Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation


{kind=link}
{kind=link}
Abstract
1. Introduction
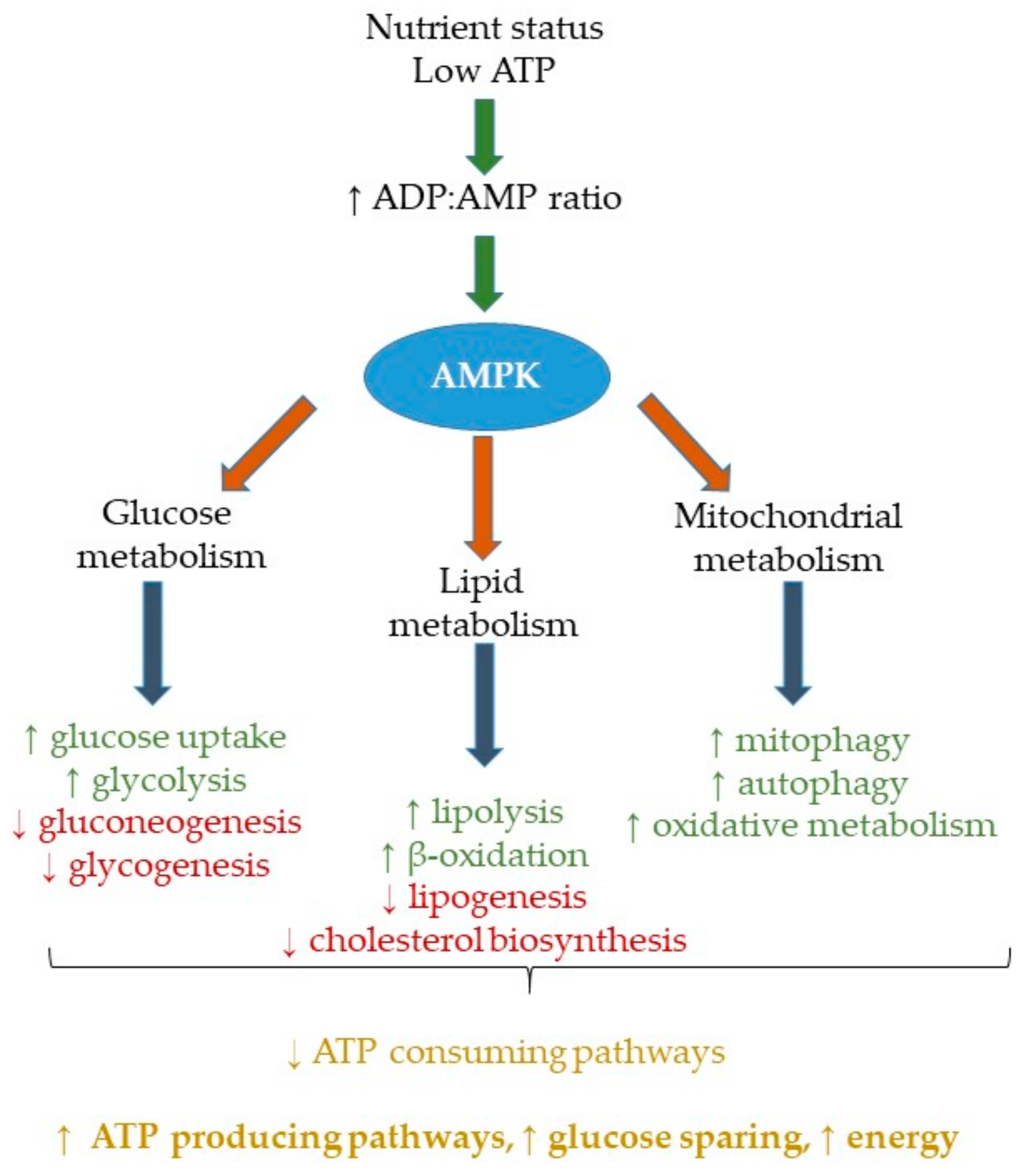
2. AMPK Activation, Metabolism and Nutrient Status
2.1. Nutrient Status and Impaired AMPK Action
2.2. The Involvement of AMPK in Insulin Resistance
2.3. AMPK and Its Link to Cancer
3. The Intersection between Metabolism and Inflammation
3.1. AMPK and the NLRP3 Inflammasome
3.2. Cellular Metabolism and Its Effect on Immune Cell Function
3.3. Metabolic Reprogramming of T Cells
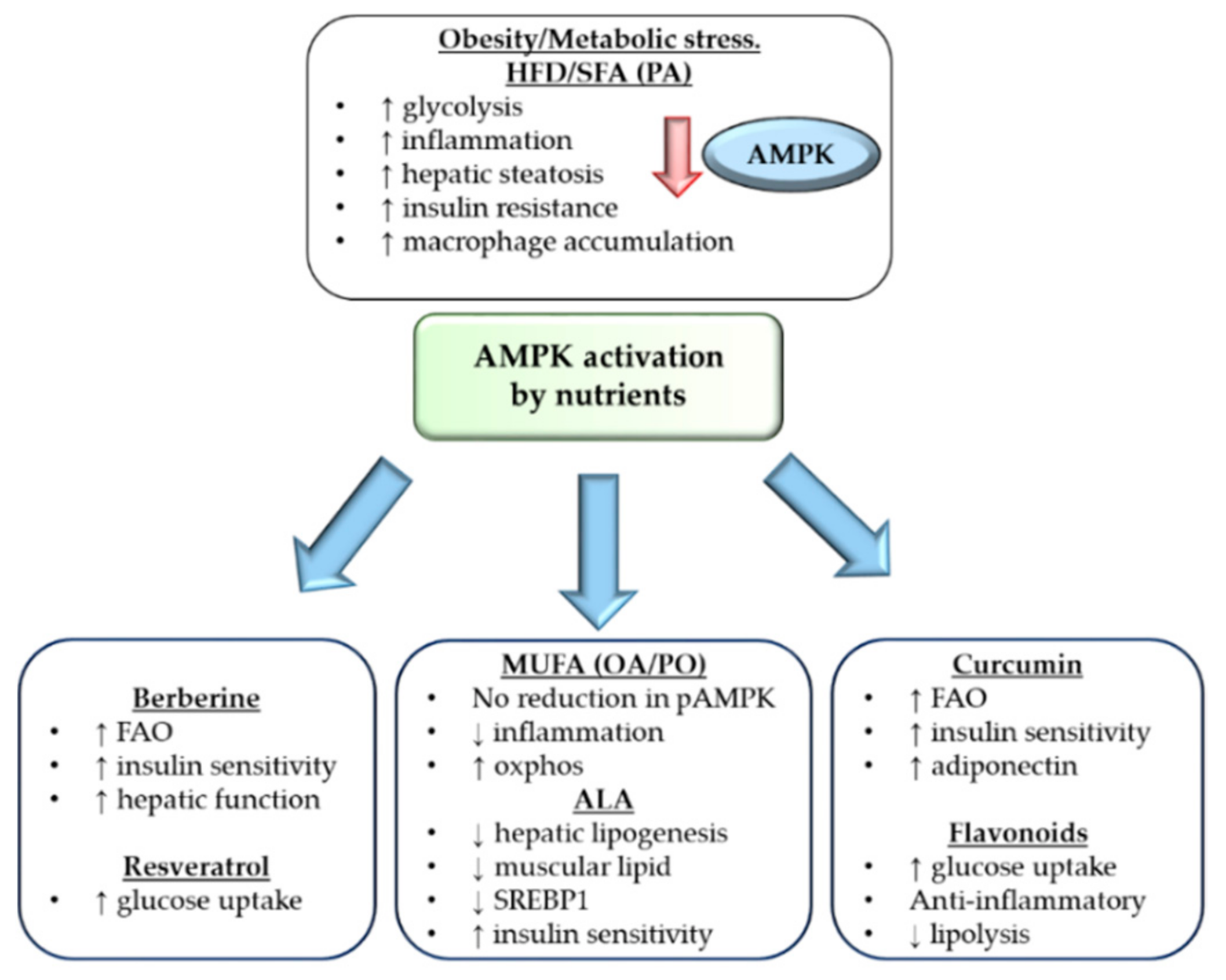
4. Modulation of AMPK Activation by Nutrients
4.1. Fatty Acids Differentially Affect AMPK Function
4.2. Reversing Metabolic Inflammation through AMPK
4.3. AMPK Regulation by Resveratrol, Berberine and Curcumin
4.4. AMPK Regulation by Flavonoids
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ADP | Adenosine diphosphate |
| AICAR | 5′-aminoimidazole-4-carboxamide ribonucleotide |
| Akt | Protein kinase B |
| ALA | A-linoleic acid |
| AMP | Adenosine monophosphate |
| AMPK | AMP-Activated Protein Kinase |
| AMPKK | AMPK kinase |
| ATP | Adenosine triphosphate |
| BMDM | Bone marrow derived macrophages |
| CaMKK | Ca2+/calmodulin-dependent protein kinase kinases |
| CC | Compound C |
| CVD | Cardiovascular disease |
| FA | Fatty acid |
| FAO | Fatty acid oxidation |
| FBP | Fructose-1,6-bisphosphate |
| FNDC5 | Fibronectin type III domain-containing 5 |
| Glut1 | Glucose transporter 1 |
| HIF-1α | Hypoxia-inducible factor 1-α |
| HFD | High fat diet |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin 6 |
| IL-10 | Interleukin 10 |
| LKB1 | Liver kinase B1 |
| LPS | Lipopolysaccharide |
| Mme | Metabolically activated macrophages |
| mTOR | Mammalian target of rapamycin |
| MUFA | Monounsaturated fatty acids |
| OA | Oleic acid |
| OxPHOS | Oxidative phosphorylation |
| P | Phosphorylation |
| PA | Palmitic acid |
| PO | Palmitoleic acid |
| PPAR-γ | Peroxisome proliferator-activated receptor γ |
| PUFA | Polyunsaturated fatty acid |
| ROS | Reactive oxygen species |
| SAT | Subcutaneous adipose tissue |
| SFA | Saturated fatty acid |
| SIRT1 | Sirtuin |
| SREBP-1 | Sterol regulatory element-binding protein 1 |
| T2D | Type 2 diabetes |
| TCA | Tricarboxylic acid |
| Thr | Threonine |
| TNF-α | Tumour necrosis factor α |
| VAT | Visceral adipose tissue |
| α | Alpha |
| β | Beta |
| γ | Gamma |
References
- Hardie, D.G. The AMP-activated protein kinase pathway—New players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1? Secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Connor, E.B.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]
- Stein, S.C.; Woods, A.; Jones, J.A.; Davison, M.D.; Carling, D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 2000, 345, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Chen, Z.P.; Scott, J.W.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef] [PubMed]
- Valentine, R.J.; Coughlan, K.A.; Ruderman, N.B.; Saha, A.K. Insulin inhibits AMPK activity and phosphorylates AMPK Ser485/491 through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch. Biochem. Biophys. 2014, 562, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M.; Holloway, G.P. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.P.; van Denderen, B.J.; et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, K.; Sharma, E.; Amara, V.R.; Pamulapati, H.; Dhawale, V.S. Metformin improves metabolic memory in high fat diet (HFD)-induced renal dysfunction. J. Biol. Chem. 2016, 291, 21848–21856. [Google Scholar] [CrossRef] [PubMed]
- Yavari, A.; Stocker, C.J.; Ghaffari, S.; Wargent, E.T.; Steeples, V.; Czibik, G.; Pinter, K.; Bellahcene, M.; Woods, A.; Martínez de Morentin, P.B.; et al. Chronic Activation of γ2 AMPK Induces Obesity and Reduces β Cell Function. Cell Metab. 2016, 23, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical Decrease of an Adipose-Specific Protein, Adiponectin, in Obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Declèves, A.-E.; Mathew, A.V.; Cunard, R.; Sharma, K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 2011, 22, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B.Z. Macrophage α1 AMP-activated protein kinase (α1 AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, E.P.; Desjardins, E.M.; Crane, J.D.; Smith, B.K.; Green, A.E.; Ducommun, S.; Henriksen, T.I.; Rebalka, I.A.; Razi, A.; Sakamoto, K.; et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016, 24, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, I.; Woltersdorf, W.W.; da Silva Xavier, G.; Rowe, R.L.; Cross, S.E.; Korbutt, G.S.; Rajotte, R.V.; Smith, R.; Rutter, G.A. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: Impact on glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1023–E1031. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Xavier, G.; Leclerc, I.; Varadi, A.; Tsuboi, T.; Moule, S.K.; Rutter, G.A. Role of AMP-activated protein kinase in the regulation by glucose of islet β cell gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 4023–4028. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, E.P.; Balasubramanian, P.; Lee, Y.-H.; Weng, C.; Kershaw, E.E.; Granneman, J.G. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic β3-adrenergic receptor activation. J. Lipid Res. 2014, 55, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK phosphorylates desnutrin/ATGL and HSL to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [PubMed]
- Habinowski, S.A.; Witters, L.A. The Effects of AICAR on Adipocyte Differentiation of 3T3-L1 Cells. Biochem. Biophys. Res. Commun. 2001, 286, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B. The role of AMP-activated protein kinase in regulating white adipose tissue metabolism. Mol. Cell. Endocrinol. 2013, 366, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A.; Viollet, B.; Andreelli, F.; Kahn, A.; Vaulont, S.; Sul, H.S. Induced Adiposity and Adipocyte Hypertrophy in Mice Lacking the AMP-Activated Protein Kinase-alpha2 Subunit. Diabetes 2004, 53, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Wang, H.; Guo, X.; Li, H.; Wang, Y.; Xu, X.; Tan, L.; Mashek, M.T.; Zhang, C.; et al. AMP-activated protein kinase α1 protects against diet-induced insulin resistance and obesity. Diabetes 2012, 61, 3114–3125. [Google Scholar] [PubMed]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin. Cancer Res. 2015, 21, 3836–3840. [Google Scholar] [CrossRef] [PubMed]
- Carretero, J.; Medina, P.P.; Blanco, R.; Smit, L.; Tang, M.; Roncador, G.; Maestre, L.; Conde, E.; Lopez-Rios, F.; Clevers, H.C.; et al. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene 2007, 26, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Fowkes, F.G.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Mills, G.B. AMPK: A Contextual Oncogene or Tumor Suppressor? Cancer Res. 2013, 73, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Metformin for cancer prevention. Front. Med. 2011, 5, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Pilon, G.; Dallaire, P.; Marette, A. Inhibition of Inducible Nitric-oxide Synthase by Activators of AMP-activated Protein Kinase. J. Biol. Chem. 2004, 279, 20767. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A.A.; Connaughton, R.M.; Lyons, C.L.; McMorrow, A.M.; Roche, H.M. Fatty acids and chronic low grade inflammation associated with obesity and the metabolic syndrome. Eur. J. Pharmacol. 2016, 785, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.M.; Lyons, C.L.; Finucane, O.M.; Roche, H.M. Interactions between differential fatty acids and inflammatory stressors-impact on metabolic health. Prostaglandins Leukot. Essent. Fat. Acids 2015, 92, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 6, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Dillon, E.; Guo, W.; Finucane, O.; McMorrow, A.; Murphy, A.; Lyons, C.; Jones, D.; Ryan, M.; Gibney, M.; et al. High-Density Lipoprotein Proteomic Composition, and not Efflux Capacity, Reflects Differential Modulation of Reverse Cholesterol Transport by Saturated and Monounsaturated Fat Diets. Circulation 2016, 133, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses In Vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Blagih, J.; Saucillo, D.C.; Tonelli, L.; Griss, T.; Rathmell, J.C.; Jones, R.G. The Liver Kinase B1 Is a Central Regulator of T Cell Development, Activation, and Metabolism. J. Immunol. 2011, 187, 4187–4198. [Google Scholar] [CrossRef] [PubMed]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKα1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Barra, N.G.; Ashkar, A.A.; Schertzer, J.D. Immunometabolism of T cells and NK cells: Metabolic control of effector and regulatory function. Inflamm. Res. 2018, 67, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, J.; Brüne, B.; Namgaladze, D. AICAR inhibits NFκB DNA binding independently of AMPK to attenuate LPS-triggered inflammatory responses in human macrophages. Sci. Rep. 2018, 8, 7081. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Nakahira, K.; Choi, A.M. Fatty acid synthesis and NLRP3-inflammasome. Oncotarget 2015, 6, 21765–21766. [Google Scholar] [CrossRef] [PubMed]
- Cacicedo, J.M.; Yagihashi, N.; Keaney, J.F., Jr; Ruderman, N.B.; Ido, Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Dicter, N.; Madar, Z.; Tirosh, O. α-Lipoic Acid Inhibits Glycogen Synthesis in Rat Soleus Muscle via Its Oxidative Activity and the Uncoupling of Mitochondria. J. Nutr. 2002, 132, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Song, K.H.; Koh, E.H.; Won, J.C.; Kim, H.S.; Park, H.S.; Kim, M.S.; Kim, S.W.; Lee, K.U.; Park, J.Y. α-Lipoic acid increases insulin sensitivity by activating AMPK in skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 332, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; de Vogel-van den Bosch, J.; Towler, M.C.; Schaart, G.; Moonen-Kornips, E.; Mensink, R.P.; Hesselink, M.K.; Hardie, D.G.; Schrauwen, P. Prevention of high-fat diet-induced muscular lipid accumulation in rats by α lipoic acid is not mediated by AMPK activation. J. Lipid Res. 2010, 51, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Park, K.G.; Min, A.K.; Koh, E.H.; Kim, H.S.; Kim, M.O.; Park, H.S.; Kim, Y.D.; Yoon, T.S.; Jang, B.K.; Hwang, J.S.; et al. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology 2008, 48, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L.; et al. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Pillon, N.J.; Sivaloganathan, D.M.; Costford, S.R.; Liu, Z.; Théret, M.; Chazaud, B.; Klip, A. Palmitoleate Reverses High Fat-induced Proinflammatory Macrophage Polarization via AMP-activated Protein Kinase (AMPK). J. Biol. Chem. 2015, 290, 16979–16988. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Q.; Geng, Z.; Zhou, B.; Zhang, F.; Han, Y.; Zhou, Y.B.; Wang, J.J.; Gao, X.Y.; Chen, Q.; Li, Y.H.; et al. FNDC5 attenuates adipose tissue inflammation and insulin resistance via AMPK-mediated macrophage polarization in obesity. Metabolism 2018, 83, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Feskens, E.J.; Bos, M.B.; Hoelen, D.W.; Heijligenberg, R.; Bromhaar, M.G.; de Groot, L.C.; de Vries, J.H.; Müller, M.; Afman, L.A. A saturated fatty acid—Rich diet induces an obesity-linked proinflammatory gene expression profile in adipose tissue of subjects at risk of metabolic syndrome. Am. J. Clin. Nutr. 2009, 90, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, Z.; Ke, L.; Li, Z.; Li, W.; Zhang, Z.; Zhou, Y.; Feng, X.; Zhu, W. Resveratrol improves glucose uptake in insulin-resistant adipocytes via Sirt1. J. Nutr. Biochem. 2018, 55, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.T.; Kwak, D.W.; Lin, S.K.; Kim, H.M.; Kim, Y.M.; Park, O.J. Resveratrol Induces Apoptosis in Chemoresistant Cancer Cells via Modulation of AMPK Signaling Pathway. Ann. N. Y. Acad. Sci. 2007, 1095, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Lee, Y.S.; Cha, S.H.; Jeong, H.W.; Choe, S.S.; Lee, M.R.; Oh, G.T.; Park, H.S.; Lee, K.U.; Lane, M.D.; et al. Berberine improves lipid dysregulation in obesity by controlling central and peripheral AMPK activity. Am. J. Physiol. Endocrinol. Metab. 2009, 296, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Li, J.Y.; Gosby, A.; To, S.W.; Cheng, Z.; Miyoshi, H.; Taketo, M.M.; Cooney, G.J.; Kraegen, E.W.; James, D.E.; et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: A mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008, 57, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, M.J.; Kim, E.J.; Yang, Y.; Lee, M.S.; Lim, J.S. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem. Pharmacol. 2011, 83, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, A.; Ha, T.; Subhan, F.; Lee, Y.S. New mechanisms and the anti-inflammatory role of curcumin in obesity and obesity-related metabolic diseases. Eur. J. Nutr. 2011, 50, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Um, M.Y.; Hwang, K.H.; Ahn, J.; Ha, T.Y. Curcumin Attenuates Diet-Induced Hepatic Steatosis by Activating AMP-Activated Protein Kinase. Basic Clin. Pharmacol. Toxicol. 2013, 113, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Na, L.X.; Zhang, Y.L.; Li, Y.; Liu, L.Y.; Li, R.; Kong, T.; Sun, C.H. Curcumin improves insulin resistance in skeletal muscle of rats. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Yang, H.; Cao, C.; Song, X.; Wallin, B.; Kivlin, R.; Lu, S.; Hu, G.; Di, W.; Wan, Y. AMPK mediates curcumin-induced cell death in CaOV3 ovarian cancer cells. Oncol. Rep. 1994, 20, 1553–1559. [Google Scholar]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Nagaoka, M.; Yonezawa, T.; Tanabe, R.; Woo, J.T.; Kato, H.; Chung, U.I.; Yagasaki, K. Regulatory mechanism for the stimulatory action of genistein on glucose uptake in vitro and in vivo. J. Nutr. Biochem. 2012, 23, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zhang, Y.; Yang, Q.; Cheng, S.; Hao, J.; Zhao, X.; Jiang, Z. Genistein Suppresses LPS-Induced Inflammatory Response through Inhibiting NF-κB following AMP Kinase Activation in RAW 264.7 Macrophages. PLoS ONE 2012, 7, e53101. [Google Scholar] [CrossRef] [PubMed]
- Palacios-González, B.; Zarain-Herzberg, A.; Flores-Galicia, I.; Noriega, L.G.; Alemán-Escondrillas, G.; Zariñan, T.; Ulloa-Aguirre, A.; Torres, N.; Tovar, A.R. Genistein stimulates fatty acid oxidation in a leptin receptor-independent manner through the JAK2-mediated phosphorylation and activation of AMPK in skeletal muscle. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Fujimori, K. Antiadipogenic Effect of Dietary Apigenin through Activation of AMPK in 3T3-L1 Cells. J. Agric. Food Chem. 2011, 59, 13346–13352. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.H.; Son, H.J.; Jang, Y.J.; Ahn, J.; Jung, C.H.; Ha, T.Y. Apigenin Ameliorates the Obesity-Induced Skeletal Muscle Atrophy by Attenuating Mitochondrial Dysfunction in the Muscle of Obese Mice. Mol. Nutr. Food Res. 2017, 61, 1700218. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Cho, Y.Y.; Choi, M.S. Apigenin Ameliorates Dyslipidemia, Hepatic Steatosis and Insulin Resistance by Modulating Metabolic and Transcriptional Profiles in the Liver of High-Fat Diet-Induced Obese Mice. Nutrients 2016, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Chung, H.Y.; Kim, N.D. Role of Apigenin in Cancer Prevention via the Induction of Apoptosis and Autophagy. J. Cancer Prev. 2016, 21, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyons, C.L.; Roche, H.M. Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. Int. J. Mol. Sci. 2018, 19, 3092. https://doi.org/10.3390/ijms19103092
Lyons CL, Roche HM. Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. International Journal of Molecular Sciences. 2018; 19(10):3092. https://doi.org/10.3390/ijms19103092
Chicago/Turabian StyleLyons, Claire L., and Helen M. Roche. 2018. "Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation" International Journal of Molecular Sciences 19, no. 10: 3092. https://doi.org/10.3390/ijms19103092
APA StyleLyons, C. L., & Roche, H. M. (2018). Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. International Journal of Molecular Sciences, 19(10), 3092. https://doi.org/10.3390/ijms19103092