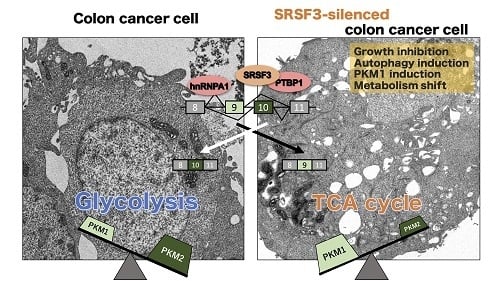
SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells

 , ,
, , 


Abstract
:
1. Introduction
2. Results
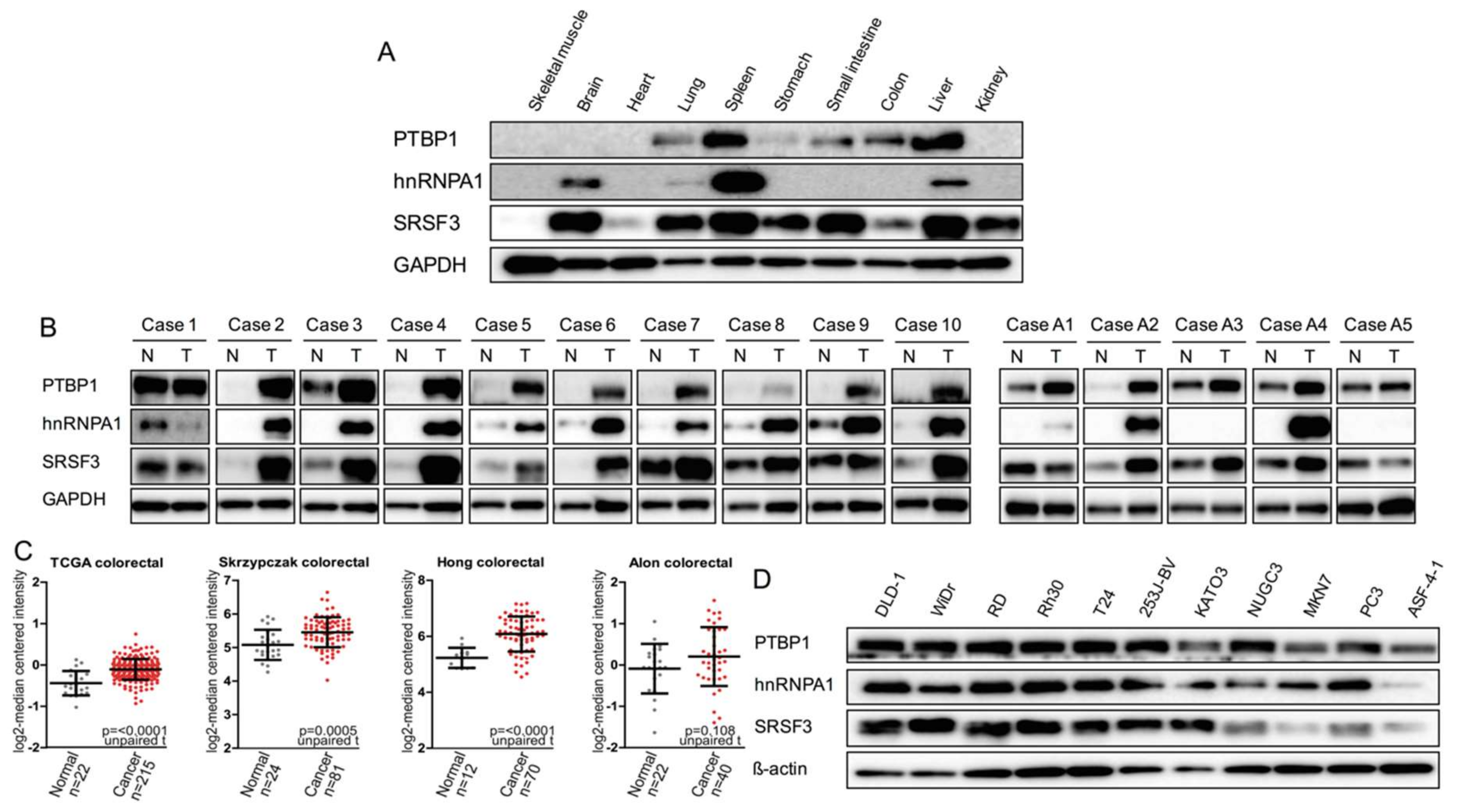
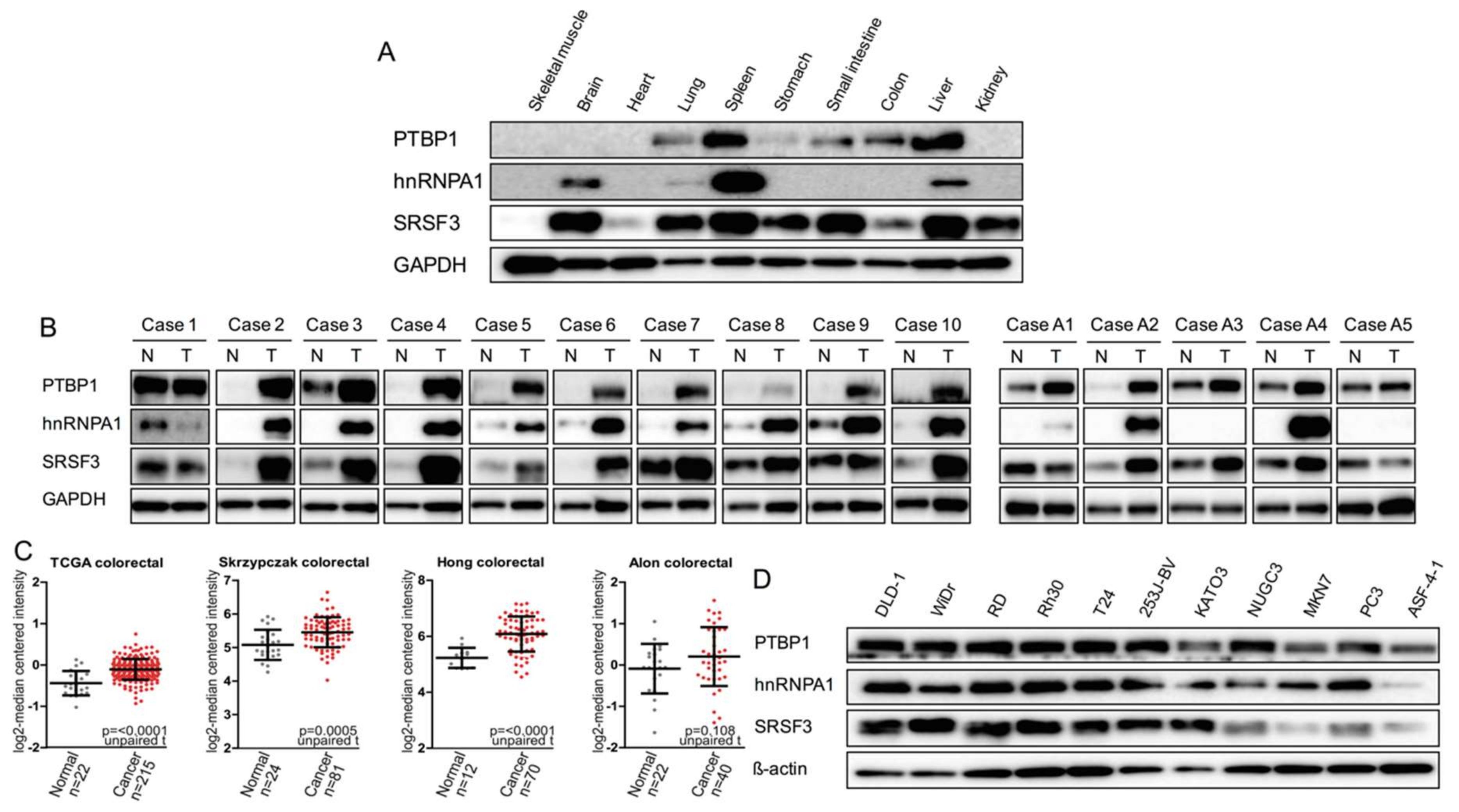
2.1. Expression of PTBP1, hnRNPA1, and SRSF3 in Mouse Normal Tissues, Human Clinical Colorectal Tumors, and Human Cancer Cell Lines
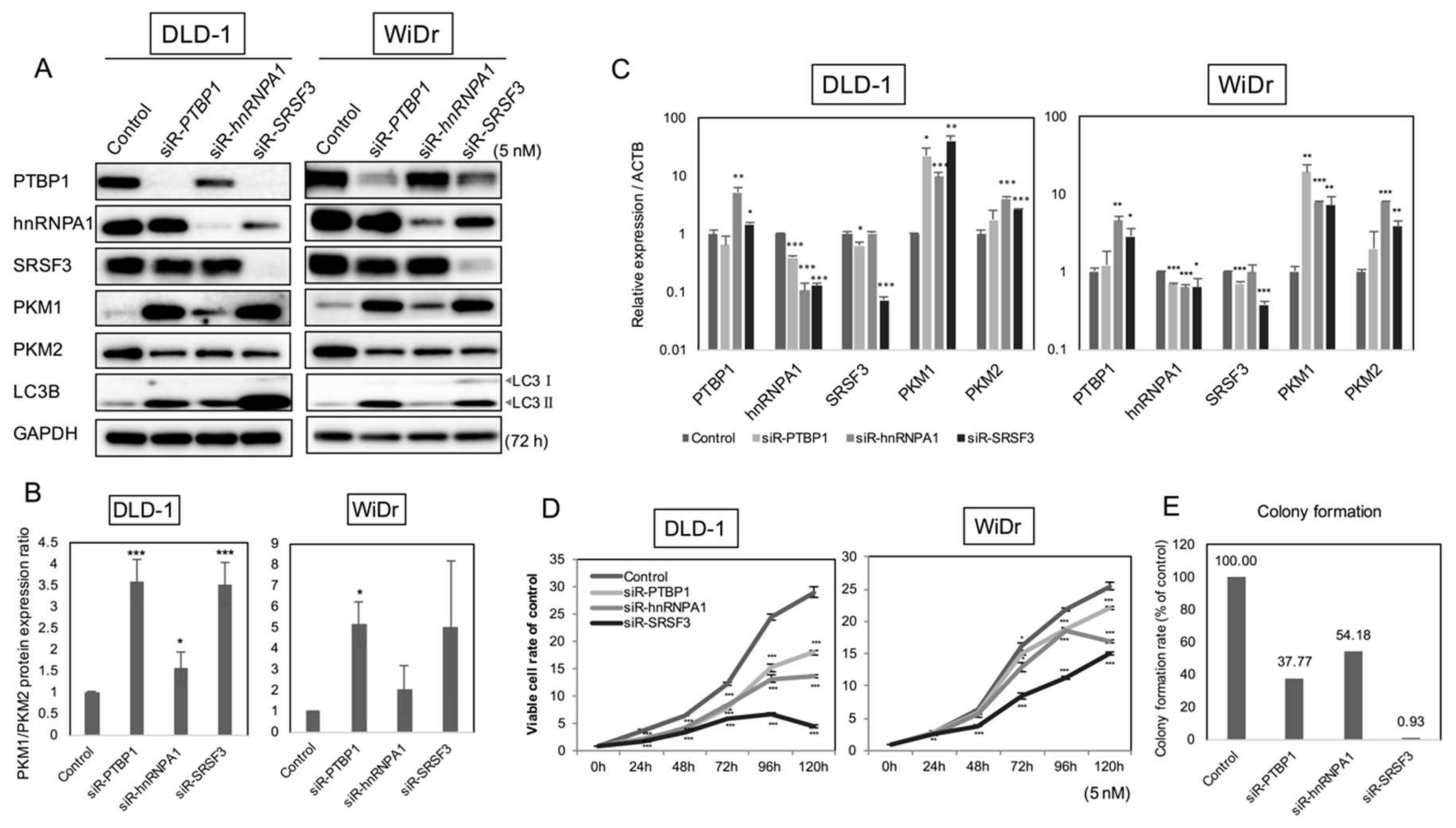
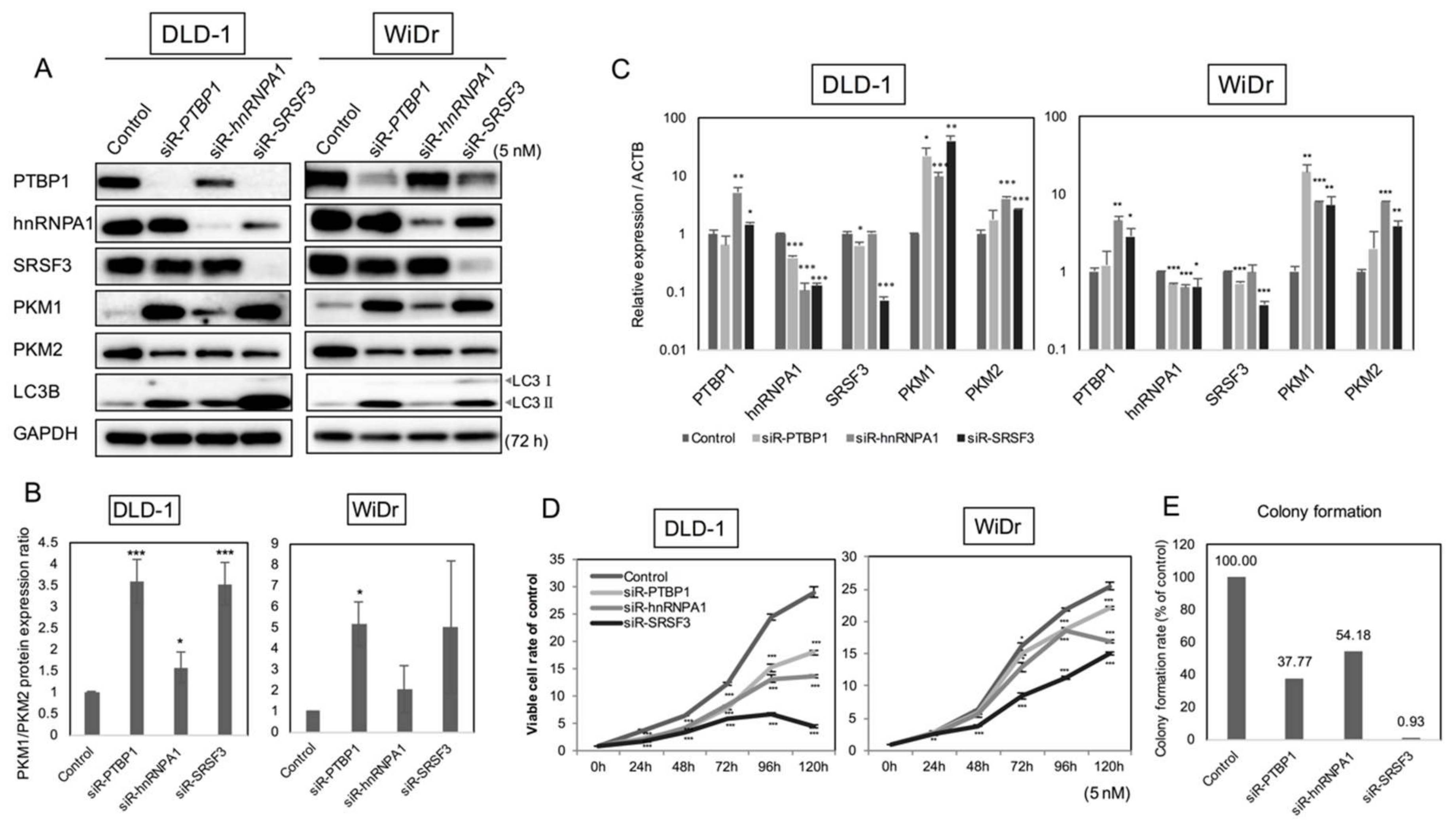
2.2. PKM Isoform Expression and Contribution of PTBP1, hnRNPA1, and SRSF3 to Tumorigenesis
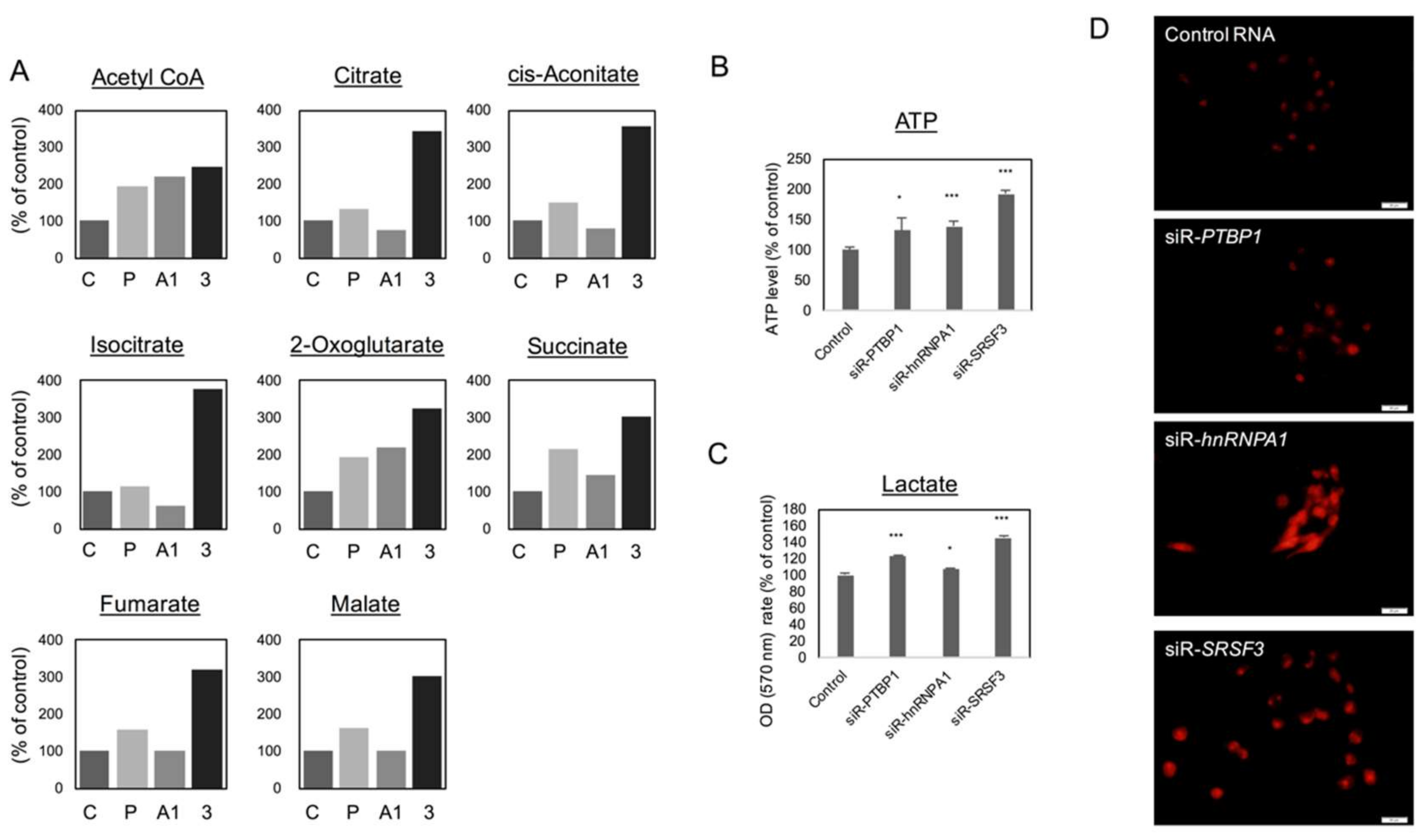
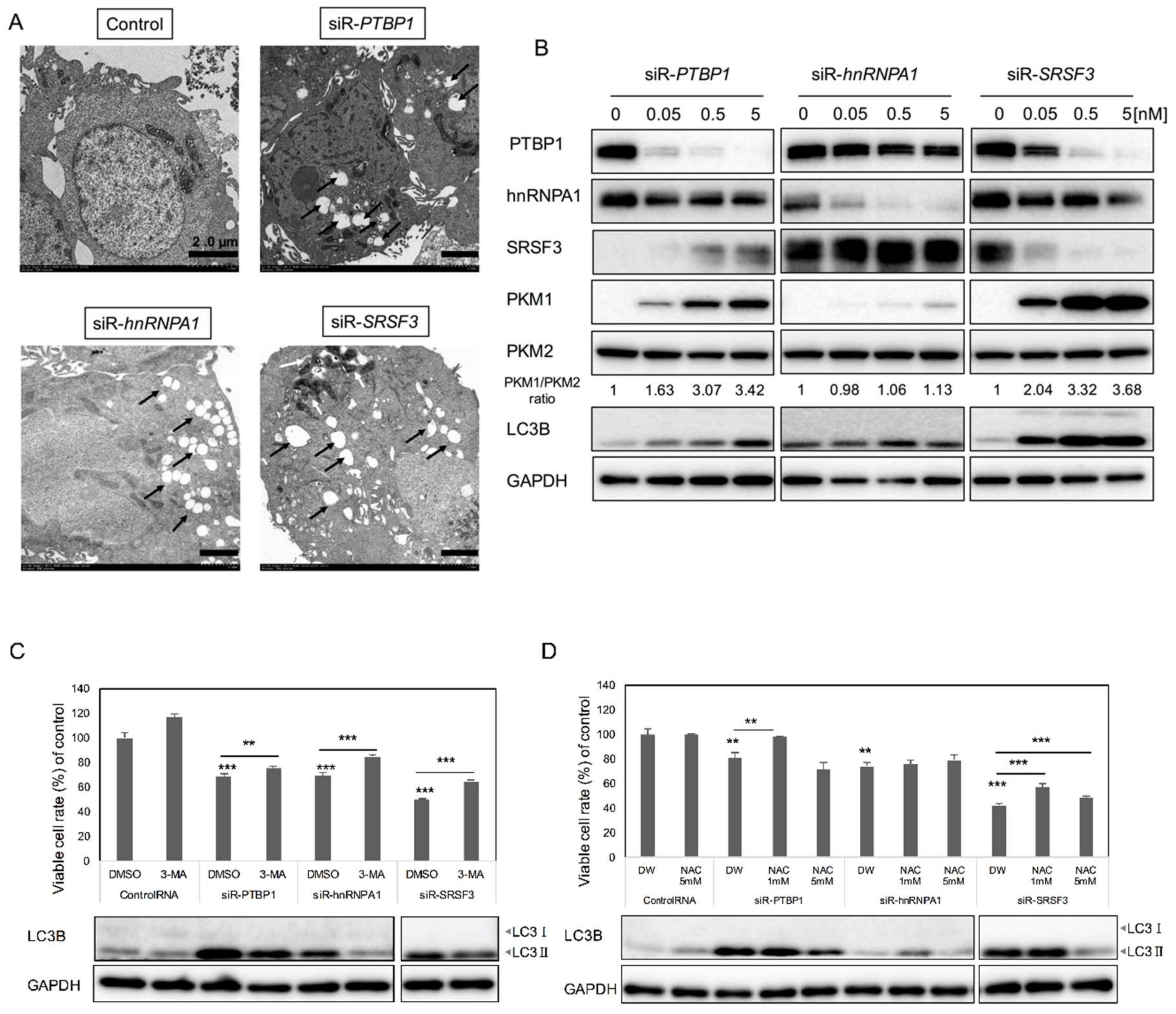
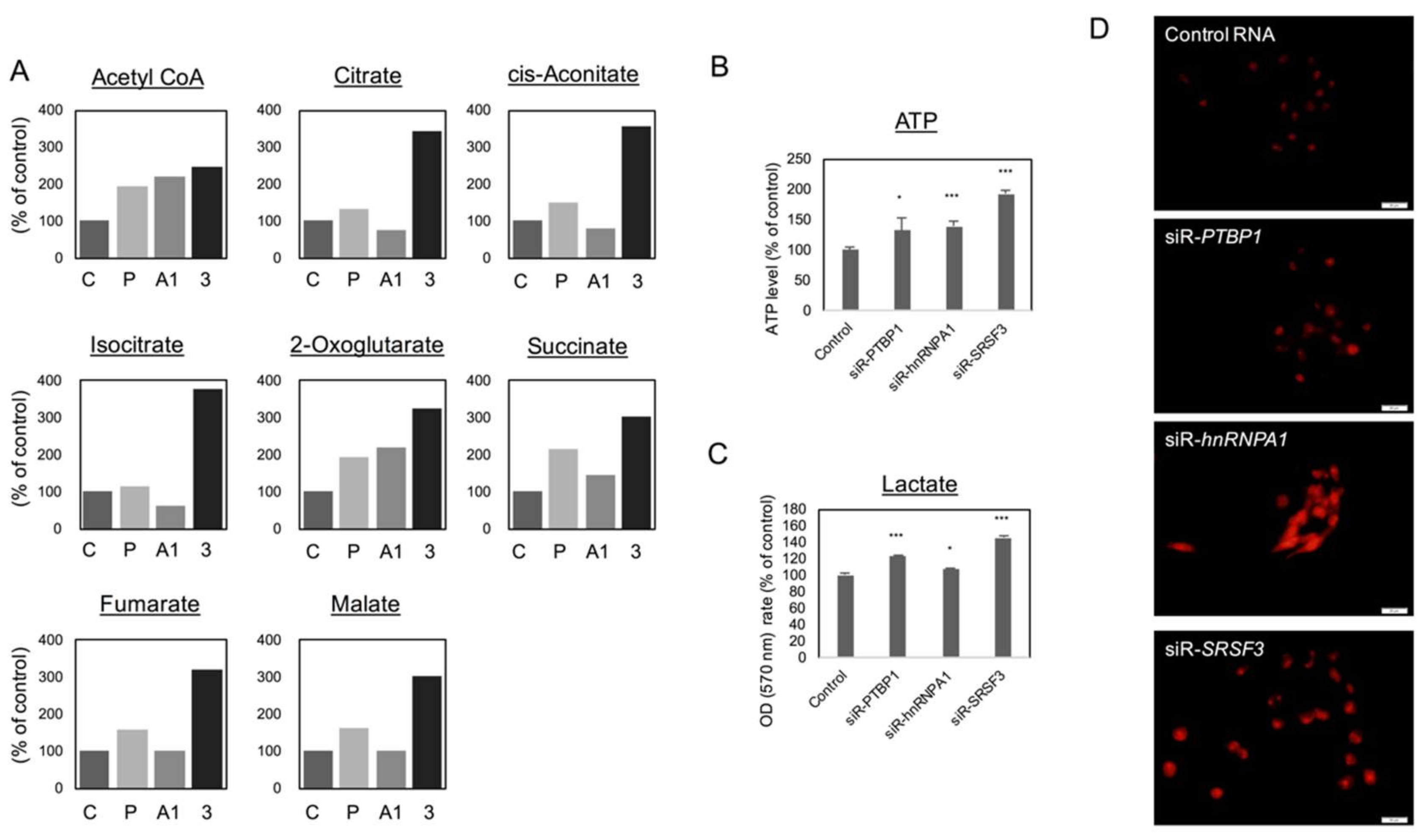
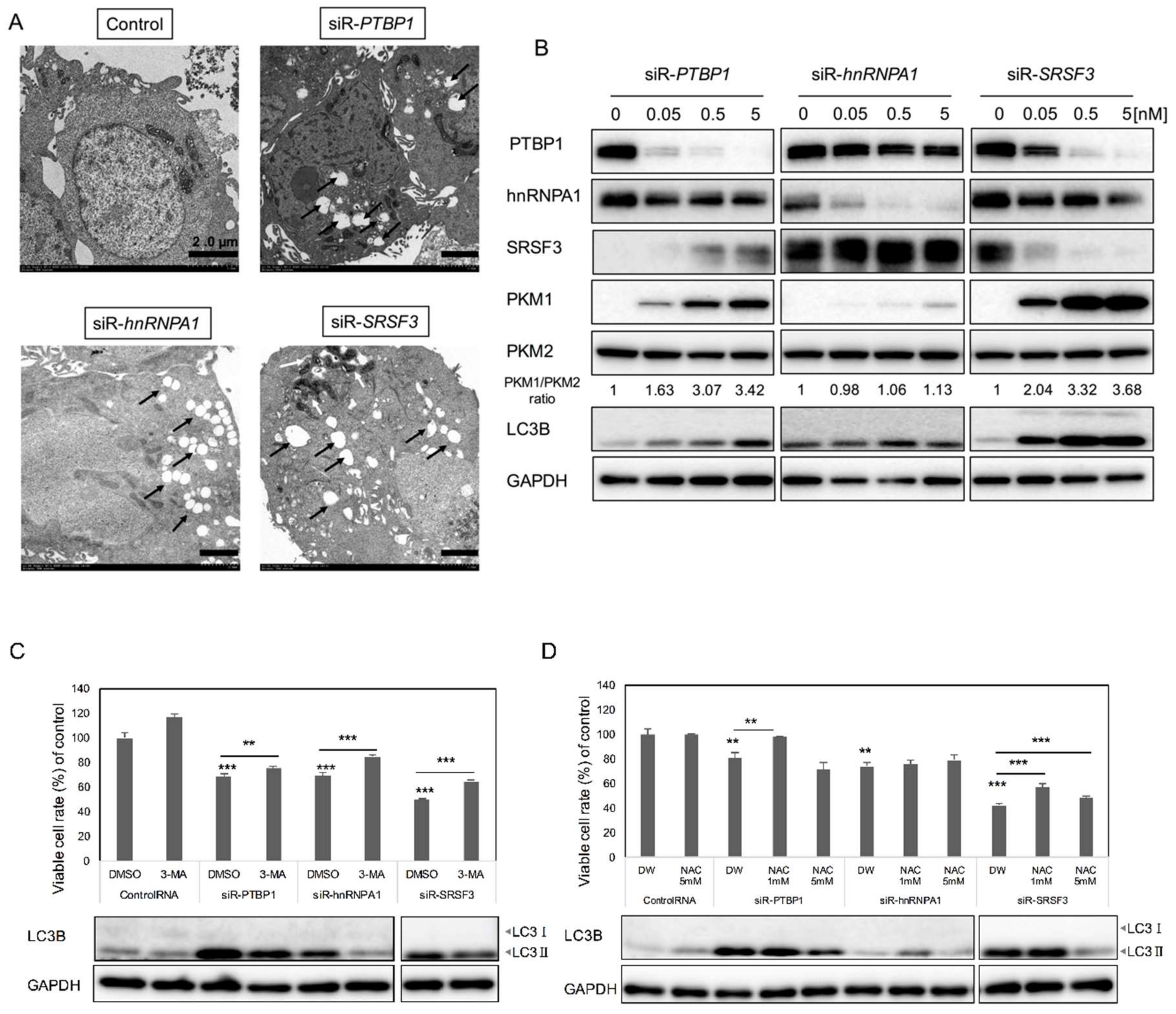
2.3. Silencing PTBP1, hnRNPA1, and SRSF3 Caused ROS-Induced Autophagic Cell Death in Colon Cancer Cells
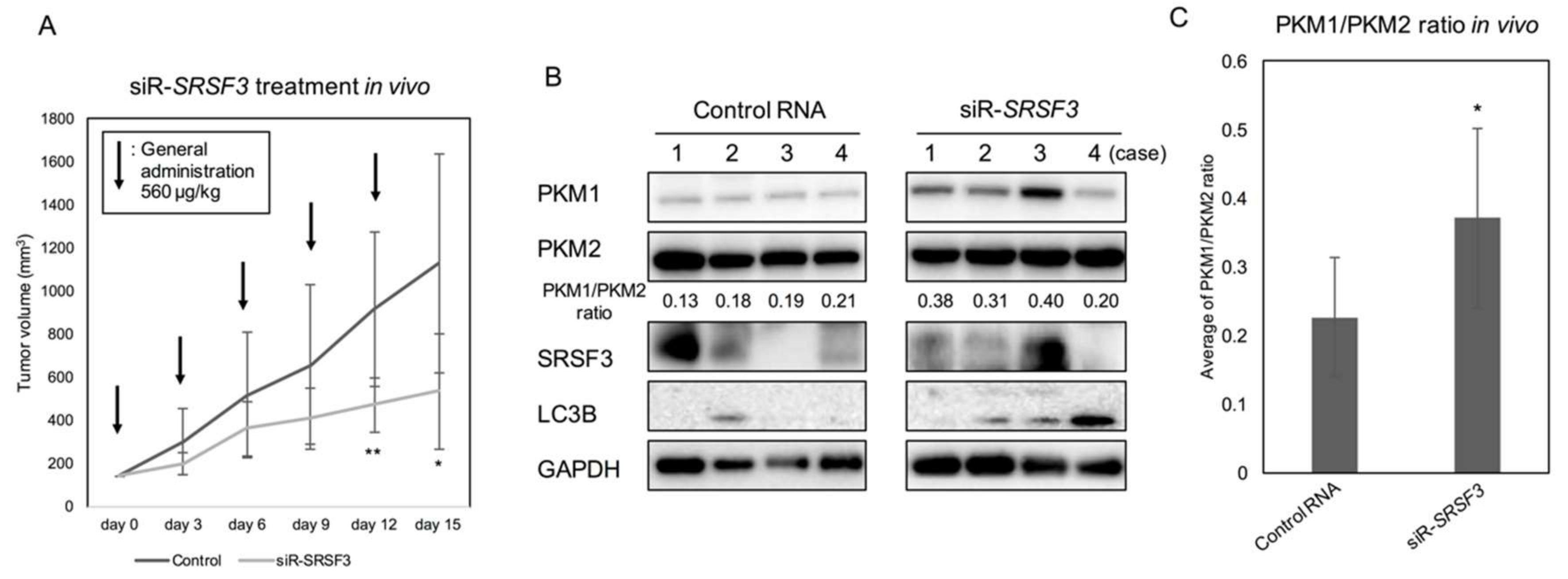
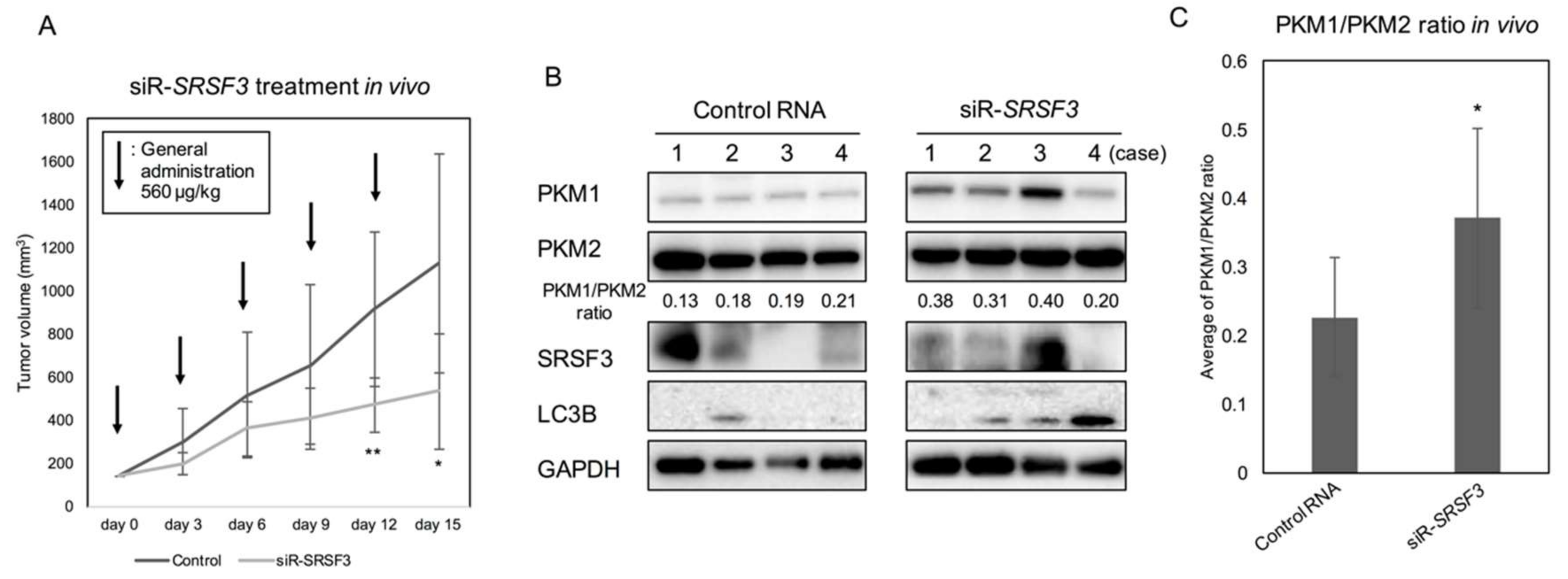
2.4. Anti-Tumor Effect by Silencing SRSF3 in Vivo
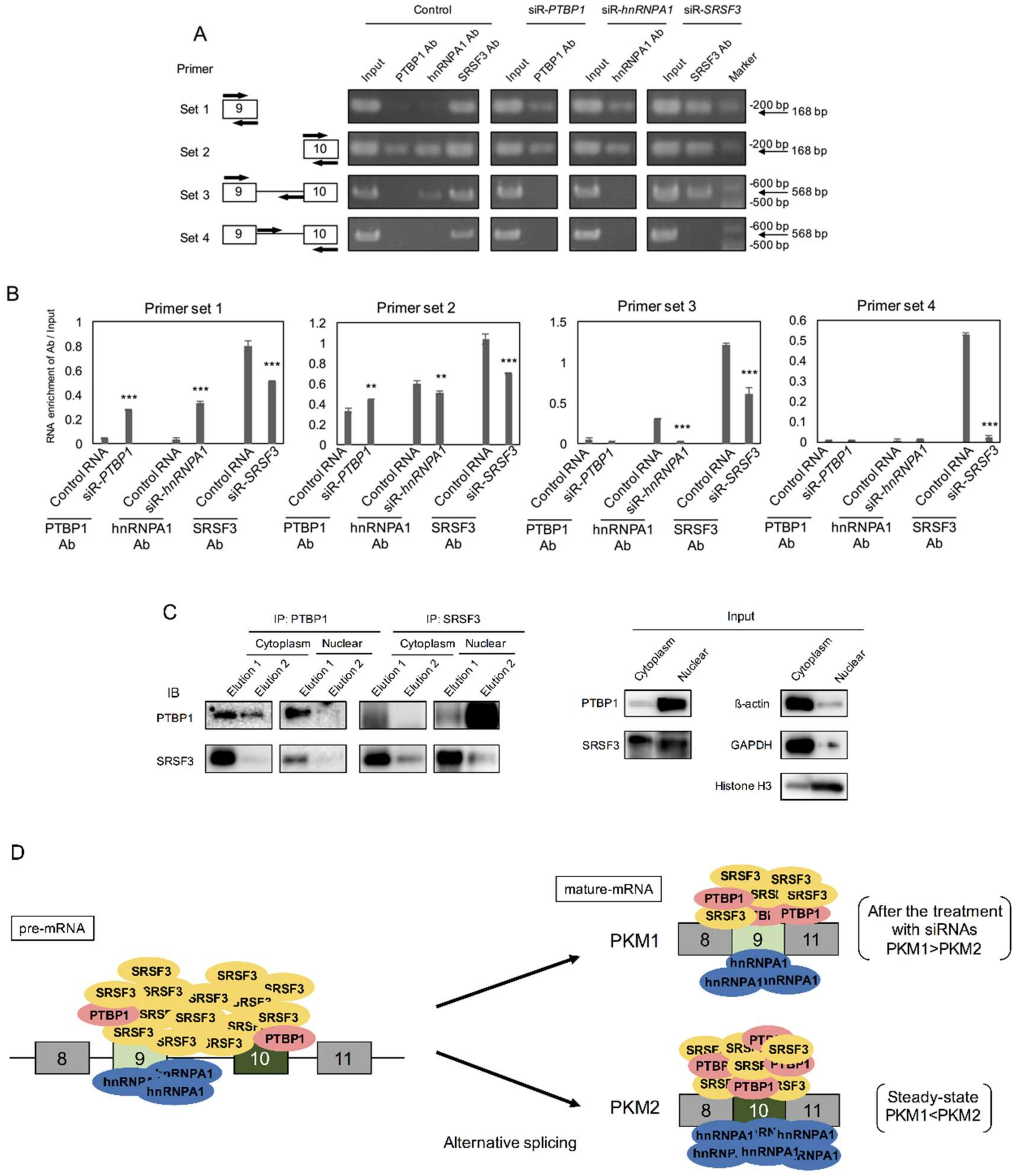
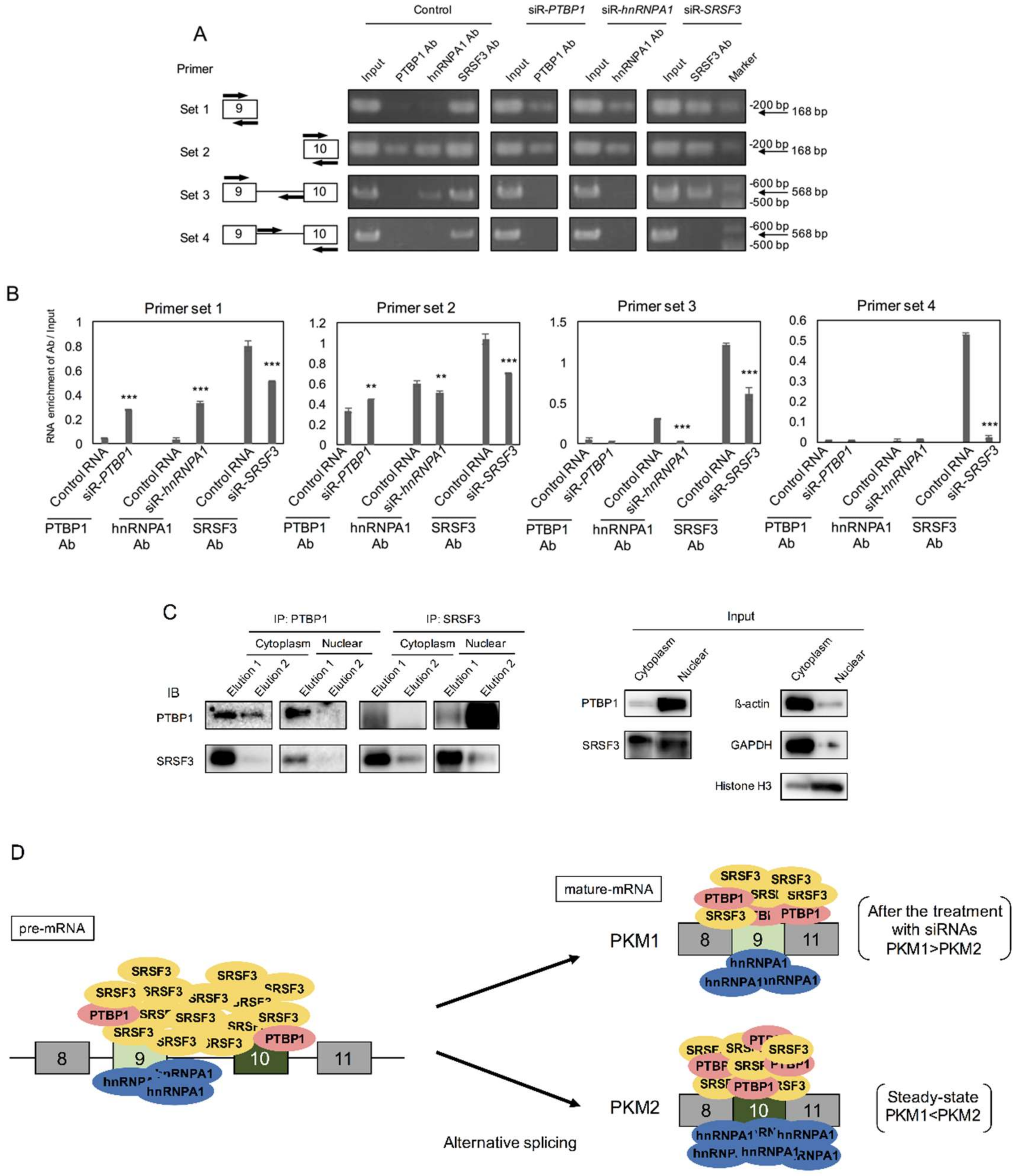
2.5. PTBP1 and SRSF3 Interacted with PKM mRNA
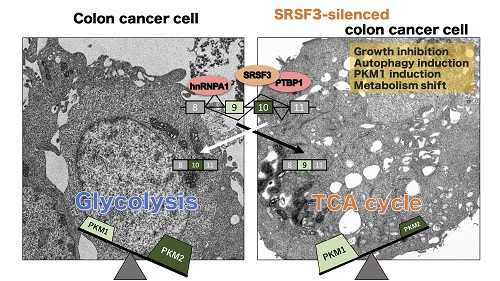
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Dataset Analysis
4.3. Cell Culture and Cell Viability
4.4. Transfection with Short-Interfering RNA for Each Splicer Cording Gene
4.5. Inhibitor Agents
4.6. Protein Extraction and Western Blot Analysis
4.7. RNA Extraction and Real-Time Reverse Transcription-PCR
4.8. Soft Agar Colony Formation Assay
4.9. Metabolome Analysis
4.10. ATP Assay
4.11. Lactate Assay
4.12. Morphological Assessment of ROS Production
4.13. Electron Microscopy Study
4.14. Human Tumor Xenograft Model
4.15. RIP Assay
4.16. Nuclear Protein Extraction and Crosslink-Immunoprecipitation
4.17. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SRSF3 | serine and arginine rich splicing factor 3 |
| SR | serine and arginine |
| PTBP1 | polypyrimidine tract binding protein 1 |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
| PKM | pyruvate kinase muscle |
| TCA | tricarboxylic acid |
| ADP | adenosine 5′-diphosphate |
| ATP | adenosine 5′-triphosphate |
| PEP | phosphoenolpyruvate |
| AS | alternative splicing |
| ESE | exonic splicing enhancer |
| OXPHOS | oxidative phosphorylation |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| ROS | reactive oxygen species |
| ESS | exonic splicing silencer |
| ISS | intronic splicing silencer |
| ISE | intronic splicing enhancer |
| FOXM1 | forkhead box protein M1 |
References
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, W.; Karl, P.; Erwin, N. Ueber den Stoffwechsel der Tumoren. Biochemische Zeitschrift 1924, 152, 319–344. [Google Scholar]
- Otto, W. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158. [Google Scholar] [CrossRef] [PubMed]
- Kiichi Imamura, T.T. Multimolecular forms of pyruvate kinase from rat and other mammalian tissues. The J. Biochem. 1972, 71, 1043–1051. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, T.; Yamada, K.; Inoue, H.; Matsuda, T.; Tanaka, T. The L- and R- type isozymes of rat pyruvate kinase are produced from a single gene by use of different promotors. J. Biol. Chem. 1987, 262, 14366–14371. [Google Scholar] [PubMed]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, J.; Feliu, J.E.; Marco, R.; Sols, A. Pyruvate kinase: Classes of regulatory isoenzymes in mammalian tissues. Eur. J. Biochem. 1973, 37, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbo, C.; Orru, S.; Salvatore, F. SRp20: An overview of its role in human diseases. Biochem. Biophys. Res. Commun. 2013, 436, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goina, E.; Skoko, N.; Pagani, F. Binding of DAZAP1 and hnRNPA1/A2 to an exonic splicing silencer in a natural BRCA1 exon 18 mutant. Mol. Cell Biol. 2008, 28, 3850–3860. [Google Scholar] [CrossRef] [PubMed]
- Xodo, L.; Paramasivam, M.; Membrino, A.; Cogoi, S. Protein hnRNPA1 binds to a critical G-rich element of KRAS and unwinds G-quadruplex structures: Implications in transcription. In Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 2008; pp. 159–160. [Google Scholar]
- Kim, J.; Park, R.Y.; Chen, J.K.; Kim, J.; Jeong, S.; Ohn, T. Splicing factor SRSF3 represses the translation of programmed cell death 4 mRNA by associating with the 5′-UTR region. Cell Death Differ. 2014, 21, 481–90. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chatterjee, D.; Jeon, H.Y.; Akerman, M.; Vander Heiden, M.G.; Cantley, L.C.; Krainer, A.R. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J. Mol. Cell Biol. 2012, 4, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Ito, Y.; Otsuki, Y.; Uno, B.; Uchiyama, K.; et al. MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer. Cancer Lett. 2015, 363, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Ito, Y.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Sugiyama, T.; Futamura, M.; Otsuki, Y.; et al. Organ-specific PTB1-associated microRNAs determine expression of pyruvate kinase isoforms. Sci. Rep. 2015, 5, 8647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-associated microRNA-1 and -133b suppress the Warburg effect in colorectal tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Taniguchi, K.; Matsuhashi, N.; Tajirika, T.; Futamura, M.; Takai, T.; Akao, Y.; Yoshida, K. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016, 107, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Koichiro, M.; Kohei, T.; Nobuhiko, S.; Yuki, K.; Teruo, I.; Kiyoshi, T.; Tomoaki, T.; Yuki, Y.; Satoshi, K.; Yukihiro, A.; et al. MiR-145 negatively regulates Warburg effect by silencing KLF4 and PTBP1 in bladder cancer cells. Oncotarget 2017, 8, 33064. [Google Scholar]
- Takai, T.; Yoshikawa, Y.; Inamoto, T.; Minami, K.; Taniguchi, K.; Sugito, N.; Kuranaga, Y.; Shinohara, H.; Kumazaki, M.; Tsujino, T.; et al. A Novel Combination RNAi toward Warburg Effect by Replacement with miR-145 and Silencing of PTBP1 Induces Apoptotic Cell Death in Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Downey, T.; Eu, K.W.; Koh, P.K.; Cheah, P.Y. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin. Exp. Metastasis 2010, 27, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzypczak, M.; Goryca, K.; Rubel, T.; Paziewska, A.; Mikula, M.; Jarosz, D.; Pachlewski, J.; Oledzki, J.; Ostrowski, J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS ONE 2010, 5, e13091. [Google Scholar] [CrossRef]
- Alon, U.; Barkai, N.; Notterman, D.A.; Gish, K.; Ybarra, S.; Mack, D.; Levine, A.J. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1999, 96, 6745–6750. [Google Scholar] [Green Version]
- Clower, C.V.; Chatterjee, D.; Wang, Z.; Cantley, L.C.; Vander Heiden, M.G.; Krainer, A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 1894–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Jia, J.; Jia, R. PTBP1 and PTBP2 impaired autoregulation of SRSF3 in cancer cells. Sci. Rep. 2015, 5, 14548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.-X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induc- tion and maintenance. Int. J. Biol. Sci. 2010, 6, 806. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Gao, W.; Ho, W.K.; Lei, W.B.; Wei, W.I.; Chan, J.Y.; Wong, T.S. The clinical association of programmed cell death protein 4 (PDCD4) with solid tumors and its prognostic significance: A meta-analysis. Chin. J. Cancer 2016, 35, 95. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Jeong, S. SRSF3 represses the expression of PDCD4 protein by coordinated regulation of alternative splicing, export and translation. Biochem. Biophys. Rse. Commun. 2016, 470, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; David, C.J.; Manley, J.L. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat. Struct. Mol. Biol. 2012, 19, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarkavelis, G.; Boussios, S.; Papadaki, A.; Katsanos, K.H.; Christodoulou, D.K.; Pentheroudakis, G. Current and future biomarkers in colorectal cancer. Ann. Gastroenterol. 2017, 30, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Kuranaga, Y.; Yamada, N.; Kashiwaya, M.; Nakamura, M.; Cui, L.; Kumazaki, M.; Shinohara, H.; Sugito, N.; Taniguchi, K.; Ito, Y.; et al. Anti-Oncogenic gem-Dihydroperoxides Induce Apoptosis in Cancer Cells by Trapping Reactive Oxygen Species. Int. J. Mol. Sci. 2016, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Sugito, N.; Taniguchi, K.; Kuranaga, Y.; Ohishi, M.; Soga, T.; Ito, Y.; Miyachi, M.; Kikuchi, K.; Hosoi, H.; Akao, Y. Cancer-Specific Energy Metabolism in Rhabdomyosarcoma Cells Is Regulated by MicroRNA. Nucleic Acid Ther. 2017, 27, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2003, 2, 488–494. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age | Sex a | Site b | Size c | Depth d | Stage e |
|---|---|---|---|---|---|---|
| 1 | 41 | M | RS | 70×45 | SS | B |
| 2 | 49 | F | R | 50×70 | SS | B |
| 3 | 42 | M | R | 30×20 | SS | B |
| 4 | 68 | M | R | 65×60 | SE | D |
| 5 | 73 | M | R | 40×40 | MP | A |
| 6 | 56 | F | S | 27×24 | SS | C |
| 7 | 64 | M | R | 28×22 | MP | A |
| 8 | 82 | F | A | 33×28 | MP | A |
| 9 | 62 | M | S | 55×40 | SS | D |
| 10 | 68 | F | R | 35×35 | SS | B |
| Case | Sex a | Site b | Size c | Grade d |
|---|---|---|---|---|
| 1 | F | S | 20 | High |
| 2 | F | S | 20 | Low |
| 3 | F | A | 15 | Low |
| 4 | M | S | 15 | Low |
| 5 | M | R | 10 | High |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuranaga, Y.; Sugito, N.; Shinohara, H.; Tsujino, T.; Taniguchi, K.; Komura, K.; Ito, Y.; Soga, T.; Akao, Y. SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3012. https://doi.org/10.3390/ijms19103012
Kuranaga Y, Sugito N, Shinohara H, Tsujino T, Taniguchi K, Komura K, Ito Y, Soga T, Akao Y. SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells. International Journal of Molecular Sciences. 2018; 19(10):3012. https://doi.org/10.3390/ijms19103012
Chicago/Turabian StyleKuranaga, Yuki, Nobuhiko Sugito, Haruka Shinohara, Takuya Tsujino, Kohei Taniguchi, Kazumasa Komura, Yuko Ito, Tomoyoshi Soga, and Yukihiro Akao. 2018. "SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells" International Journal of Molecular Sciences 19, no. 10: 3012. https://doi.org/10.3390/ijms19103012
APA StyleKuranaga, Y., Sugito, N., Shinohara, H., Tsujino, T., Taniguchi, K., Komura, K., Ito, Y., Soga, T., & Akao, Y. (2018). SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells. International Journal of Molecular Sciences, 19(10), 3012. https://doi.org/10.3390/ijms19103012