Development of Antibody Therapeutics against Flaviviruses


Abstract
1. Introduction
2. Monoclonal Antibody as Therapy
3. Potential Therapeutic Antibodies against Flavivirus
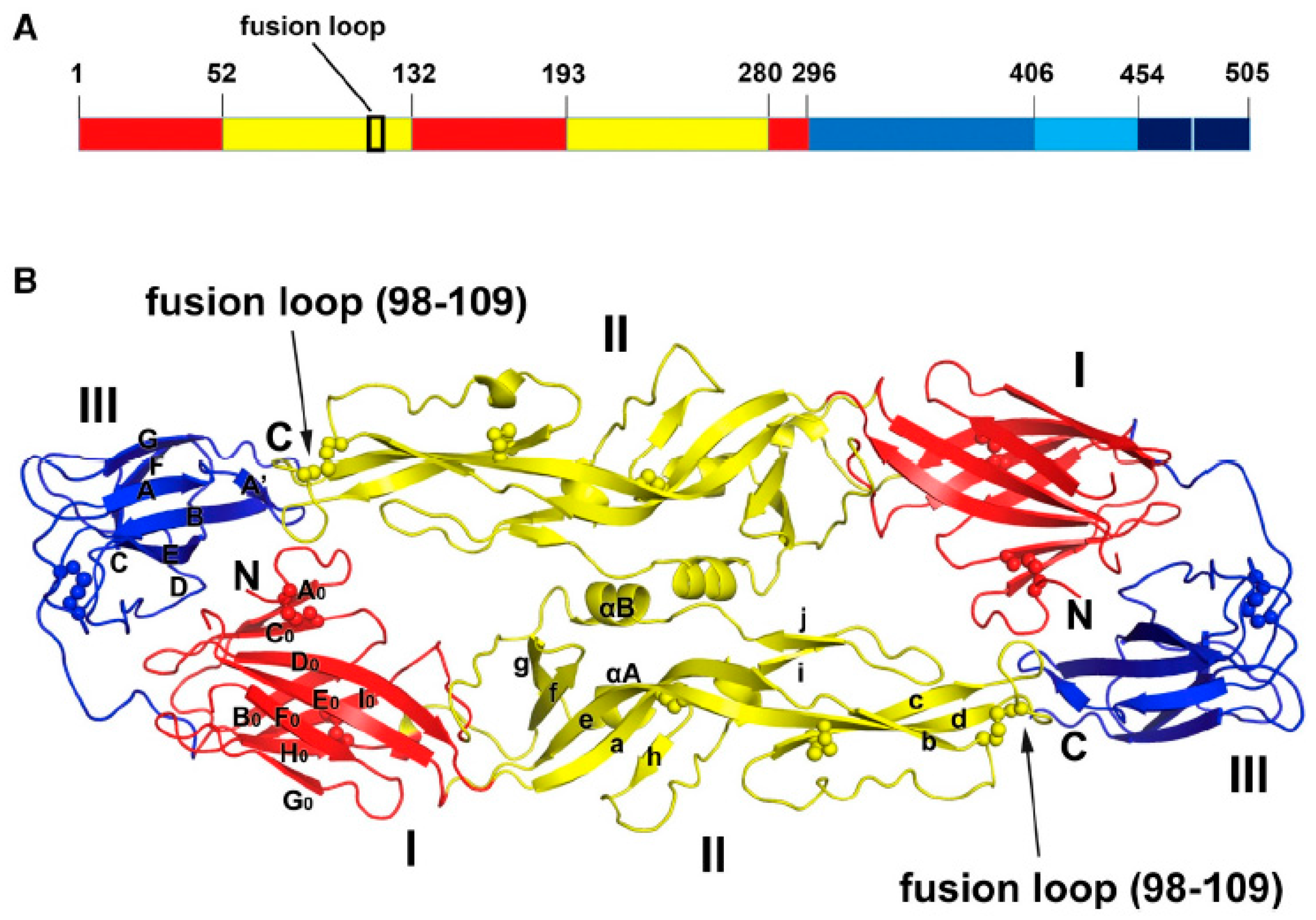
3.1. Antibodies Targeting DIII
3.2. Antibodies Targeting DI, DII
3.3. Antibodies that Recognize Quaternary Structures
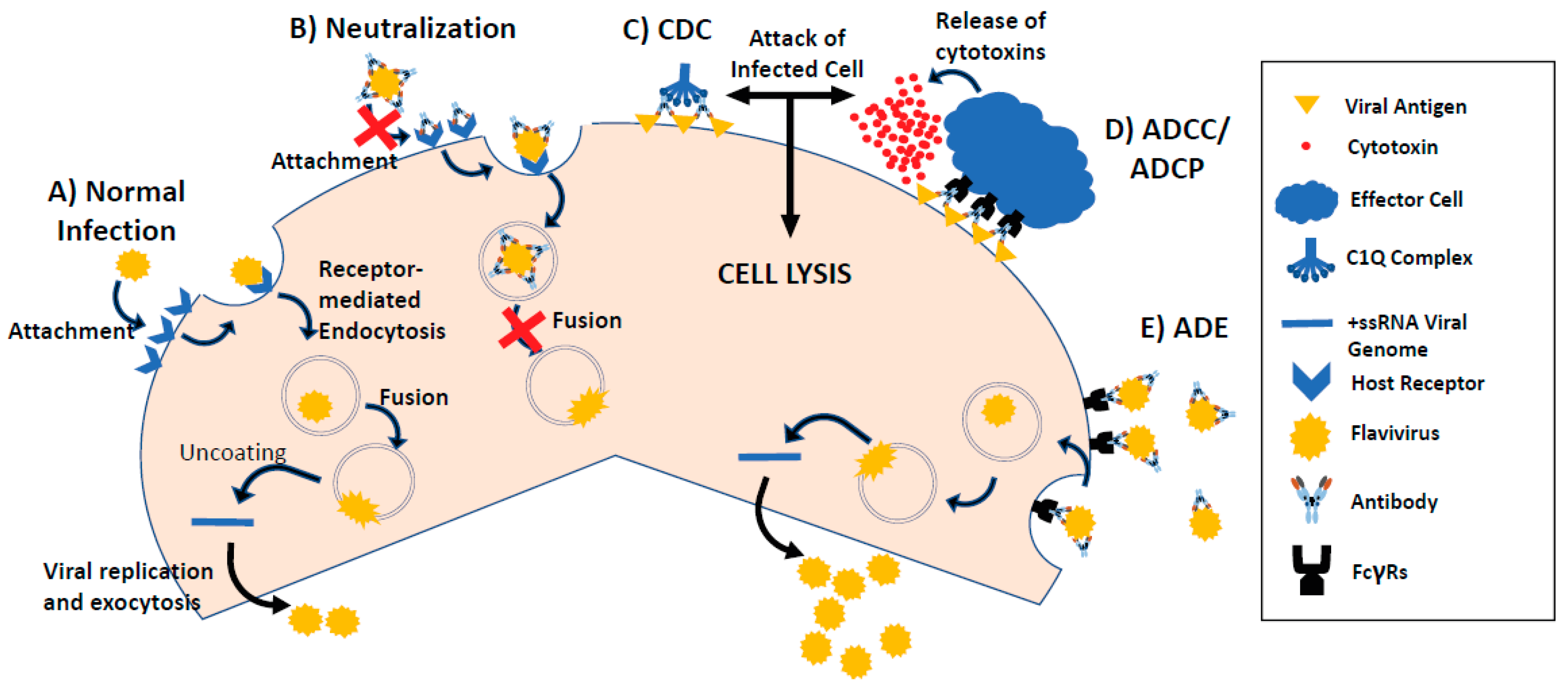
4. Antibody-Dependent Enhancement of Viral Infection
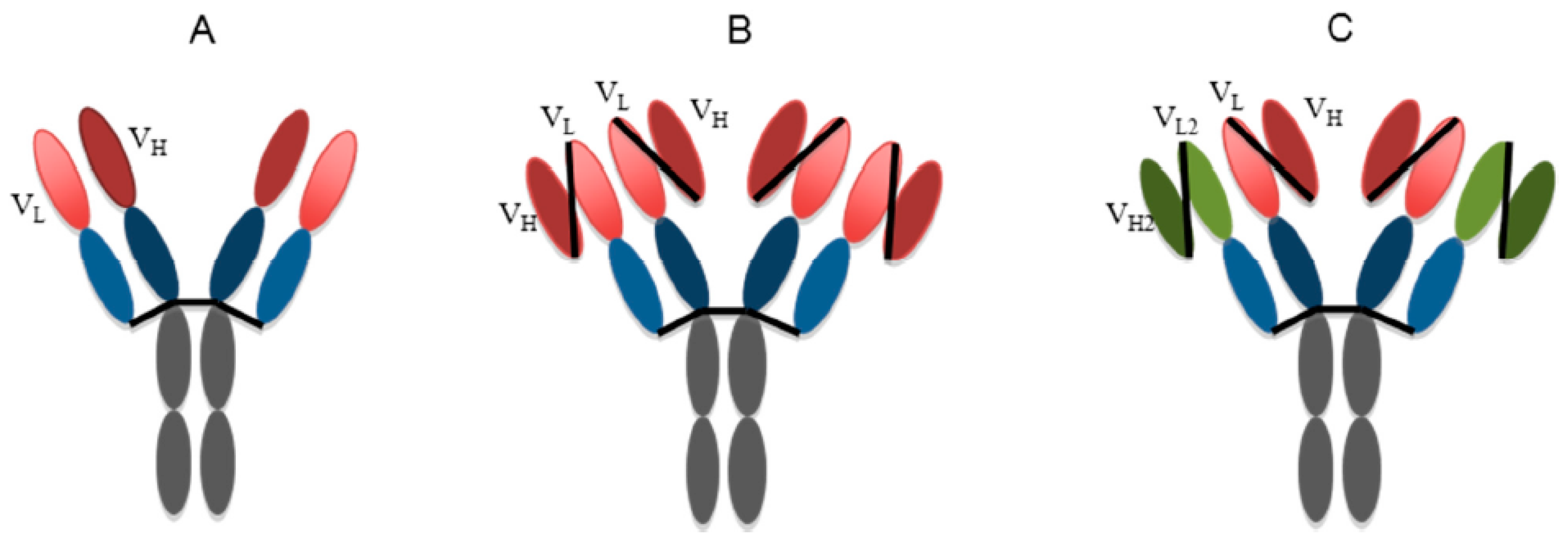
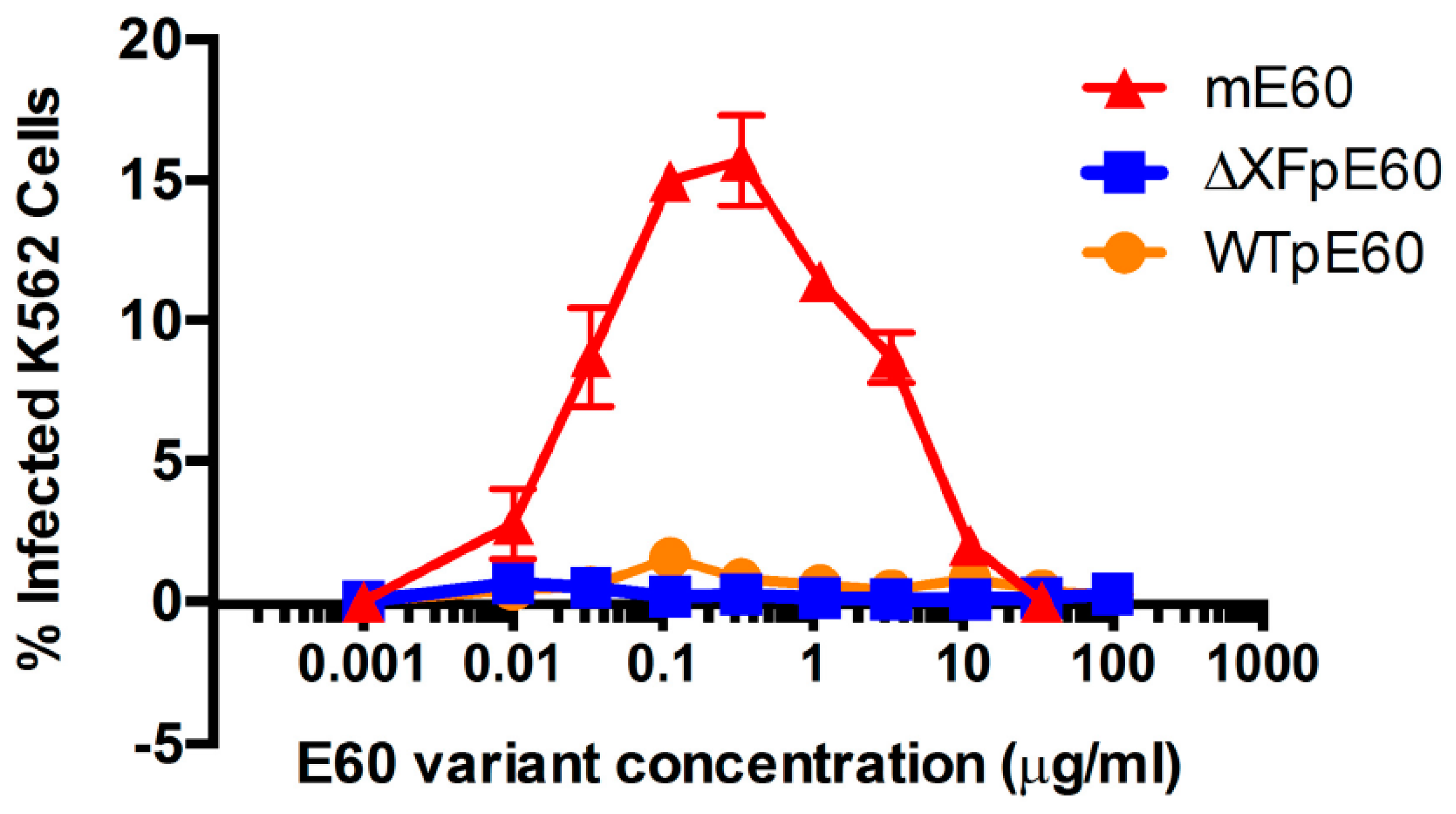
5. Plant-Produced Antibodies against Flaviviruses
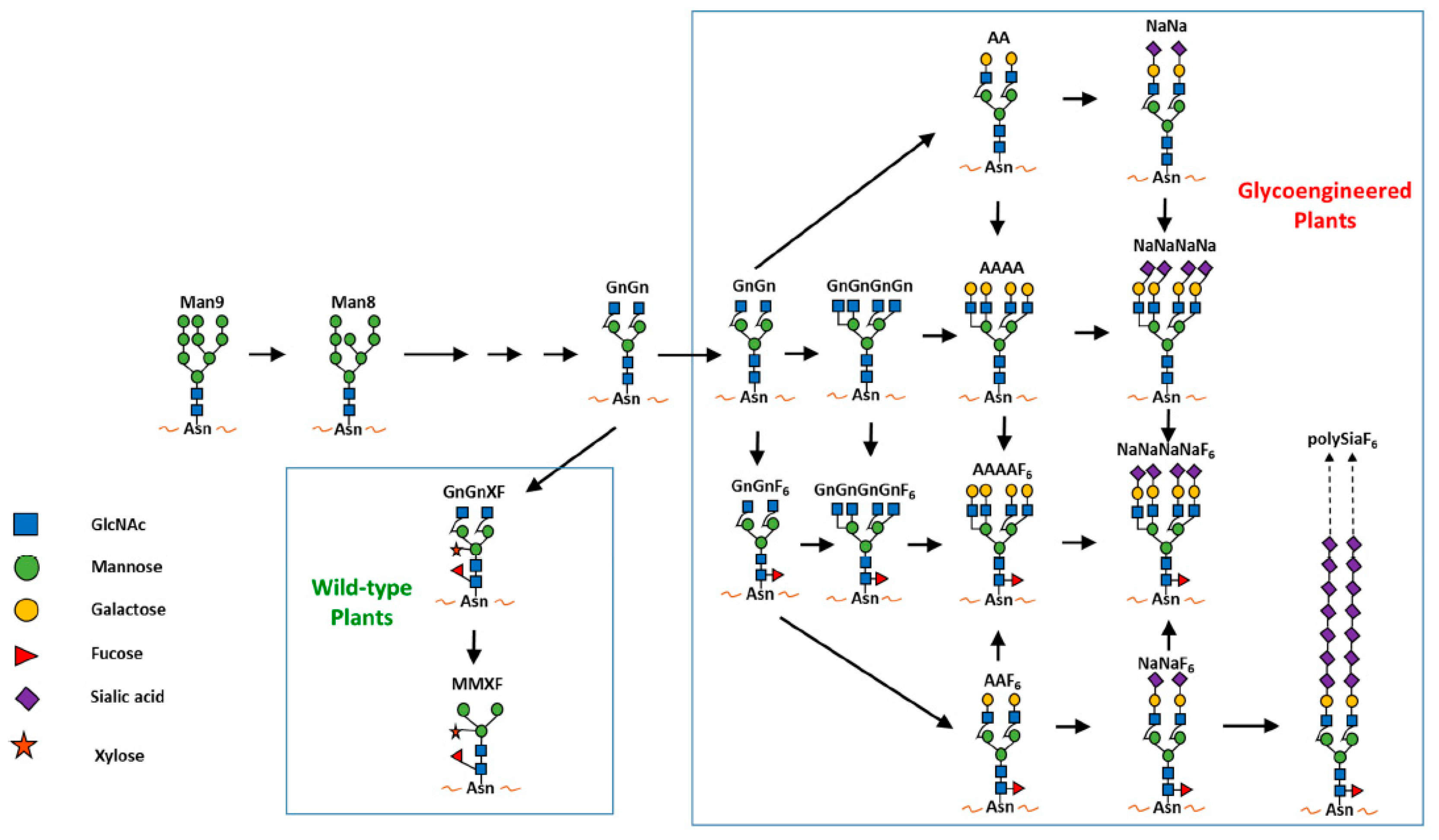
5.1. Plants as a System for the Development and Production of Antibody-Based Therapeutics
5.2. Plant-Produced Antibodies against WNV
5.3. Plant-Derived Antibodies against DENV and ZIKV
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuno, G.; Chang, G.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar] [PubMed]
- Blitvich, B.J.; Firth, A.E. Insect-specific flaviviruses: A systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, M.; Ze-Ze, L.; Vazquez, A.; Sanchez Seco, M.P.; Amaro, F.; Dottori, M. Insect-specific flaviviruses, a worldwide widespread group of viruses only detected in insects. Infect. Genet. Evol. 2016, 40, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Junglen, S.; Korries, M.; Grasse, W.; Wieseler, J.; Kopp, A.; Hermanns, K.; León-Juárez, M.; Drosten, C.; Kümmerer, B.M. Host range restriction of insect-specific flaviviruses occurs at several levels of the viral life cycle. mSphere 2017, 2, e0037-16. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.R. Historical perspectives on flavivirus research. Viruses 2017, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Mlera, L.; Melik, W.; Bloom, M.E. The role of viral persistence in flavivirus biology. Pathog. Dis. 2014, 71, 137–163. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.; Tesh, R.B.; Fisher-Hoch, S. Persistent infection with west nile virus years after initial infection. J. Infect. Dis. 2010, 201, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.G.; Faria, N.R.; Reiner, R.C., Jr.; Golding, N.; Nikolay, B.; Stasse, S.; Johansson, M.A.; Salje, H.; Faye, O.; Wint, G.R.W.; et al. Spread of yellow fever virus outbreak in Angola and the Democratic Republic of the Congo 2015-16: A modelling study. Lancet Infect. Dis. 2017, 17, 330–338. [Google Scholar] [CrossRef]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef]
- Collins, M.H.; Metz, S.W. Progress and works in progress: Update on flavivirus vaccine development. Clin. Ther. 2017, 39, 1519–1536. [Google Scholar] [CrossRef] [PubMed]
- Pang, T. Sage committee advice on dengue vaccine. Lancet Infect. Dis. 2016, 16, 880–882. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 a resolution cryo-em structure of zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Kim, B.S.; Chipman, P.R.; Rossmann, M.G.; Kuhn, R.J. Structure of west nile virus. Science 2003, 302, 248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ge, P.; Yu, X.; Brannan, J.M.; Bi, G.; Zhang, Q.; Schein, S.; Zhou, Z.H. Cryo-em structure of the mature dengue virus at 3.5-Å resolution. Nat. Struct. Mol. Biol. 2013, 20, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of dengue virus: Implications for flavivirus organization, maturation, and fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [PubMed]
- VanBlargan, L.A.; Goo, L.; Pierson, T.C. Deconstructing the antiviral neutralizing-antibody response: Implications for vaccine development and immunity. Microbiol. Mol. Biol. Rev. 2016, 80, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Bruening, J.; Weigel, B.; Pietschmann, T. Protein interactions during the flavivirus and hepacivirus life cycle. Mol. Cell. Proteom. 2017, 16, S75–S91. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. Therapeutic monoclonal antibodies approved by fda in 2016. MOJ Immunol. 2017, 5, 00145. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chien, C.R.C.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Drug therapy: Trastuzumab-mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Mellor, J.D.; Brown, M.P.; Irving, H.R.; Zalcberg, J.R.; Dobrovic, A. A critical review of the role of fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J. Hematol. Oncol. 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.H.; Hu, W.G.; Qin, X.B. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: Implications for therapy. Oncologist 2008, 13, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin g fragment c receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with her-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Sakai, K.; Arao, T.; Shimoyama, T.; Tamura, T.; Nishio, K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007, 98, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Garraud, O.; Heshmati, F.; Pozzetto, B.; Lefrere, F.; Girot, R.; Saillol, A.; Laperche, S. Plasma therapy against infectious pathogens, as of yesterday, today and tomorrow. Transfus. Clin. Biol. 2016, 23, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Sahr, F.; Ansumana, R.; Massaquoi, T.A.; Idriss, B.R.; Sesay, F.R.; Lamin, J.M.; Baker, S.; Nicol, S.; Conton, B.; Johnson, W.; et al. Evaluation of convalescent whole blood for treating ebola virus disease in freetown, sierra leone. J. Infect. 2017, 74, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Catalano, L.; Liumbruno, G.M.; Grazzini, G. Convalescent plasma: New evidence for an old therapeutic tool? Blood Transfus. 2016, 14, 152–157. [Google Scholar] [PubMed]
- Sparrow, E.; Friede, M.; Sheikh, M.; Torvaldsen, S. Therapeutic antibodies for infectious diseases. Bull. World Health Organ. 2017, 95, 235–237. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 2014, 134, e620–e638. [Google Scholar]
- Huang, K.; Incognito, L.; Cheng, X.; Ulbrandt, N.D.; Wu, H. Respiratory syncytial virus-neutralizing monoclonal antibodies motavizumab and palivizumab inhibit fusion. J. Virol. 2010, 84, 8132–8140. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Oliver, C.; Prince, G.A.; Hemming, V.G.; Pfarr, D.S.; Wang, S.C.; Dormitzer, M.; OGrady, J.; Koenig, S.; Tamura, J.K.; et al. Development of a humanized monoclonal antibody (medi-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 1997, 176, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, B.J.; Shadiack, A.M.; Carpenter, S.; Sanford, D.; Henning, L.N.; O’Connor, E.; Gonzales, N.; Mondick, J.; French, J.; Stark, G.V.; et al. Efficacy projection of obiltoxaximab for treatment of inhalational anthrax across a range of disease severity. Antimicrob. Agents Chemother. 2016, 60, 5787–5795. [Google Scholar] [CrossRef] [PubMed]
- Olson, W.C.; Jacobson, J.M. Ccr5 monoclonal antibodies for HIV-1 therapy. Curr. Opin. HIV AIDS 2009, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.; Fessel, J.; Emu, B.; Schrader, S.; Kumar, P.; Richmond, G.; Weinheimer, S.; Marsolais, C. Long-acting ibalizumab in patients with multi-drug resistant HIV-1: A 24-week study. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, WA, USA, 13–16 February 2017; p. 449LB. [Google Scholar]
- Lee, W.S.; Kent, S.J. Anti-hiv-1 antibody-dependent cellular cytotoxicity: Is there more to antibodies than neutralization? Curr. Opin. HIV AIDS 2007. [Google Scholar] [CrossRef] [PubMed]
- Czuczman, M.S.; Grillo-Lopez, A.J.; White, C.A.; Saleh, M.; Gordon, L.; LoBuglio, A.F.; Jonas, C.; Klippenstein, D.; Dallaire, B.; Varns, C. Treatment of patients with low-grade B-cell lymphoma with the combination of chimeric anti-CD20 monoclonal antibody and CHOP chemotherapy. J. Clin. Oncol. 1999, 17, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Dadachova, E.; Pirofski, L.A. Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2004, 2, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, J.; Das, S.; Fatima, Z.; Hameed, S. Multidrug resistance: An emerging crisis. Interdiscip. Perspect. Infect. Dis. 2014, 2014, 541340. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lai, H. The growing potential of plant-made monoclonal antibodies. Drug Target Rev. 2015, 2, 41–44. [Google Scholar]
- Pierson, T.C.; Diamond, M.S. Molecular mechanisms of antibody-mediated neutralisation of flavivirus infection. Expert Rev. Mol. Med. 2008, 10, e12. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 2003, 60, 421–467. [Google Scholar] [PubMed]
- Pierson, T.C.; Fremont, D.H.; Kuhn, R.J.; Diamond, M.S. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: Implications for vaccine development. Cell Host Microbe 2008, 4, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Beltramello, M.; Williams, K.L.; Simmons, C.P.; Macagno, A.; Simonelli, L.; Quyen, N.T.H.; Sukupolvi-Petty, S.; Navarro-Sanchez, E.; Young, P.R.; de Silva, A.M.; et al. The human immune response to dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 2010, 8, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis-virus at 2 angstrom resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef] [PubMed]
- Nybakken, G.E.; Nelson, C.A.; Chen, B.R.; Diamond, M.S.; Fremont, D.H. Crystal structure of the west nile virus envelope glycoprotein. J. Virol. 2006, 80, 11467–11474. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.P.; Song, J.; Lu, X.S.; Deng, Y.Q.; Musyoki, A.M.; Cheng, H.J.; Zhang, Y.F.; Yuan, Y.; Song, H.; Haywood, J.; et al. Structures of the zika virus envelope protein and its complex with a flavivirus broadly protective antibody. Cell Host Microbe 2016, 19, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.M.; Summers, P.L.; Dubois, D.R.; Cohen, W.H.; Gentry, M.K.; Timchak, R.L.; Burke, D.S.; Eckels, K.H. Monoclonal-antibodies for dengue virus prm glycoprotein protect mice against lethal dengue infection. Am. J. Trop. Med. Hyg. 1989, 41, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Nybakken, G.E.; Thompson, B.S.; Engle, M.J.; Marri, A.; Fremont, D.H.; Diamond, M.S. Antibodies against west nile virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J. Virol. 2006, 80, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Nybakken, G.E.; Oliphant, T.; Johnson, S.; Burke, S.; Diamond, M.S.; Fremont, D.H. Structural basis of west nile virus neutralization by a therapeutic antibody. Nature 2005, 437, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Stettler, K.; Beltramello, M.; Espinosa, D.A.; Graham, V.; Cassotta, A.; Bianchi, S.; Vanzetta, F.; Minola, A.; Jaconi, S.; Mele, F.; et al. Specificity, cross-reactivity, and function of antibodies elicited by zika virus infection. Science 2016, 353, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.N.; Tharakaraman, K.; Rowley, K.J.; Costa, V.V.; Chan, K.R.; Wong, Y.H.; Ong, L.C.; Tan, H.C.; Koch, T.; Cain, D.; et al. Structure-guided design of an anti-dengue antibody directed to a non-immunodominant epitope. Cell 2015, 162, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Bozzacco, L.; Keeffe, J.R.; Khouri, R.; Olsen, P.C.; Gazumyan, A.; Schaefer-Babajew, D.; Avila-Rios, S.; Nogueira, L.; Patel, R.; et al. Recurrent potent human neutralizing antibodies to zika virus in Brazil and Mexico. Cell 2017, 169, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.Y.; Fernandez, E.; Dowd, K.A.; Speer, S.D.; Platt, D.J.; Gorman, M.J.; Govero, J.; Nelson, C.A.; Pierson, T.C.; Diamond, M.S.; et al. Structural basis of zika virus-specific antibody protection. Cell 2016, 166, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Sukupolvi-Petty, S.; Austin, S.K.; Engle, M.; Brien, J.D.; Dowd, K.A.; Williams, K.L.; Johnson, S.; Rico-Hesse, R.; Harris, E.; Pierson, T.C.; et al. Structure and function analysis of therapeutic monoclonal antibodies against dengue virus type 2. J. Virol. 2010, 84, 9227–9239. [Google Scholar] [CrossRef] [PubMed]
- Tharakaraman, K.; Robinson, L.N.; Hatas, A.; Chen, Y.L.; Siyue, L.; Raguram, S.; Sasisekharan, V.; Wogan, G.N.; Sasisekharan, R. Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increased in vivo potency. Proc. Natl. Acad. Sci. USA 2013, 110, E1555–E1564. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Nybakken, G.E.; Chipman, P.R.; Zhang, W.; Diamond, M.S.; Fremont, D.H.; Kuhn, R.J.; Rossmann, M.G. West nile virus in complex with the fab fragment of a neutralizing monoclonal antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 12400–12404. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T.; Engle, M.; Nybakken, G.E.; Doane, C.; Johnson, S.; Huang, L.; Gorlatov, S.; Mehlhop, E.; Marri, A.; Chung, K.M.; et al. Development of a humanized monoclonal antibody with therapeutic potential against west nile virus. Nat. Med. 2005, 11, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S. Progress on the development of therapeutics against west nile virus. Antivir. Res. 2009, 83, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Teoh, E.P.; Kukkaro, P.; Teo, E.W.; Lim, A.P.; Tan, T.T.; Yip, A.; Schul, W.; Aung, M.; Kostyuchenko, V.A.; Leo, Y.S.; et al. The structural basis for serotype-specific neutralization of dengue virus by a human antibody. Sci. Transl. Med. 2012, 4, 139ra83. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.A.; Austin, S.K.; Shrestha, B.; Dowd, K.A.; Mukherjee, S.; Nelson, C.A.; Johnson, S.; Mabila, M.N.; Christian, E.A.; Rucker, J.; et al. Potent dengue virus neutralization by a therapeutic antibody with low monovalent affinity requires bivalent engagement. PLoS Pathog. 2014, 10, e1004072. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Brien, J.D.; Sukupolvi-Petty, S.; Austin, S.K.; Edeling, M.A.; Kim, T.; O’Brien, K.M.; Nelson, C.A.; Johnson, S.; Fremont, D.H.; et al. The development of therapeutic antibodies that neutralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog. 2010, 6, e1000823. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.K.; Dowd, K.A.; Shrestha, B.; Nelson, C.A.; Edeling, M.A.; Johnson, S.; Pierson, T.C.; Diamond, M.S.; Fremont, D.H. Structural basis of differential neutralization of DENV-1 genotypes by an antibody that recognizes a cryptic epitope. PLoS Pathog. 2012, 8, e1002930. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Parrish, C.R.; Murray, J.M.; Wright, P.J. Localization of a neutralizing epitope on the envelope protein of dengue virus type-2. Virology 1994, 202, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Lok, S.M.; Ng, M.L.; Aaskov, J. Amino acid and phenotypic changes in dengue 2 virus associated with escape from neutralisation by igm antibody. J. Med. Virol. 2001, 65, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Thullier, P.; Demangel, C.; Bedouelle, H.; Megret, F.; Jouan, A.; Deubel, V.; Mazie, J.C.; Lafaye, P. Mapping of a dengue virus neutralizing epitope critical for the infectivity of all serotypes: Insight into the neutralization mechanism. J. Gen. Virol. 2001, 82, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Lok, S.M.; Kostyuchenko, V.; Nybakken, G.E.; Holdaway, H.A.; Battisti, A.J.; Sukupolvi-Petty, S.; Sedlak, D.; Fremont, D.H.; Chipman, P.R.; Roehrig, J.T.; et al. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat. Struct. Mol. Biol. 2008, 15, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, J.J.B.; Sanchez, M.E.N.; Fretes, N.; Urvoas, A.; Staropoli, I.; Kikuti, C.M.; Coffey, L.L.; Arenzana-Seisdedos, F.; Bedouelle, H.; Rey, F.A. Mechanism of dengue virus broad cross-neutralization by a monoclonal antibody. Structure 2012, 20, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Low, J.G.H.; Ooi, E.E.; Vasudevan, S.G. Current status of dengue therapeutics research and development. J. Infect. Dis. 2017, 215, S96–S102. [Google Scholar] [CrossRef] [PubMed]
- Throsby, M.; Geuijen, C.; Goudsmit, J.; Bakker, A.Q.; Korimbocus, J.; Kramer, R.A.; Clijsters-van der Horst, M.; de Jong, M.; Jongeneelen, M.; Thijsse, S.; et al. Isolation and characterization of human monoclonal antibodies from individuals infected with west nile virus. J. Virol. 2006, 80, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Wahala, W.M.P.B.; Kraus, A.A.; Haymore, L.B.; Accavitti-Loper, M.A.; de Silva, A.M. Dengue virus neutralization by human immune sera: Role of envelope protein domain III-reactive antibody. Virology 2009, 392, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T.; Nybakken, G.E.; Austin, S.K.; Xu, Q.; Bramson, J.; Loeb, M.; Throsby, M.; Fremont, D.H.; Pierson, T.C.; Diamond, M.S. Induction of epitope-specific neutralizing antibodies against west nile virus. J. Virol. 2007, 81, 11828–11839. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Jumnainsong, A.; Onsirisakul, N.; Fitton, P.; Vasanawathana, S.; Limpitikul, W.; Puttikhunt, C.; Edwards, C.; Duangchinda, T.; Supasa, S.; et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science 2010, 328, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Yang, H.B.; Liu, X.Q.; Dai, L.P.; Ma, T.; Qi, J.X.; Wong, G.; Peng, R.C.; Liu, S.; Li, J.F.; et al. Molecular determinants of human neutralizing antibodies isolated from a patient infected with zika virus. Sci. Transl. Med. 2016, 8, 369ra179. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; de Alwis, A.R.; Kose, N.; Harris, E.; Ibarra, K.D.; Kahle, K.M.; Pfaff, J.M.; Xiang, X.; Doranz, B.J.; de Silva, A.M.; et al. The potent and broadly neutralizing human dengue virus-specific monoclonal antibody 1C19 reveals a unique cross-reactive epitope on the bc loop of domain II of the envelope protein. mBio 2013, 4, e00873-13. [Google Scholar] [CrossRef] [PubMed]
- Fibriansah, G.; Tan, J.L.; Smith, S.A.; de Alwis, A.R.; Ng, T.S.; Kostyuchenko, V.A.; Ibarra, K.D.; Wang, J.; Harris, E.; de Silva, A.; et al. A potent anti-dengue human antibody preferentially recognizes the conformation of e protein monomers assembled on the virus surface. EMBO Mol. Med. 2014, 6, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-Q.; Dai, J.-X.; Ji, G.-H.; Jiang, T.; Wang, H.-J.; Yang, H.-O.; Tan, W.-L.; Liu, R.; Yu, M.; Ge, B.-X.; et al. A broadly flavivirus cross-neutralizing monoclonal antibody that recognizes a novel epitope within the fusion loop of e protein. PLoS ONE 2011, 6, e16059. [Google Scholar] [CrossRef] [PubMed]
- De Alwis, R.; Smith, S.A.; Olivarez, N.P.; Messer, W.B.; Huynh, J.P.; Wahala, W.M.P.B.; White, L.J.; Diamond, M.S.; Baric, R.S.; Crowe, J.E.; et al. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc. Natl. Acad. Sci. USA 2012, 109, 7439–7444. [Google Scholar] [CrossRef] [PubMed]
- Crill, W.D.; Chang, G.J. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. J. Virol. 2004, 78, 13975–13986. [Google Scholar] [CrossRef] [PubMed]
- Stiasny, K.; Kiermayr, S.; Holzmann, H.; Heinz, F.X. Cryptic properties of a cluster of dominant flavivirus cross-reactive antigenic sites. J. Virol. 2006, 80, 9557–9568. [Google Scholar] [CrossRef] [PubMed]
- Costin, J.M.; Zaitseva, E.; Kahle, K.M.; Nicholson, C.O.; Rowe, D.K.; Graham, A.S.; Bazzone, L.E.; Hogancamp, G.; Sierra, M.F.; Fong, R.H.; et al. Mechanistic study of broadly neutralizing human monoclonal antibodies against dengue virus that target the fusion loop. J. Virol. 2013, 87, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.Y.; Tsai, W.Y.; Lin, S.R.; Kao, C.L.; Hu, H.P.; King, C.C.; Wu, H.C.; Chang, G.J.; Wang, W.K. Antibodies to envelope glycoprotein of dengue virus during the natural course of infection are predominantly cross-reactive and recognize epitopes containing highly conserved residues at the fusion loop of domain II. J. Virol. 2008, 82, 6631–6643. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Wongwiwat, W.; Supasa, S.; Zhang, X.K.; Dai, X.H.; Rouvinski, A.; Jumnainsong, A.; Edwards, C.; Quyen, N.T.H.; Duangchinda, T.; et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat. Immunol. 2015, 16, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.R.; Dowd, K.A.; Engle, M.; Tesh, R.B.; Johnson, S.; Pierson, T.C.; Diamond, M.S. Poorly neutralizing cross-reactive antibodies against the fusion loop of west nile virus envelope protein protect in vivo via fc gamma receptor and complement-dependent effector mechanisms. J. Virol. 2011, 85, 11567–11580. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.R.; Moesker, B.; Goudsmit, J.; Jongeneelen, M.; Austin, S.K.; Oliphant, T.; Nelson, S.; Pierson, T.C.; Wilschut, J.; Throsby, M.; et al. Human monoclonal antibodies against west nile virus induced by natural infection neutralize at a postattachment step. J. Virol. 2009, 83, 6494–6507. [Google Scholar] [CrossRef] [PubMed]
- Gallichotte, E.N.; Widman, D.G.; Yount, B.L.; Wahala, W.M.; Durbin, A.; Whitehead, S.; Sariol, C.A.; Crowe, J.E.; de Silva, A.M.; Baric, R.S. A new quaternary structure epitope on dengue virus serotype 2 is the target of durable type-specific neutralizing antibodies. mBio 2015, 6, e01461-15. [Google Scholar] [CrossRef] [PubMed]
- Fibriansah, G.; Ibarra, K.D.; Ng, T.S.; Smith, S.A.; Tan, J.L.; Lim, X.N.; Ooi, J.S.G.; Kostyuchenko, V.A.; Wang, J.Q.; de Silva, A.M.; et al. Cryo-em structure of an antibody that neutralizes dengue virus type 2 by locking e protein dimers. Science 2015, 349, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Goh, B.C.; Lim, S.Y.; Teo, E.W.; Lim, A.P.C.; Dedon, P.C.; Hanson, B.J.; MacAry, P.A.; Lescar, J. Structural mimicry of the dengue virus envelope glycoprotein revealed by the crystallographic study of an idiotype-anti-idiotype fab complex. J. Virol. 2017, 91, e00406-17. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Dejnirattisai, W.; Cao, B.; Scheaffer, S.M.; Supasa, P.; Wongwiwat, W.; Esakky, P.; Drury, A.; Mongkolsapaya, J.; Moley, K.H.; et al. Human antibodies to the dengue virus e-dimer epitope have therapeutic activity against zika virus infection. Nat. Immunol. 2017, 18, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.S.; Miller, A.; Sapparapu, G.; Fernandez, E.; Klose, T.; Long, F.; Fokine, A.; Porta, J.C.; Jiang, W.; Diamond, M.S.; et al. A human antibody against zika virus crosslinks the e protein to prevent infection. Nat. Commun. 2017, 8, 14722. [Google Scholar] [CrossRef] [PubMed]
- Fibriansah, G.; Tan, J.L.; Smith, S.A.; de Alwis, R.; Ng, T.S.; Kostyuchenko, V.A.; Jadi, R.S.; Kukkaro, P.; de Silva, A.M.; Crowe, J.E.; et al. A highly potent human antibody neutralizes dengue virus serotype 3 by binding across three surface proteins. Nat. Commun. 2015, 6, 6341. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Vogt, M.R.; Goudsmit, J.; Holdaway, H.A.; Aksyuk, A.A.; Chipman, P.R.; Kuhn, R.J.; Diamond, M.S.; Rossmann, M.G. Neutralization of west nile virus by cross-linking of its surface proteins with fab fragments of the human monoclonal antibody CR4354. Proc. Natl. Acad. Sci. USA 2010, 107, 18950–18955. [Google Scholar] [CrossRef] [PubMed]
- Sapparapu, G.; Fernandez, E.; Kose, N.; Cao, B.; Fox, J.M.; Bombardi, R.G.; Zhao, H.Y.; Nelson, C.A.; Bryan, A.L.; Barnes, T.; et al. Neutralizing human antibodies prevent zika virus replication and fetal disease in mice. Nature 2016, 540, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Rouvinski, A.; Guardado-Calvo, P.; Barba-Spaeth, G.; Duquerroy, S.; Vaney, M.C.; Kikuti, C.M.; Navarro Sanchez, M.E.; Dejnirattisai, W.; Wongwiwat, W.; Haouz, A.; et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 2015, 520, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, J.A.; Plante, J.A.; Plante, K.S.; Young, E.F.; McGowan, E.; Gallichotte, E.N.; Widman, D.G.; Heise, M.T.; de Silva, A.M.; Baric, R.S. Dengue virus envelope dimer epitope monoclonal antibodies isolated from dengue patients are protective against zika virus. mBio 2016, 7, e01123-16. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Engle, M.; Fuchs, A.; Keller, T.; Johnson, S.; Gorlatov, S.; Diamond, M.S.; Chen, Q. Monoclonal antibody produced in plants efficiently treats west nile virus infection in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Dent, M.; Hurtado, J.; Paul, A.M.; Sun, H.; Lai, H.; Yang, M.; Esqueda, A.; Bai, F.; Steinkellner, H.; Chen, Q. Plant-produced anti-dengue virus monoclonal antibodies exhibit reduced antibody-dependent enhancement of infection activity. J. Gen. Virol. 2016, 97, 3280–3290. [Google Scholar] [CrossRef] [PubMed]
- Messer, W.B.; Yount, B.L.; Royal, S.R.; de Alwis, R.; Widman, D.G.; Smith, S.A.; Crowe, J.E.; Pfaff, J.M.; Kahle, K.M.; Doranz, B.J.; et al. Functional transplant of a dengue virus serotype 3 (denv3)-specific human monoclonal antibody epitope into denv1. J. Virol. 2016, 90, 5090–5097. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B.; Chow, J.S.; Marchette, N.J. Immunological enhancement of dengue virus replication. Nat. New Biol. 1973, 243, 24–25. [Google Scholar] [PubMed]
- Halstead, S.B. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J. Infect. Dis. 1979, 140, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Foo, S.S.; Bruzzone, R.; Vu Dinh, L.; King, N.J.C.; Mahalingam, S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol. Rev. 2015, 268, 340–364. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.M.; Carlin, E.R.; Jenkins, M.M.; Tan, A.L.; Barcellona, C.M.; Nicholson, C.O.; Michael, S.F.; Isern, S. Dengue virus antibodies enhance zika virus infection. Clin. Transl. Immunol. 2016, 5, e117. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Supasa, P.; Wongwiwat, W.; Rouvinski, A.; Barba-Spaeth, G.; Duangchinda, T.; Sakuntabhai, A.; Cao-Lormeau, V.M.; Malasit, P.; Rey, F.A.; et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat. Immunol. 2016, 17, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Castanha, P.M.S.; Nascimento, E.J.M.; Braga, C.; Cordeiro, M.T.; de Carvalho, O.V.; de Mendonca, L.R.; Azevedo, E.A.N.; Franca, R.F.O.; Dhalia, R.; Marques, E.T.A. Dengue virus-specific antibodies enhance brazilian zika virus infection. J. Infect. Dis. 2017, 215, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Priyamvada, L.; Quicke, K.M.; Hudson, W.H.; Onlamoon, N.; Sewatanon, J.; Edupuganti, S.; Pattanapanyasat, K.; Chokephaibulkit, K.; Mulligan, M.J.; Wilson, P.C.; et al. Human antibody responses after dengue virus infection are highly cross-reactive to zika virus. Proc. Natl. Acad. Sci. USA 2016, 113, 7852–7857. [Google Scholar] [CrossRef] [PubMed]
- Bardina, S.V.; Bunduc, P.; Tripathi, S.; Duehr, J.; Frere, J.J.; Brown, J.A.; Nachbagauer, R.; Foster, G.A.; Krysztof, D.; Tortorella, D.; et al. Enhancement of zika virus pathogenesis by preexisting antiflavivirus immunity. Science 2017, 356, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.S.; Li, M.Z.; Chong, C.S.; Ng, L.C.; Tan, C.H. Aedes (stegomyia) albopictus (skuse): A potential vector of zika virus in singapore. PLoS Negl. Trop. Dis. 2013, 7, e2348. [Google Scholar] [CrossRef] [PubMed]
- Takada, A.; Kawaoka, Y. Antibody-dependent enhancement of viral infection: Molecular mechanisms and in vivo implications. Rev. Med. Virol. 2003, 13, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Mehlhop, E.; Ansarah-Sobrinho, C.; Johnson, S.; Engle, M.; Fremont, D.H.; Pierson, T.C.; Diamond, M.S. Complement protein C1q inhibits antibody-dependent enhancement of flavivirus infection in an igg subclass-specific manner. Cell Host Microbe 2007, 2, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Mehlhop, E.; Nelson, S.; Jost, C.A.; Gorlatov, S.; Johnson, S.; Fremont, D.H.; Diamond, M.S.; Pierson, T.C. Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of west nile virus. Cell Host Microbe 2009, 6, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Haslwanter, D.; Blaas, D.; Heinz, F.X.; Stiasny, K. A novel mechanism of antibody-mediated enhancement of flavivirus infection. PLoS Pathog. 2017, 13, e1006643. [Google Scholar] [CrossRef] [PubMed]
- Wines, B.D.; Powell, M.S.; Parren, P.W.H.I.; Barnes, N.; Hogarth, P.M. The IgG Fc contains distinct Fc receptor (FcR) binding sites: The leukocyte receptors fc gamma RI and Fc gamma riia bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J. Immunol. 2000, 164, 5313–5318. [Google Scholar] [CrossRef] [PubMed]
- Hezareh, M.; Hessell, A.J.; Jensen, R.C.; van de Winkel, J.G.J.; Parren, P.W.H.I. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001, 75, 12161–12168. [Google Scholar] [CrossRef] [PubMed]
- Hessell, A.J.; Hangartner, L.; Hunter, M.; Havenith, C.E.; Beurskens, F.J.; Bakker, J.M.; Lanigan, C.M.; Landucci, G.; Forthal, D.N.; Parren, P.W.; et al. Fc receptor but not complement binding is important in antibody protection against hiv. Nature 2007, 449, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.R.; Ong, E.Z.; Mok, D.Z.L.; Ooi, E.E. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert Rev. Anti-Infect. Ther. 2015, 13, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q. Turning a new leaf. Eur. Biopharm. Rev. 2011, 2, 64–68. [Google Scholar]
- Chen, Q. Expression and manufacture of pharmaceutical proteins in genetically engineered horticultural plants. In Transgenic Horticultural Crops: Challenges and Opportunities—Essays by Experts; Mou, B., Scorza, R., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2011; pp. 83–124. [Google Scholar]
- Chen, Q. Expression and purification of pharmaceutical proteins in plants. Biol. Eng. Trans. 2008, 1, 291–321. [Google Scholar] [CrossRef]
- Chen, Q.; Davis, K. The potential of plants as a system for the development and production of human biologics. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, C.; Santi, L. Plant-made biologics. Biomed Res. Int. 2014, 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Kwong, A.T.; Holtz, B.R.; Erwin, R.L.; Marcel, S.; McDonald, K.A. Techno-economic analysis of a transient plant-based platform for monoclonal antibody production. mAbs 2016, 8, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Tuse, D.; Tu, T.; McDonald, K. Manufacturing economics of plant-made biologics: Case studies in therapeutic and industrial enzymes. Biomed Res. Int. 2014, 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lai, H. Plant-Derived Monoclonal Antibodies as Human Biologics for Infectious Disease and Cancer in Plant-Derived Pharmaceuticals: Principles and Applications for Developing Countries; Hefferon, K.L., Ed.; CABI: Cryodon, UK, 2014; pp. 42–75. [Google Scholar]
- Chen, Q.; He, J.; Phoolcharoen, W.; Mason, H.S. Geminiviral vectors based on bean yellow dwarf virus for production of vaccine antigens and monoclonal antibodies in plants. Hum. Vaccines 2011, 7, 331–338. [Google Scholar] [CrossRef]
- Klimyuk, V.; Pogue, G.; Herz, S.; Butler, J.; Haydon, H. Production of recombinant antigens and antibodies in Nicotiana benthamiana using ‘magnifection’ technology: Gmp-compliant facilities for small- and large-scale manufacturing. Curr. Top. Microbiol. Immunol. 2014, 375, 127–154. [Google Scholar] [PubMed]
- Peyret, H.; Lomonossoff, G.P. When plant virology met agrobacterium: The rise of the deconstructed clones. Plant Biotechnol. J. 2015, 13, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Lico, C.; Chen, Q.; Santi, L. Viral vectors for production of recombinant proteins in plants. J. Cell. Physiol. 2008, 216, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Bendandi, M.; Marillonnet, S.; Kandzia, R.; Thieme, F.; Nickstadt, A.; Herz, S.; Frode, R.; Inoges, S.; Lopez-Diaz de Cerio, A.; Soria, E.; et al. Rapid, high-yield production in plants of individualized idiotype vaccines for non-hodgkin’s lymphoma. Ann. Oncol. 2010, 21, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
- Santi, L.; Batchelor, L.; Huang, Z.; Hjelm, B.; Kilbourne, J.; Arntzen, C.J.; Chen, Q.; Mason, H.S. An efficient plant viral expression system generating orally immunogenic norwalk virus-like particles. Vaccine 2008, 26, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Phoolcharoen, W.; Bhoo, S.H.; Lai, H.; Ma, J.; Arntzen, C.J.; Chen, Q.; Mason, H.S. Expression of an immunogenic ebola immune complex in Nicotiana benthamiana. Plant Biotechnol. J. 2011, 9, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Phoolcharoen, W.; Dye, J.M.; Kilbourne, J.; Piensook, K.; Pratt, W.D.; Arntzen, C.J.; Chen, Q.; Mason, H.S.; Herbst-Kralovetz, M.M. A nonreplicating subunit vaccine protects mice against lethal ebola virus challenge. Proc. Natl. Acad. Sci. USA 2011, 108, 20695–20700. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; He, J.; Engle, M.; Diamond, M.S.; Chen, Q. Robust production of virus-like particles and monoclonal antibodies with geminiviral replicon vectors in lettuce. Plant Biotechnol. J. 2012, 10, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, Q.; Hjelm, B.; Arntzen, C.; Mason, H. A DNA replicon system for rapid high-level production of virus-like particles in plants. Biotechnol. Bioeng. 2009, 103, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Phoolcharoen, W.; Lai, H.; Piensook, K.; Cardineau, G.; Zeitlin, L.; Whaley, K.; Arntzen, C.J.; Mason, H.; Chen, Q. High-level rapid production of full-size monoclonal antibodies in plants by a single-vector DNA replicon system. Biotechnol. Bioeng. 2010, 106, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Loos, A.; Steinkellner, H. Plant glyco-biotechnology on the way to synthetic biology. Front. Plant Sci. 2014, 5, 523. [Google Scholar] [CrossRef] [PubMed]
- Shaaltiel, Y.; Tekoah, Y. Plant specific n-glycans do not have proven adverse effects in humans. Nat. Biotechnol. 2016, 34, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q. Glycoengineering of plants yields glycoproteins with polysialylation and other defined n-glycoforms. Proc. Natl. Acad. Sci. USA 2016, 113, 9404–9406. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, S.; Halim, A.; Schulz, M.A.; Frodin, M.; Rahman, S.H.; Vester-Christensen, M.B.; Behrens, C.; Kristensen, C.; Vakhrushev, S.Y.; et al. Engineered cho cells for production of diverse, homogeneous glycoproteins. Nat. Biotechnol. 2015, 33, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Strasser, R.; Altmann, F.; Steinkellner, H. Controlled glycosylation of plant-produced recombinant proteins. Curr. Opin. Biotechnol. 2014, 30, 95–100. [Google Scholar] [CrossRef] [PubMed]
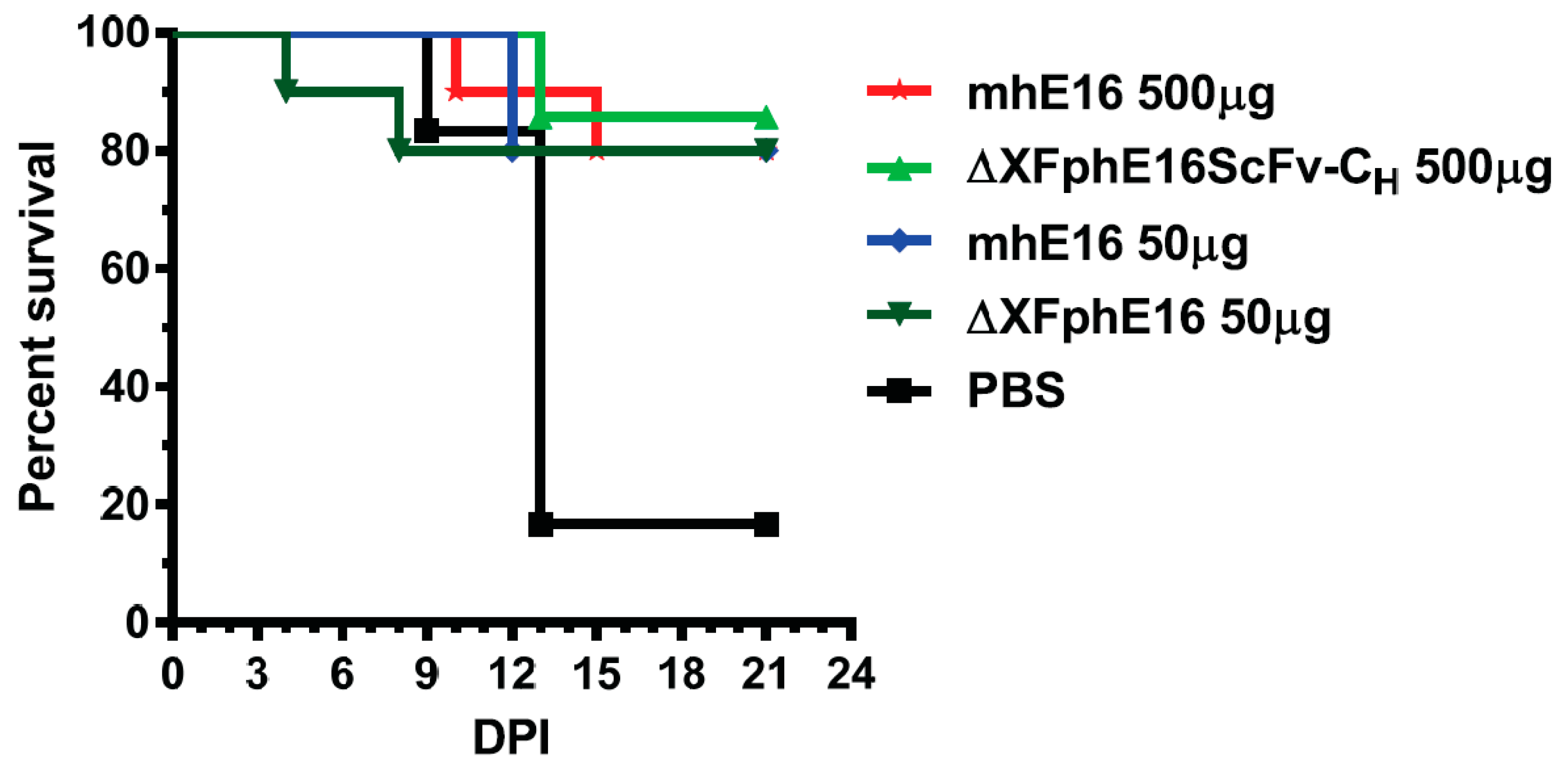
- Lai, H.; He, J.; Hurtado, J.; Stahnke, J.; Fuchs, A.; Mehlhop, E.; Gorlatov, S.; Loos, A.; Diamond, M.S.; Chen, Q. Structural and functional characterization of an anti-west nile virus monoclonal antibody and its single-chain variant produced in glycoengineered plants. Plant Biotechnol. J. 2014, 12, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Olinger, G.G.; Pettitt, J.; Kim, D.; Working, C.; Bohorov, O.; Bratcher, B.; Hiatt, E.; Hume, S.D.; Johnson, A.K.; Morton, J.; et al. Delayed treatment of ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc. Natl. Acad. Sci. USA 2012, 109, 18030–18035. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Chen, Q. Bioprocessing of plant-derived virus-like particles of norwalk virus capsid protein under current good manufacture practice regulations. Plant Cell Rep. 2012, 31, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, L.; Pettitt, J.; Scully, C.; Bohorova, N.; Kim, D.; Pauly, M.; Hiatt, A.; Ngo, L.; Steinkellner, H.; Whaley, K.J.; et al. Enhanced potency of a fucose-free monoclonal antibody being developed as an ebola virus immunoprotectant. Proc. Natl. Acad. Sci. USA 2011, 108, 20690–20694. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wong, G.; Audet, J.; Bello, A.; Fernando, L.; Alimonti, J.B.; Fausther-Bovendo, H.; Wei, H.; Aviles, J.; Hiatt, E.; et al. Reversion of advanced ebola virus disease in nonhuman primates with zmapp. Nature 2014, 514, 47–53. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lai, H.; Engle, M.; Gorlatov, S.; Gruber, C.; Steinkellner, H.; Diamond, M.S.; Chen, Q. Generation and analysis of novel plant-derived antibody-based therapeutic molecules against west nile virus. PLoS ONE 2014, 9, e93541. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Pardridge, W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.E.A.; Quam, M.B.; Wilder-Smith, A. Epidemiology of dengue: Past, present and future prospects. Clin. Epidemiol. 2013, 5, 299–309. [Google Scholar] [PubMed]
- Rothman, A.L. Dengue: Defining protective versus pathologic immunity. J. Clin. Investig. 2004, 113, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Dengue. Lancet 2007, 370, 1644–1652. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengue antibody-dependent enhancement: Knowns and unknowns. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M. Antibody-dependent of enhancement of infection and the pathogenesis of viral disease. Clin. Infect. Dis. 1994, 19, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R.; Harrison, L.M.; Salas, R.A.; Tovar, D.; Nisalak, A.; Ramos, C.; Boshell, J.; de Mesa, M.T.; Nogueira, R.M.; da Rosa, A.T. Origins of dengue type 2 viruses associated with increased pathogenicity in the americas. Virology 1997, 230, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.R.; Whitehead, S.S. Immune response to dengue virus and prospects for a vaccine. Annu. Rev. Immunol. 2011, 29, 587–619. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.L.; Harris, E. Global spread and persistence of dengue. Ann. Rev. Microbiol. 2008, 62, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A.; Gubler, D.J. Geographic expansion of dengue: The impact of international travel. Med. Clin. N. Am. 2008, 92, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.J.; Williams, K.L.; Lachica, R.; Flores, D.; Kyle, J.L.; Mehlhop, E.; Johnson, S.; Diamond, M.S.; Beatty, P.R.; Harris, E. Lethal antibody enhancement of dengue disease in mice is prevented by fc modification. PLoS Pathog. 2010, 6, e1000790. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Sukupolvi-Petty, S.; Beltramello, M.; Johnson, S.; Sallusto, F.; Lanzavecchia, A.; Diamond, M.S.; Harris, E. Therapeutic efficacy of antibodies lacking fcgamma receptor binding against lethal dengue virus infection is due to neutralizing potency and blocking of enhancing antibodies [corrected]. PLoS Pathog. 2013, 9, e1003157. [Google Scholar] [CrossRef]
- Zheng, K.; Bantog, C.; Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011, 3, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Forrer, K.; Helk, B.; Trout, B.L. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol. J. 2010, 6, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Arango, M.; Perez, A.B.; Fonte, L.; Sierra, B.; Rodriguez-Roche, R.; Aguirre, E.; Fiterre, I.; Guzman, M.G. Antibodies from patients with dengue viral infection mediate cellular cytotoxicity. J. Clin. Virol. 2006, 37, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Laoprasopwattana, K.; Libraty, D.H.; Endy, T.P.; Nisalak, A.; Chunsuttiwat, S.; Ennis, F.A.; Rothman, A.L.; Green, S. Antibody-dependent cellular cytotoxicity mediated by plasma obtained before secondary dengue virus infections: Potential involvement in early control of viral replication. J. Infect. Dis. 2007, 195, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Peng, Y.; Berkowitz, S.A.; Engen, J.R. Post-translational modifications differentially affect igg1 conformation and receptor binding. Mol. Cell. Proteom. 2010, 9, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Prog. 2005, 21, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T.; Nybakken, G.E.; Engle, M.; Xu, Q.; Nelson, C.A.; Sukupolvi-Petty, S.; Marri, A.; Lachmi, B.-E.; Olshevsky, U.; Fremont, D.H.; et al. Antibody recognition and neutralization determinants on domains I and II of west nile virus envelope protein. J. Virol. 2006, 80, 12149–12159. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, J.; Chen, Q. Plant-produced anti-dengue monoclonal antibodies protect mice against lethal challenges of dengue virus infection. Manuscript in preparation.
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the western hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Attar, N. Zika virus circulates in new regions. Nat. Rev. Microbiol. 2016, 14, 62. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-barré syndrome outbreak associated with zika virus infection in french polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Samarasekera, U.; Triunfol, M. Concern over zika virus grips the world. Lancet 2016, 387, 521–524. [Google Scholar] [CrossRef]
- Barba-Spaeth, G.; Dejnirattisai, W.; Rouvinski, A.; Vaney, M.-C.; Medits, I.; Sharma, A.; Simon-Lorière, E.; Sakuntabhai, A.; Cao-Lormeau, V.-M.; Haouz, A.; et al. Structural basis of potent zika–dengue virus antibody cross-neutralization. Nature 2016, 536, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, Q. Monoclonal antibodies from plants potently neutralizes zika virus without enhancing dengue virus infection. Manuscript in preparation.
- Huisman, W.; Martina, B.E.E.; Rimmelzwaan, G.F.; Gruters, R.A.; Osterhaus, A.D.M.E. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Name | Target Virus | Epitope | Development Stage | References |
|---|---|---|---|---|
| hE16, pE16 | WNV | Lateral ridge of DIII | Phase II trial | [57,66,103] |
| CR4374 | WNV | E protein DIII | Preclinical/mouse model | [77] |
| CR4354 | WNV | E protein Hinge between DI, DII | Preclinical/mouse model | [77,92] |
| CR4348 | WNV | E protein DII | Preclinical/mouse model | [77,92] |
| Plant-made E60 | DENV1–4 | E protein DII fusion loop | Preclinical/mouse model | [104] |
| DENV1-E105, E106 | DENV1 | E protein DIII lateral ridge | Preclinical/mouse model | [69] |
| 1F4 | DENV1 | E protein DI, DI-DII hinge | Preclinical/mouse model | [83] |
| 1C19 | DENV1–4 | E protein DII BC loop | Preclinical/mouse model | [11,82] |
| HM14c10 | DENV1 | E protein dimer-dimer interface | Preclinical/mouse model | [67,95] |
| 2D22 | DENV2 | E protein dimer-dimer interface | Preclinical/mouse model | [85,93,94] |
| 5J7 | DENV3 | Across three E protein | Preclinical/mouse model | [98,105] |
| Ab513 | DENV1–4 | E protein DIII | Preclinical/mouse model | [59] |
| 2A10G6 | DENV1–4, WNV, Zika, YFV, TBEV | E protein fusion loop | Preclinical/mouse model | [54,84] |
| ZKA64 | Zika | E protein DIII | Preclinical/mouse model | [58] |
| ZV54 | Zika | E protein DIII Lateral Ridge | Preclinical/mouse model | [61] |
| ZV67 | Zika | E protein DIII Lateral Ridge | Preclinical/mouse model | [61] |
| Z23 | Zika | E protein DIII | Preclinical/mouse model | [81] |
| Z3L1 | Zika | E protein DI, DII | Preclinical/mouse model | [81] |
| Z004 | Zika/DENV1 | Lateral Ridge in DIII | Preclinical/mouse model | [60] |
| ZIKV-117 | Zika | E protein dimer-dimer interface | Preclinical/mouse model | [97,100] |
| EDE1-B10 | Dengue/Zika | E protein dimer-dimer interface | Preclinical/mouse model | [96] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Chen, Q.; Lai, H. Development of Antibody Therapeutics against Flaviviruses. Int. J. Mol. Sci. 2018, 19, 54. https://doi.org/10.3390/ijms19010054
Sun H, Chen Q, Lai H. Development of Antibody Therapeutics against Flaviviruses. International Journal of Molecular Sciences. 2018; 19(1):54. https://doi.org/10.3390/ijms19010054
Chicago/Turabian StyleSun, Haiyan, Qiang Chen, and Huafang Lai. 2018. "Development of Antibody Therapeutics against Flaviviruses" International Journal of Molecular Sciences 19, no. 1: 54. https://doi.org/10.3390/ijms19010054
APA StyleSun, H., Chen, Q., & Lai, H. (2018). Development of Antibody Therapeutics against Flaviviruses. International Journal of Molecular Sciences, 19(1), 54. https://doi.org/10.3390/ijms19010054