MicroRNA-204 Is Necessary for Aldosterone-Stimulated T-Type Calcium Channel Expression in Cardiomyocytes

 , and
, and 
Abstract
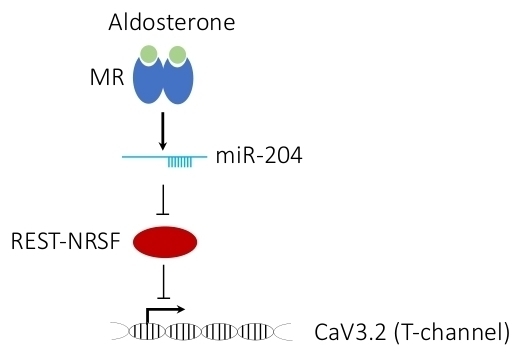
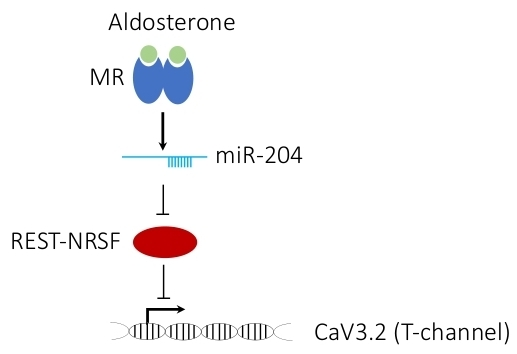
1. Introduction
2. Results
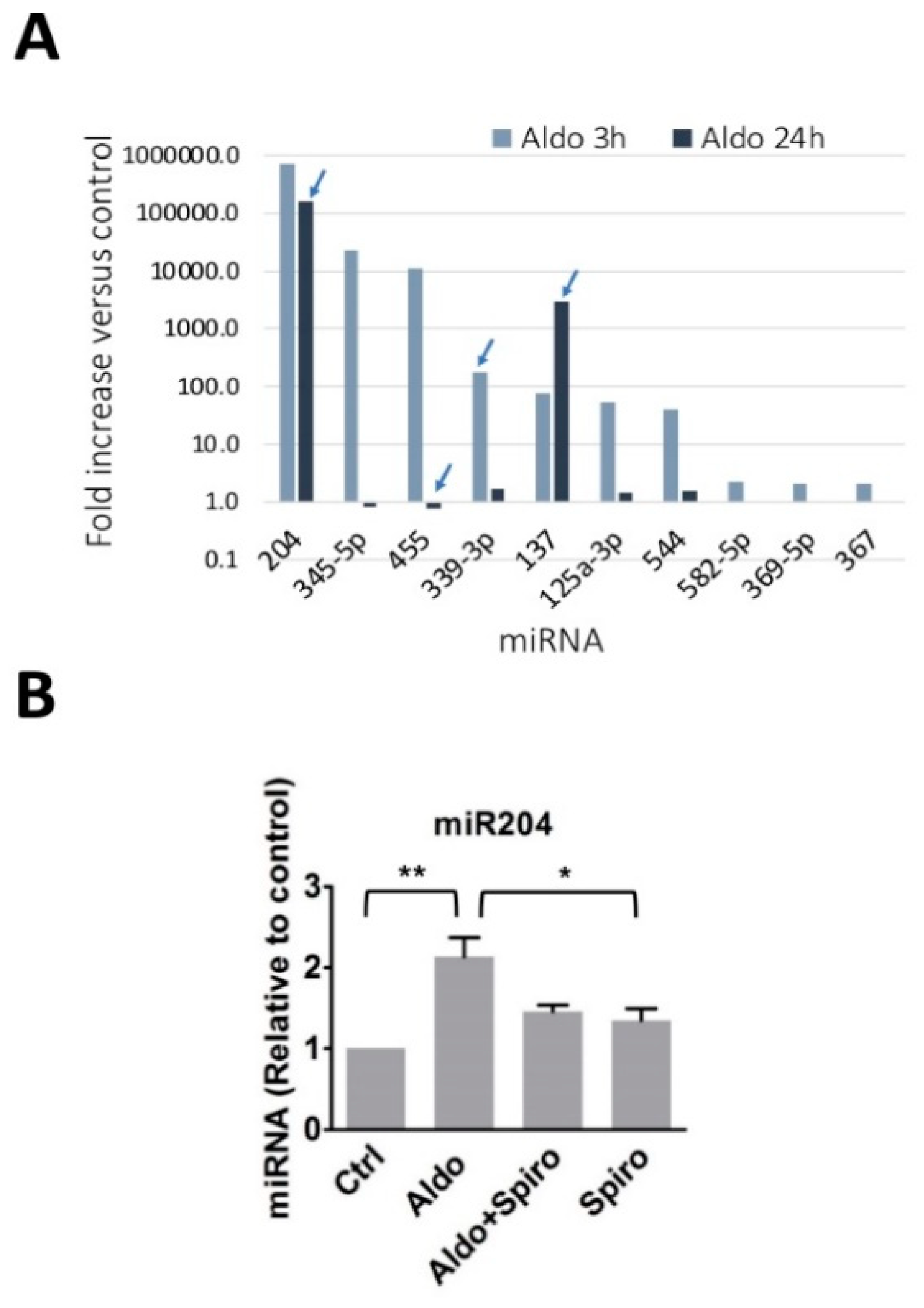
2.1. Aldosterone Stimulates miR-204 Expression
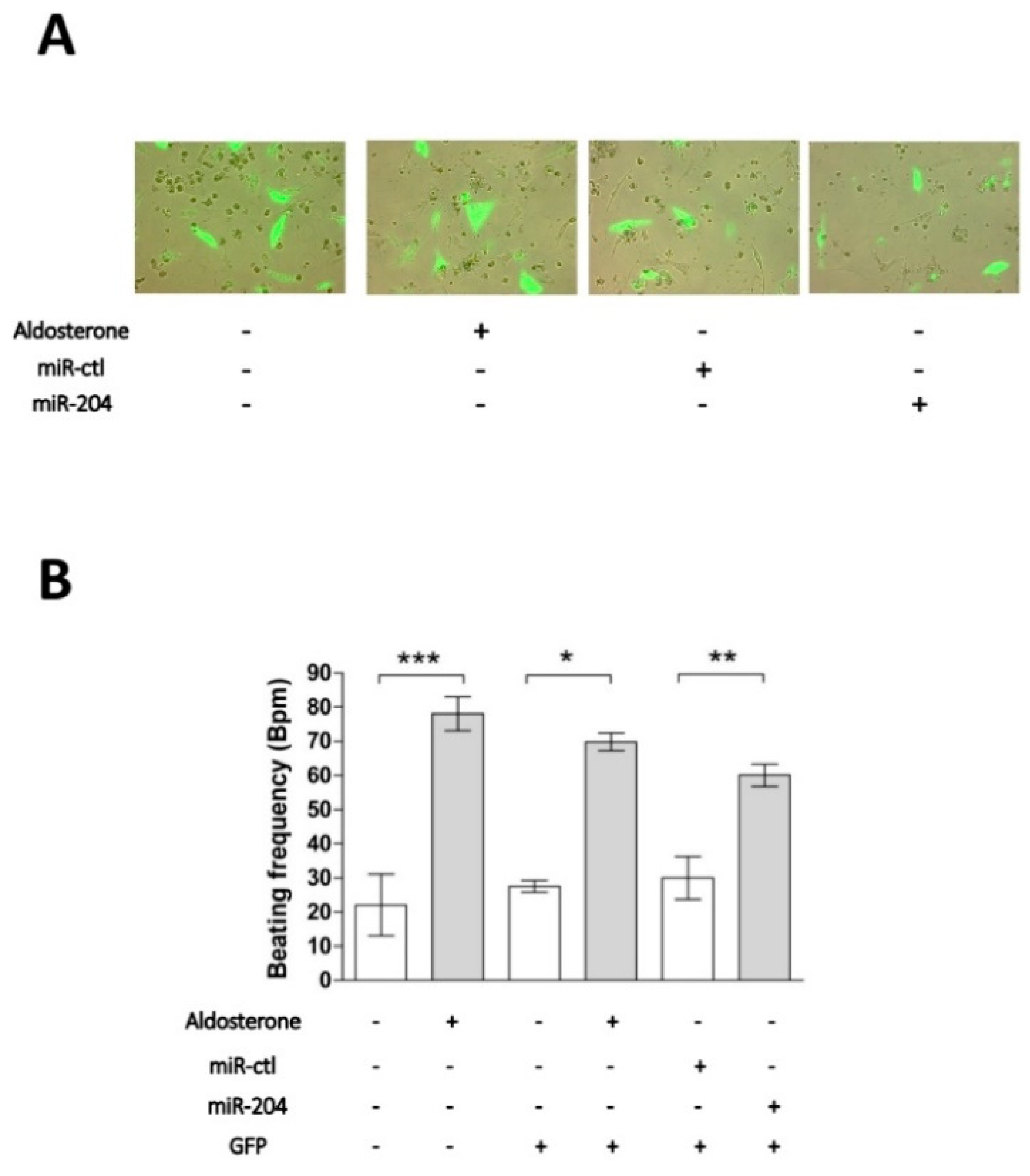
2.2. Chronotropic Action of miR-204 in Isolated Cardiomyocytes
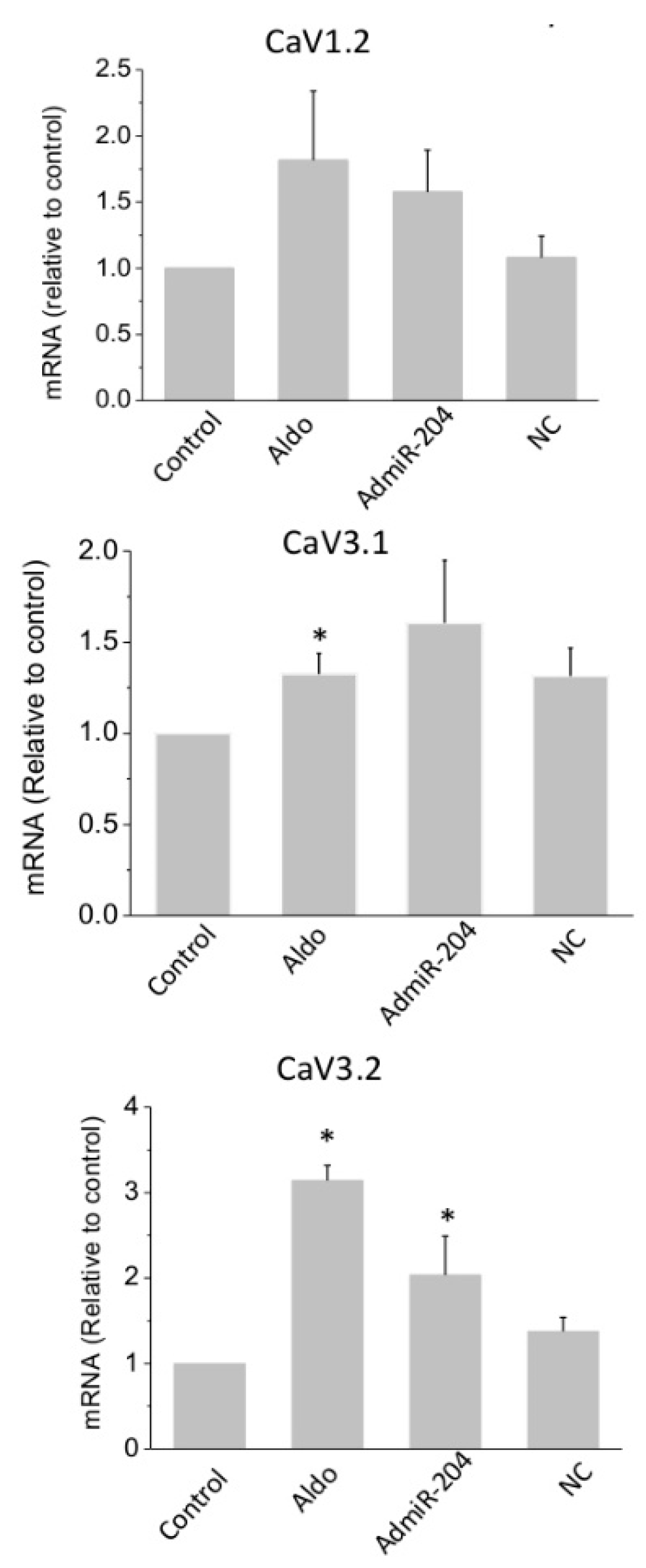
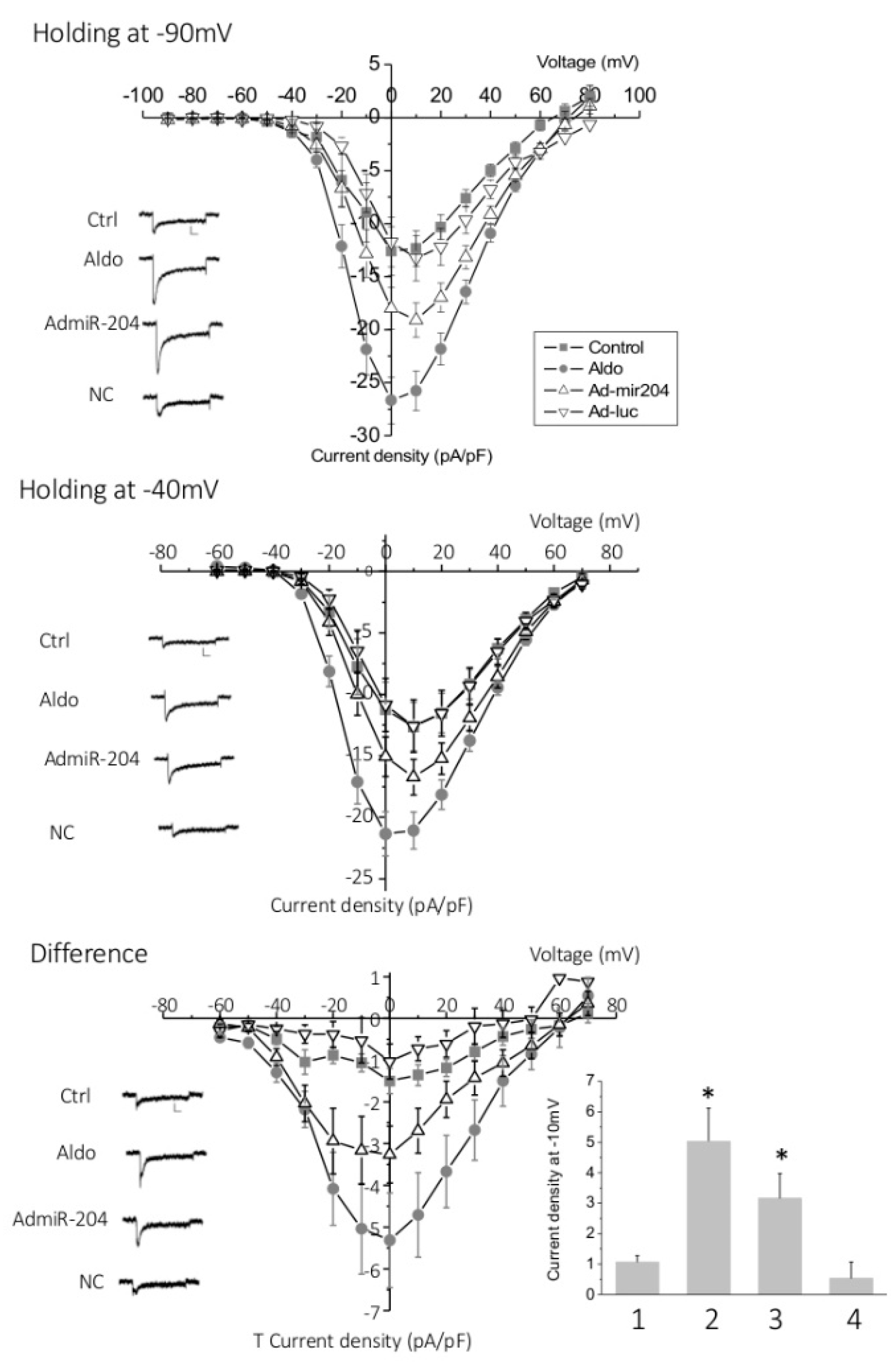
2.3. T-Type Ca2+ Channel Expression in miR-204-Overexpressing Cardiomyocytes
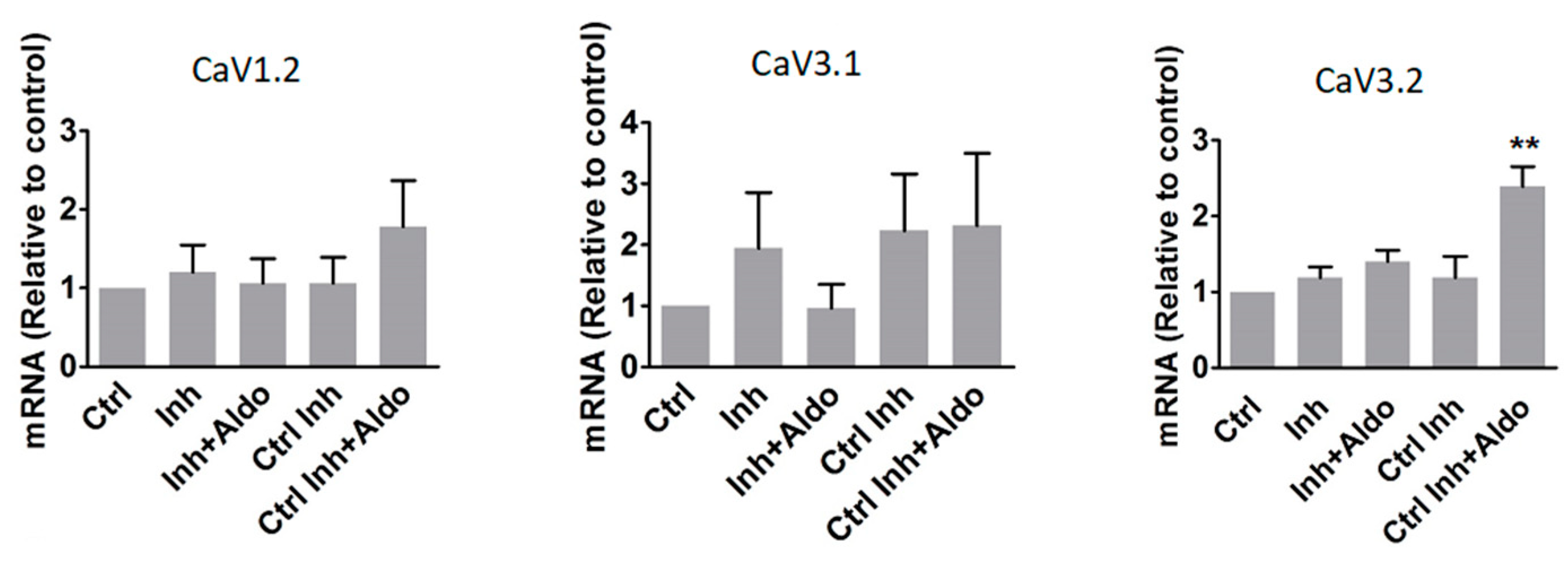
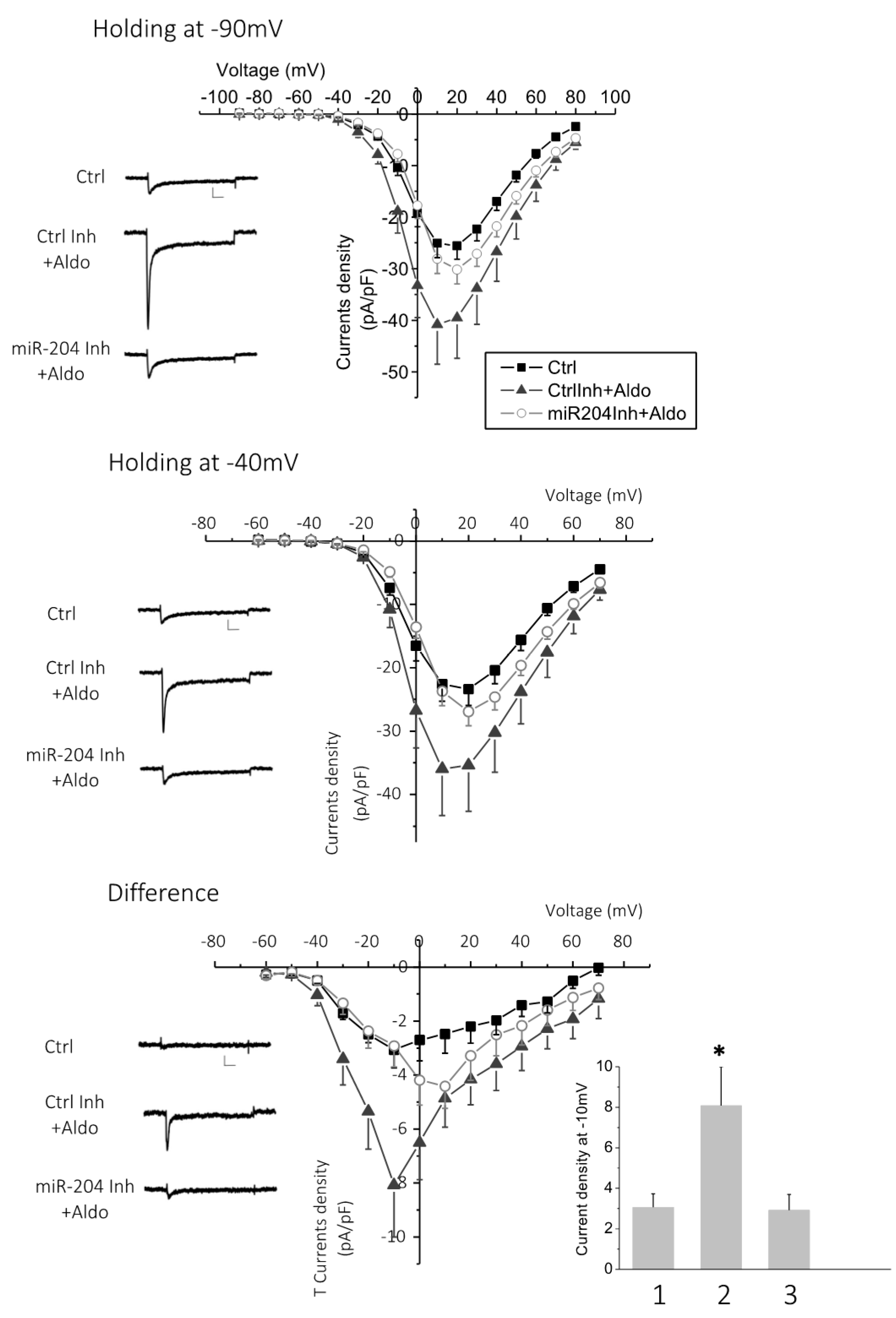
2.4. MicroRNA-204 Is Necessary for the Aldosterone-Induced Ca2+ Channel Upregulation
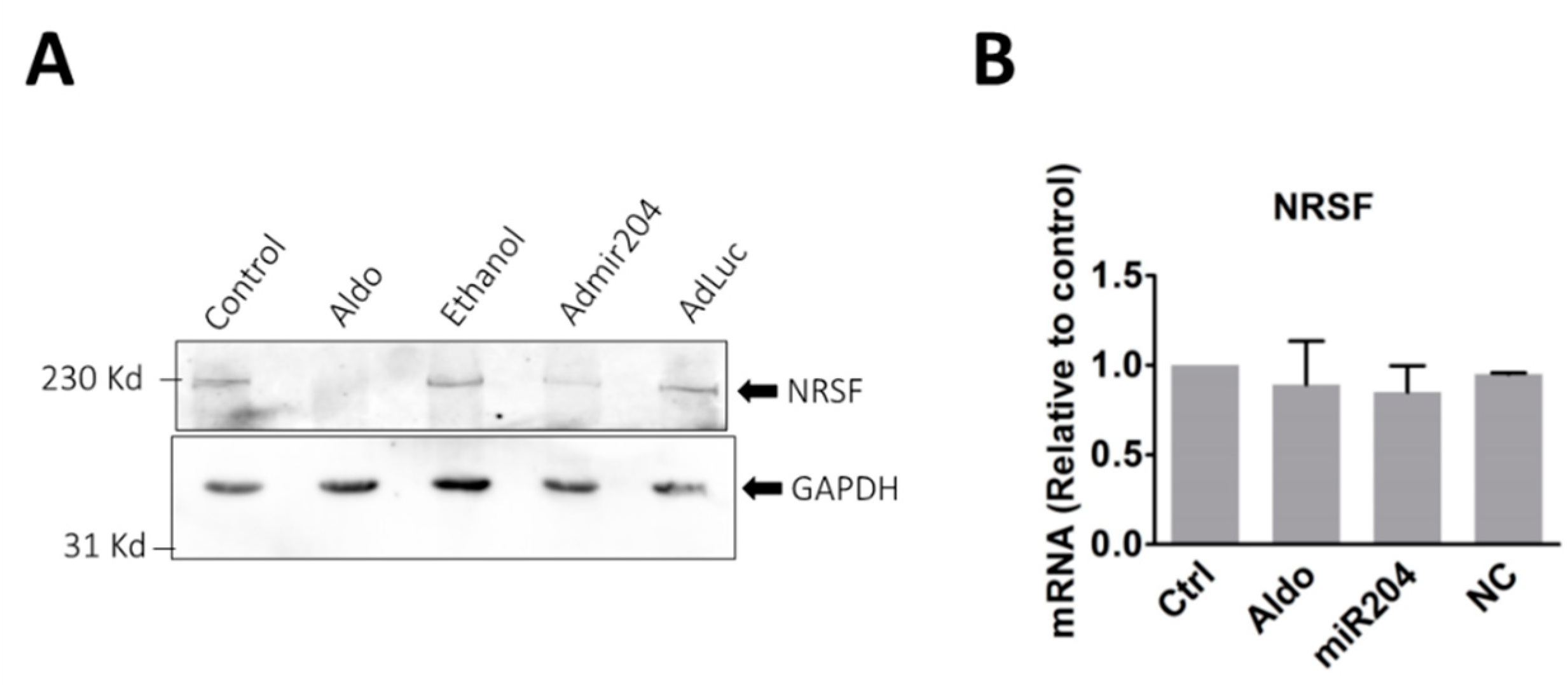
2.5. MicroRNA-204 Mimics the Aldosterone-Induced Downregulation of NRSF
3. Materials and Methods
3.1. Experimental Animals
3.2. Cell Culture, Stimulation, and Transfection
3.3. MicroRNA Preparation and Screening by Microarray
3.4. RNA Isolation and Real-Time Quantitative Reverse Transcription (RT)-Polymerase Chain Reaction (PCR)
3.5. Protein Isolation and Western Blot
3.6. Patch Clamp
3.7. Data Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Duprez, D.A.; Bauwens, F.R.; De Buyzere, M.L.; De Backer, T.L.; Kaufman, J.M.; Van Hoecke, J.; Vermeulen, A.; Clement, D.L. Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am. J. Cardiol. 1993, 71, 17a–20a. [Google Scholar] [CrossRef]
- Swedberg, K.; Eneroth, P.; Kjekshus, J.; Wilhelmsen, L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990, 82, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Sacchetto, A.; Visentin, P.; Canali, C.; Graniero, G.R.; Palatini, P.; Pessina, A.C. Changes in left ventricular anatomy and function in hypertension and primary aldosteronism. Hypertension 1996, 27, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Bathgate-Siryk, A.; Dabul, S.; Pandya, K.; Walklett, K.; Rengo, G.; Cannavo, A.; De Lucia, C.; Liccardo, D.; Gao, E.; Leosco, D.; et al. Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension 2014, 63, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Koch, W.J. Adrenal beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J. Am. Coll. Cardiol. 2011, 57, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Boixel, C.; Gavillet, B.; Rougier, J.S.; Abriel, H. Aldosterone increases voltage-gated sodium current in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2257–H2266. [Google Scholar] [CrossRef] [PubMed]
- Perrier, E.; Kerfant, B.G.; Lalevee, N.; Bideaux, P.; Rossier, M.F.; Richard, S.; Gómez, A.M.; Benitah, J.P. Mineralocorticoid receptor antagonism prevents the electrical remodeling that precedes cellular hypertrophy after myocardial infarction. Circulation 2004, 110, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Benitah, J.P.; Vassort, G. Aldosterone upregulates Ca2+ current in adult rat cardiomyocytes. Circ. Res. 1999, 85, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Lalevee, N.; Rebsamen, M.C.; Barrere-Lemaire, S.; Perrier, E.; Nargeot, J.; Benitah, J.P.; Rossier, M.F. Aldosterone increases T-type calcium channel expression and in vitro beating frequency in neonatal rat cardiomyocytes. Cardiovasc. Res. 2005, 67, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Maturana, A.; Lenglet, S.; Python, M.; Kuroda, S.; Rossier, M.F. Role of the T-Type Calcium Channel Ca(V)3.2 in the Chronotropic Action of Corticosteroids in Isolated Rat Ventricular Myocytes. Endocrinology 2009, 150, 3726–3734. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Yang, J.; Yao, Y.; Li, H.; Chen, Y.; Zhang, J.; Huang, C. Aldosterone modulates I(f) current through gene expression in cultured neonatal rat ventricular myocytes. Mol. Med. Rep. 2011, 4, 569–573. [Google Scholar] [PubMed]
- Rossier, M.F.; Python, M.; Maturana, A.D. Contribution of Mineralocorticoid and Glucocorticoid Receptors to the Chronotropic and Hypertrophic Actions of Aldosterone in Neonatal Rat Ventricular Myocytes. Endocrinology 2010, 151, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, G.F.; Marban, E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 1999, 42, 270–283. [Google Scholar] [CrossRef]
- Ferron, L.; Ruchon, Y.; Renaud, J.F.; Capuano, V. T-type Ca2+ signalling regulates aldosterone-induced CREB activation and cell death through PP2A activation in neonatal cardiomyocytes. Cardiovasc. Res. 2011, 90, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Irisawa, H.; Kameyama, M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 1988, 395, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Vassort, G.; Talavera, K.; Alvarez, J.L. Role of T-type Ca2+ channels in the heart. Cell Calcium 2006, 40, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Iijima, T. Pathophysiological significance of T-type Ca2+ channels: Properties and functional roles of T-type Ca2+ channels in cardiac pacemaking. J. Pharm. Sci. 2005, 99, 197–204. [Google Scholar] [CrossRef]
- Chiang, C.S.; Huang, C.H.; Chieng, H.L.; Chang, Y.T.; Chang, D.R.; Chen, J.J.; Chen, Y.C.; Chen, Y.H.; Shin, H.S.; Campbell, K.P.; et al. The Ca(v)3.2 T-Type Ca2+ Channel Is Required for Pressure Overload-Induced Cardiac Hypertrophy in Mice. Circ. Res. 2009, 2009. 104, 522–530. [Google Scholar] [CrossRef]
- Horiba, M.; Muto, T.; Ueda, N.; Opthof, T.; Miwa, K.; Hojo, M.; Lee, J.K.; Kamiya, K.; Kodama, I.; Yasui, K. T-type Ca2+ channel blockers prevent cardiac cell hypertrophy through an inhibition of calcineurin-NFAT3 activation as well as L-type Ca2+ channel blockers. Life Sciences 2008, 82, 554–560. [Google Scholar] [CrossRef] [PubMed]
- BenMohamed, F.; Ferron, L.; Ruchon, Y.; Gouadon, E.; Renaud, J.F.; Capuano, V. Regulation of T-type Cav3.1 channels expression by synthetic glucocorticoid dexamethasone in neonatal cardiac myocytes. Mol. Cell. Biochem. 2010, 335, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Zardo, G.; Ciolfi, A.; Vian, L.; Billi, M.; Racanicchi, S.; Grignani, F.; Nervi, C. Transcriptional targeting by microRNA-Polycomb complexes A novel route in cell fate determination. Cell Cycle 2012, 11, 3543–3549. [Google Scholar] [CrossRef] [PubMed]
- Hata, A. Functions of microRNAs in cardiovascular biology and disease. Annu. Rev. Physiol. 2013, 75, 69–93. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.; Bourajjaj, M.; Gladka, M.; Kortland, M.; van Oort, R.J.; Pinto, Y.M.; Molkentin, J.D.; De Windt, L.J. Conditional Dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 2008, 118, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Hong, C.; Chen, I.Y.; Lypowy, J.; Abdellatif, M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clinic. Invest. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.Q.; Murtaza, I.; Wang, K.; Jiao, J.Q.; Gao, J.; Li, P.F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. PNAS 2009, 106, 12103–12108. [Google Scholar] [CrossRef] [PubMed]
- Drawnel, F.M.; Wachten, D.; Molkentin, J.D.; Maillet, M.; Aronsen, J.M.; Swift, F.; Sjaastad, I.; Liu, N.; Catalucci, D.; Mikoshiba, K.; et al. Mutual antagonism between IP3RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell Biol. 2012, 199, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Saito, Y.; Takano, M.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Takahashi, N.; Adachi, Y.; Takemura, G.; Horie, M.; et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003, 22, 6310–6321. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.; Pearman, CM.; Wang, Y.; Nakao, S.; Logantha, S.J.R.J.; Cox, C.; Bennett, H.; Zhang, Y.; Johnsen, A.B.; Linscheid, N.; et al. Targeting miR-423-5p Reverses Exercise Training-Induced HCN4 Channel Remodeling and Sinus Bradycardia. Circ. Res. 2017, 121, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Barana, A.; Matamoros, M.; Dolz-Gaitón, P.; Pérez-Hernández, M.; Amorós, I.; Núñez, M.; Sacristán, S.; Pedraz, Á.; Pinto, Á.; Fernández-Avilés, F.; et al. Chronic atrial fibrillation increases microRNA-21 in human atrial myocytes decreasing L-type calcium current. Circ. Arrhythm Electrophysiol. 2014, 7, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Daimi, H.; Lozano-Velasco, E.; Haj Khelil, A.; Chibani, J.B.; Barana, A.; González de la Fuente, M.; Caballero, R.; Aranega, A.; Franco, D. Regulation of SCN5A by microRNAs: MiR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm 2015, 12, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Paylakhi, S.H.; Moazzeni, H.; Yazdani, S.; Rassouli, P.; Arefian, E.; Jaberi, E.; Arash, E.H.; Gilani, A.S.; Fan, J.B.; April, C.; et al. FOXC1 in human trabecular meshwork cells is involved in regulatory pathway that includes miR-204, MEIS2, and ITG β1. Exp. Eye Res. 2013, 111, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Chen, J.Q.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nature Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.P.; Syed, M.; Marañon, R.O.; Hall, M.E.; Reckelhoff, J.F.; Yanes Cardozo, L.L.; Romero, D.G. Role and Regulation of MicroRNAs in Aldosterone-Mediated Cardiac Injury and Dysfunction in Male Rats. Endocrinology 2017, 158, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.J.; Liang, D.D.; Zhang, H.; Liu, Y.; Zhang, D.S.; Liu, Y.; Pan, L.; Chen, X.; Doevendans, P.A.; Sun, Y.; et al. MicroRNA-204 is required for differentiation of human-derived cardiomyocyte progenitor cells. J. Mol. Cell. Card. 2012, 53, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.D.; Li, J.; Wu, Y.H.; Zhen, L.X.; Li, C.M.; Qi, M.; Wang, L.; Deng, F.; Huang, J.; Lv, F.; et al. miRNA-204 drives cardiomyocyte proliferation via targeting Jarid2. Inter. J. Card. 2015, 201, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J.; et al. TRPM3 and miR-204 Establish a Regulatory Circuit that Controls Oncogenic Autophagy in Clear Cell Renal Cell Carcinoma. Cancer Cell 2014, 26, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, L.A.; Reichelt, M.E.; Delbridge, L.M.D.; Young, M.J. Mineralocorticoid receptors and the heart, multiple cell types and multiple mechanisms: A focus on the cardiomyocyte. Clin. Sci. 2013, 125, 409–421. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Oligonucleotide Sequence |
|---|---|
| α1C | Fw: 5′-AGCAACTTCCCTCAGACGTTTG |
| Rev: 5′-GCTTCTCATGGGACGGTGAT | |
| α1G | Fw: 5′-ACGCTGAGTCTCTCTGGTTTGTC |
| Rev: 5′-TGCTTACGTGGGACTTTTCAGA | |
| α1H | Fw: 5′-GGCGAAGAAGGCAAAGATGA |
| Rev: 5′-GCGTGACACTGGGCATGTT | |
| NRSF | Fw: 5′-GGCCAAACCCTTCCGTTGT |
| Rev: 5′- TGGCTTGCTTCTCTGCACT | |
| GAPDH | Fw: 5′-CAACTCCCTCAAGATTGTCAGCAA |
| Rev: 5′-GGCATGGACTGTGGTCATGA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koyama, R.; Mannic, T.; Ito, J.; Amar, L.; Zennaro, M.-C.; Rossier, M.F.; Maturana, A.D. MicroRNA-204 Is Necessary for Aldosterone-Stimulated T-Type Calcium Channel Expression in Cardiomyocytes. Int. J. Mol. Sci. 2018, 19, 2941. https://doi.org/10.3390/ijms19102941
Koyama R, Mannic T, Ito J, Amar L, Zennaro M-C, Rossier MF, Maturana AD. MicroRNA-204 Is Necessary for Aldosterone-Stimulated T-Type Calcium Channel Expression in Cardiomyocytes. International Journal of Molecular Sciences. 2018; 19(10):2941. https://doi.org/10.3390/ijms19102941
Chicago/Turabian StyleKoyama, Riko, Tiphaine Mannic, Jumpei Ito, Laurence Amar, Maria-Christina Zennaro, Michel Florian Rossier, and Andrés Daniel Maturana. 2018. "MicroRNA-204 Is Necessary for Aldosterone-Stimulated T-Type Calcium Channel Expression in Cardiomyocytes" International Journal of Molecular Sciences 19, no. 10: 2941. https://doi.org/10.3390/ijms19102941
APA StyleKoyama, R., Mannic, T., Ito, J., Amar, L., Zennaro, M.-C., Rossier, M. F., & Maturana, A. D. (2018). MicroRNA-204 Is Necessary for Aldosterone-Stimulated T-Type Calcium Channel Expression in Cardiomyocytes. International Journal of Molecular Sciences, 19(10), 2941. https://doi.org/10.3390/ijms19102941