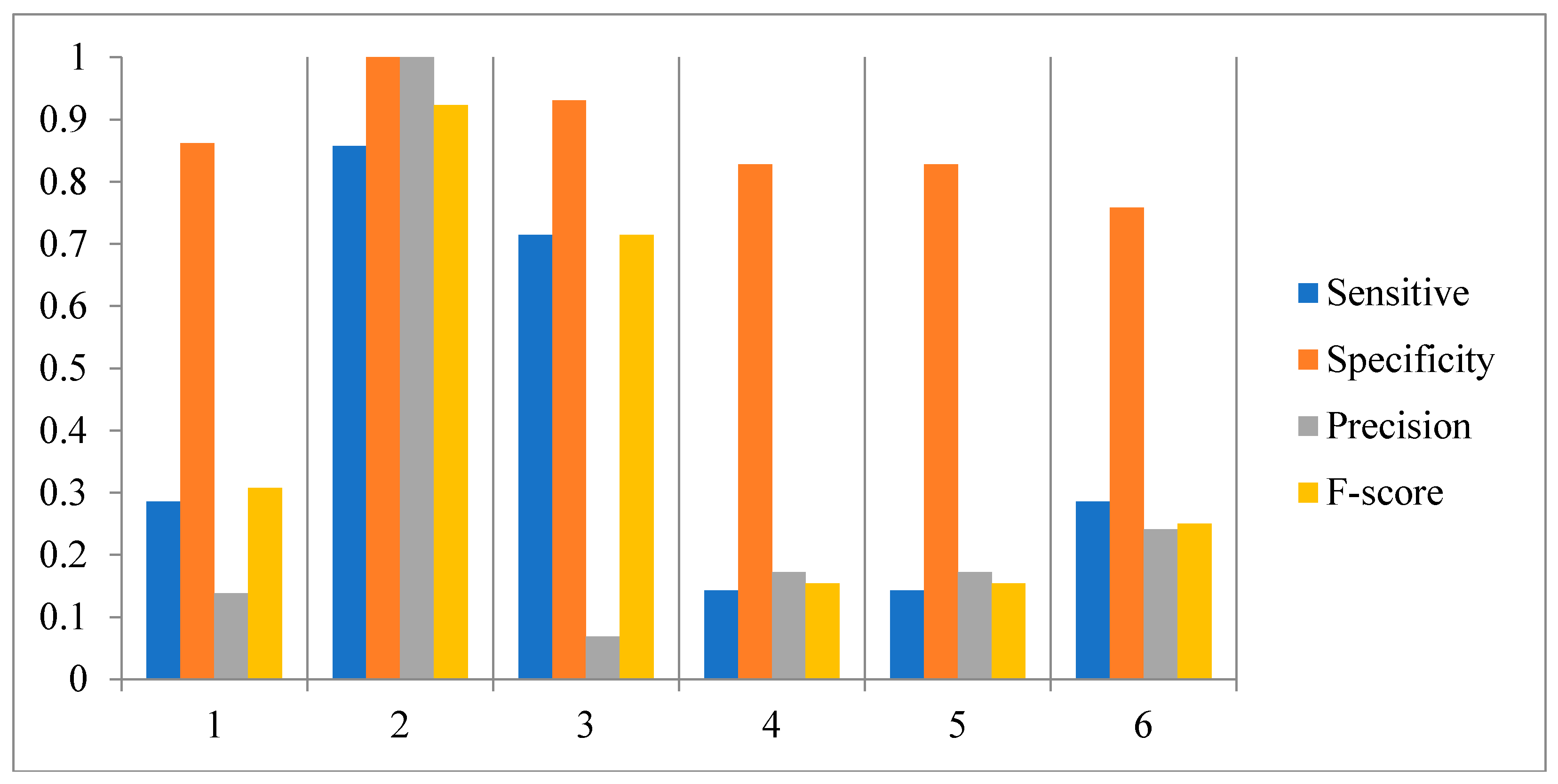{kind=link}
{kind=link}
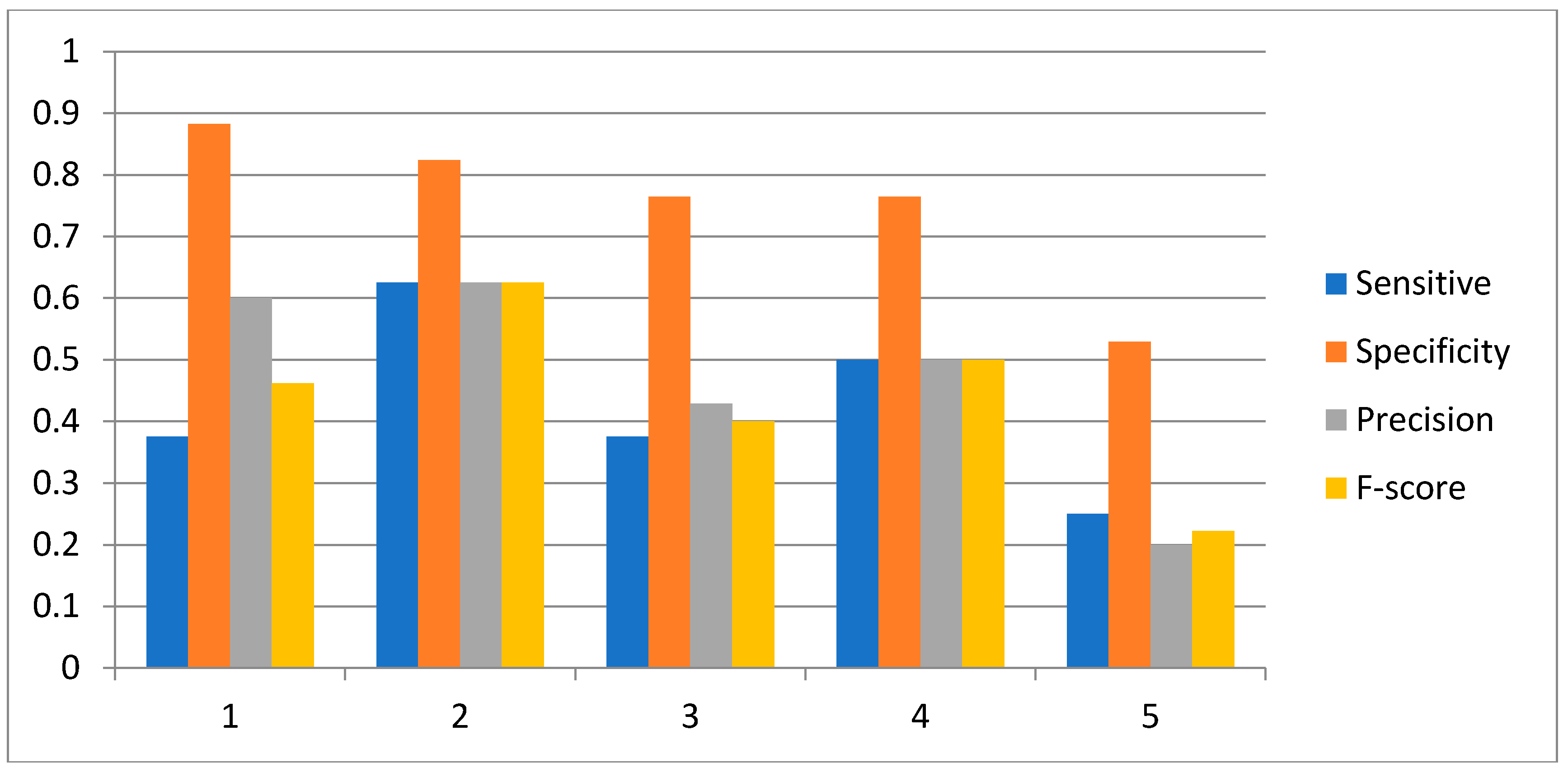{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Sensitivity | Specificity | F-Score |
|---|---|---|---|
| HSCVFNT | 0.857 | 1.00 | 0.923 |
| BN | 0.5714 | 0.931 | 0.6154 |
| S-system | 0.857 | 0.862 | 0.706 |
| TDSS | 1.00 | 0.896 | 0.875 |
| Method | Precision | Sensitivity | F-Score |
|---|---|---|---|
| HSCVFNT | 0.857 | 0.75 | 0.8 |
| HRNN | 0.667 | 0.75 | 0.70 |
| MMHO-DBN | 1.0000 | 0.5000 | 0.6667 |
| TDARACNE | 0.7142 | 0.6250 | 0.6667 |
| TDLASSO | 0.4000 | 0.2500 | 0.3077 |
| DBmcmc | 0.5000 | 0.2500 | 0.3333 |
| DBN-ZC | 0.6000 | 0.3750 | 0.4615 |
| Method | Precision | Sensitivity | F-Score |
|---|---|---|---|
| HSCVFNT | 0.625 | 0.625 | 0.625 |
| MMHO-DBN | 0.6667 | 0.2500 | 0.363 |
| TDARACNE | 0.5000 | 0.1250 | 0.2000 |
| TDLASSO | 0.2500 | 0.1250 | 0.1667 |
| DBmcmc | 0.1700 | 0.1200 | 0.1407 |
| Method | Sensitivity | Specificity | F-Score | Hit Ratio | |||
|---|---|---|---|---|---|---|---|
| mean | SD | mean | SD | mean | SD | ||
| HSCVFNT-BA | 0.8286 | 0.0602 | 0.9897 | 0.017 | 0.8856 | 0.0541 | 65% |
| HSCVFNT-PSO | 0.8143 | 0.069 | 0.9793 | 0.029 | 0.8589 | 0.0772 | 50% |
| HSCVFNT-DE | 0.8286 | 0.0614 | 0.9655 | 0.039 | 0.8449 | 0.0834 | 45% |
| HSCVFNT-GA | 0.7714 | 0.0999 | 0.9517 | 0.037 | 0.7845 | 0.0995 | 35% |
| Method | Precision | Sensitivity | F-Score | Hit Ratio | |||
|---|---|---|---|---|---|---|---|
| mean | SD | mean | SD | mean | SD | ||
| HSCVFNT-BA | 0.817 | 0.064 | 0.736 | 0.042 | 0.774 | 0.045 | 55% |
| HSCVFNT-PSO | 0.796 | 0.096 | 0.694 | 0.091 | 0.740 | 0.086 | 45% |
| HSCVFNT-DE | 0.788 | 0.117 | 0.702 | 0.095 | 0.737 | 0.096 | 55% |
| HSCVFNT-GA | 0.791 | 0.072 | 0.681 | 0.091 | 0.730 | 0.076 | 40% |
| Method | Precision | Sensitivity | F-Score | Hit Ratio | |||
|---|---|---|---|---|---|---|---|
| mean | SD | mean | SD | mean | SD | ||
| HSCVFNT-BA | 0.569 | 0.066 | 0.612 | 0.040 | 0.589 | 0.047 | 50% |
| HSCVFNT-PSO | 0.539 | 0.064 | 0.575 | 0.087 | 0.553 | 0.066 | 40% |
| HSCVFNT-DE | 0.545 | 0.067 | 0.600 | 0.053 | 0.569 | 0.051 | 35% |
| HSCVFNT-GA | 0.516 | 0.059 | 0.588 | 0.060 | 0.547 | 0.048 | 20% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Chen, Y.; Zhang, W.; Lv, J.; Bao, W.; Huang, D.-S. HSCVFNT: Inference of Time-Delayed Gene Regulatory Network Based on Complex-Valued Flexible Neural Tree Model. Int. J. Mol. Sci. 2018, 19, 3178. https://doi.org/10.3390/ijms19103178
Yang B, Chen Y, Zhang W, Lv J, Bao W, Huang D-S. HSCVFNT: Inference of Time-Delayed Gene Regulatory Network Based on Complex-Valued Flexible Neural Tree Model. International Journal of Molecular Sciences. 2018; 19(10):3178. https://doi.org/10.3390/ijms19103178
Chicago/Turabian StyleYang, Bin, Yuehui Chen, Wei Zhang, Jiaguo Lv, Wenzheng Bao, and De-Shuang Huang. 2018. "HSCVFNT: Inference of Time-Delayed Gene Regulatory Network Based on Complex-Valued Flexible Neural Tree Model" International Journal of Molecular Sciences 19, no. 10: 3178. https://doi.org/10.3390/ijms19103178
APA StyleYang, B., Chen, Y., Zhang, W., Lv, J., Bao, W., & Huang, D.-S. (2018). HSCVFNT: Inference of Time-Delayed Gene Regulatory Network Based on Complex-Valued Flexible Neural Tree Model. International Journal of Molecular Sciences, 19(10), 3178. https://doi.org/10.3390/ijms19103178