Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics

 and
and
Abstract
:1. Introduction
2. Fibrinogen Aα-Chain Amyloidosis
3. Aα-Chain Amyloidosis in Asia
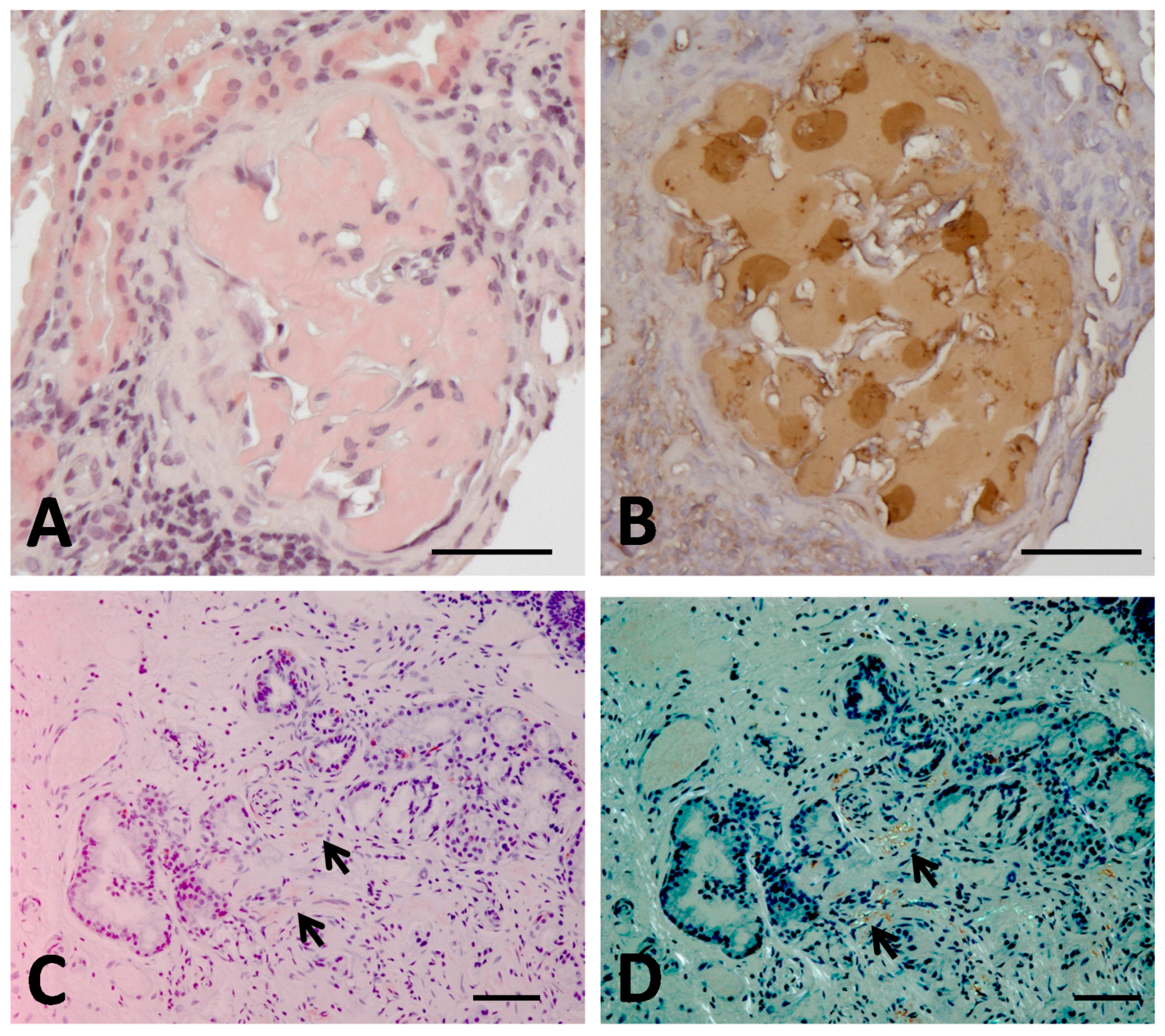
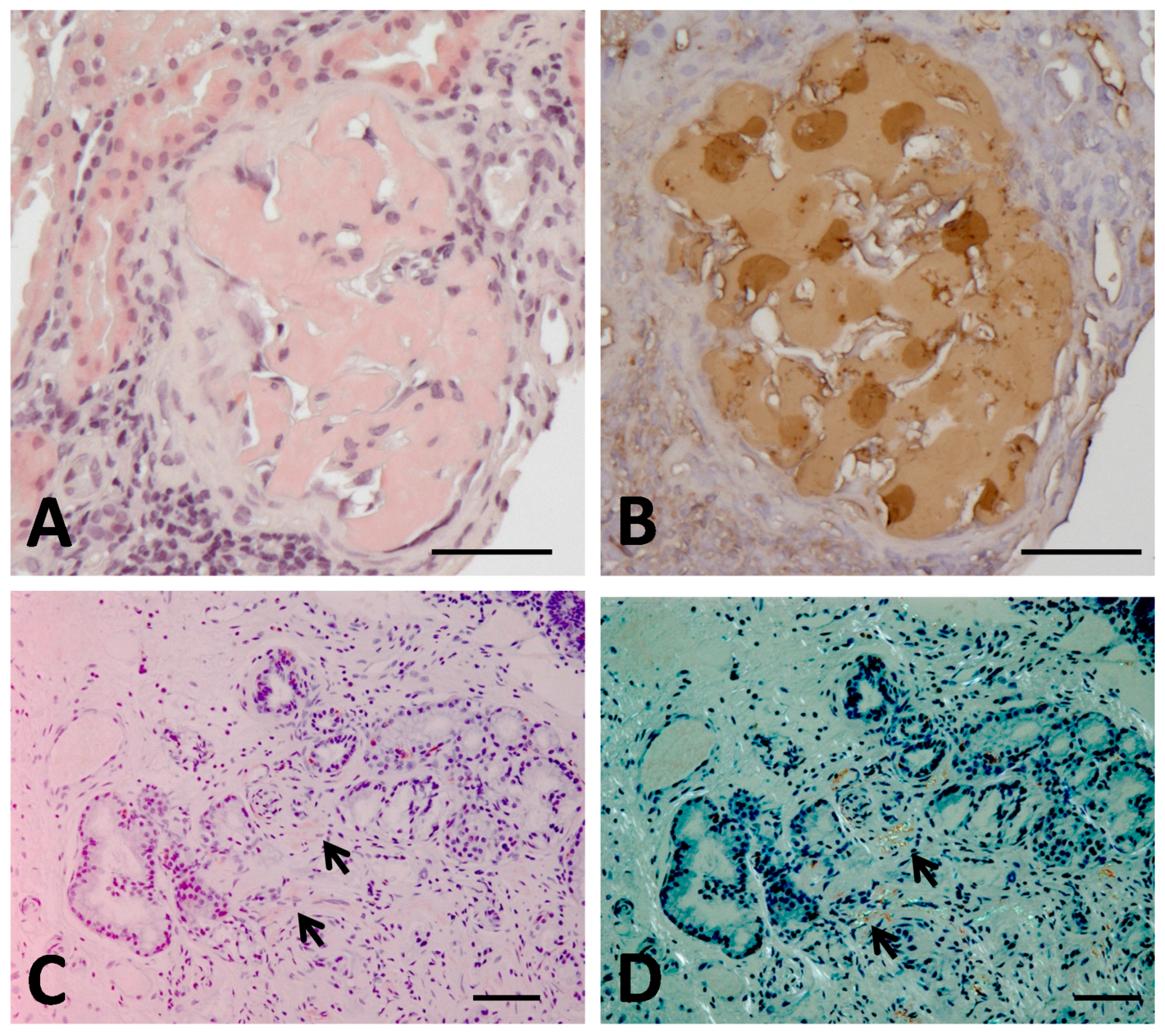
4. The First Case of Aα-Chain Amyloidosis in Japan: Clinical and Molecular Features
5. Discussion
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| Aα-chain amyloidosis | Fibrinogen Aα-chain amyloidosis |
| ESRD | End-stage renal disease |
| LMD | Laser microdissection |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
References
- Ostertag, B. Familiere amyloid-erkrankung. Z. Menschl. Vererbungs Konstit. Pehre 1950, 30, 105–115. [Google Scholar]
- Benson, M.D. Ostertag revisited: The inherited systemic amyloidosis without neuropathy. Amyloid 2005, 12, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.C.; Dwulet, F.E.; Liepnieks, J.; Benson, M.D. Variant apolipoprotein AI as a major constituent of a hemian hereditary amyloid. Biochem. Biophys. Res. Commun. 1988, 156, 762–768. [Google Scholar] [CrossRef]
- Benson, M.D.; Liepnieks, J.; Uemichi, T.; Wheeler, G.; Correa, R. Hereditary renal amyloidosis associated with a mutant fibrinogen α-chain. Nat. Genet. 1993, 3, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hawkins, P.N.; Booth, D.R.; Vigushin, D.M.; Tennent, G.A.; Soutar, A.K.; Totty, N.; Nguyen, O.; Blake, C.C.; Terry, C.J.; et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993, 362, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Liepnieks, J.; Yazaki, M.; Yamashita, T.; Hamidi Asl, K.; Guenther, B.; Kluve-Beckerman, B. A new human hereditary amyloidosis: The result of a stop-codo mutation in the apolipoprotein AII gene. Genomics 2001, 72, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Dasari, S.; Hasadsri, L.; Theis, J.D.; Vrana, J.A.; Gertz, M.A.; Muppa, P.; Zimmermann, M.T.; Grogg, K.L.; Dispenzieri, A.; et al. Novel type of renal amyloidosis derived from apolipoprotein C-II. J. Am. Soc. Nephrol. 2017, 28, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Valleix, S.; Verona, G.; Jourde-Chiche, N.; Nédelec, B.; Mangione, P.P.; Bridoux, F.; Mangé, A.; Dogan, A.; Goujon, J.M.; Lhomme, M.; et al. D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat. Commun. 2016, 7, 10353. [Google Scholar] [CrossRef] [PubMed]
- Uemichi, T.; Liepnieks, J.J.; Benson, M.D. Hereditary renal amyloidosis with a novel variant fibrinogen. J. Clin. Investig. 1994, 93, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Uemichi, T.; Liepnieks, J.J.; Yamada, T.; Gertz, M.A.; Bang, N.; Benosn, M.D. A frame shift mutation in the Fibrinogen A α-chain gene in a kindred with renal amyloidosis. Blood 1996, 87, 4197–4203. [Google Scholar] [PubMed]
- Hamidi Asl, L.; Liepnieks, J.J.; Uemichi, T.; Rebibou, J.M.; Justrabo, E.; Droz, D.; Mousson, C.; Chalopin, J.M.; Benson, M.D.; Delpech, M.; et al. Renal amyloidosis with a frame shift mutation in Fibrinogen Aα-chain gene producing a novel amyloid protein. Blood 1997, 90, 4799–4805. [Google Scholar] [PubMed]
- Kang, H.G.; Bybee, A.; Ha, I.S.; Park, M.S.; Gilbertoson, J.A.; Cheong, H.I.; Choi, Y.; Hawkins, P.N. Hereditary amyloidosis in early childhood associated with a novel insertion-deletion (indel) in the fibrinogen Aα chain gene. Kidney Int. 2005, 68, 1994–1996. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Lachmann, H.J.; Rowczenio, D.; Gilbertoson, J.A.; Zeng, C.H.; Liu, Z.H.; Li, L.S.; Wechalekar, A.; Hawkins, P.N. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen Aα-chain amyloidosis. J. Am. Soc. Nephrol. 2009, 20, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Yoshinaga, T.; Sekijima, Y.; Nishio, S.; Kanizawa, Y.; Kametani, F.; Miyashita, K.; Hachiya, N.; Higuchi, K.; Ikeda, S. The first pure form of Ostertag-type amyloidosis in Japan: A sporadic case of hereditary fibrinogen Aα-chain amyloidosis associated with a novel frameshift variant. Amyloid 2015, 22, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.; Stensland, M.; de Souza, G.A.; Strøm, E.H.; Gilbertson, J.A.; Taylor, G.; Rendell, N.; Minogue, S.; Efebera, Y.A.; Lachmann, H.J.; et al. Renal amyloidosis associated with 5 novel variants in the fibrinogen A alpha cahin protein. Kidney Int. Rep. 2017, 2, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Garnier, C.; Briki, F.; Nedelec, B.; Le Pogamp, P.; Dogan, A.; Rioux-Leclercq, N.; Goude, R.; Beugnet, C.; Martin, L.; Delpech, M.; et al. VLITL is a major cross-b-sheet signal for fibrinogen Aα-chain frameshift variants. Blood 2017, 130, 2799–2807. [Google Scholar] [CrossRef] [PubMed]
- Stangou, A.J.; Banner, N.R.; Hendry, B.M.; Rela, M.; Portmann, B.; Wendon, J.; Monaghan, M.; MacCarthy, P.; Buxton-Thomas, M.; Mathias, C.; et al. Hereditary fibrinogen Aα-chain amyloidosis: Phenotypic characterization of a systemic disease and the role of liver transplantation. Blood 2010, 115, 2998–3007. [Google Scholar] [CrossRef] [PubMed]
- Mourad, G.; Delabre, J.P.; Garigue, V. Cardiac amyloidosis with the E526V mutation of the fibrinogen Aα-chain. N. Engl. J. Med. 2008, 358, 2847–2848. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Booth, D.R.; Booth, S.E.; Bybee, A.; Gilbertson, J.A.; Gillmore, J.D.; Pepys, M.K.; Hawkins, P.N. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N. Engl. J. Med. 2002, 346, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Machado, J.R.; da Silva, M.V.; de Menezes Neves, P.D.M.; de Oliveira, F.A.; Corrêa, R.R.M.; Rodrigues, W.V.D.; Benson, M.D.; dos Reis, M.A. Fibrinogen A α-chain amyloidosis: Report of the first case in Latin America. Amyloid 2013, 20, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Tavares, I.; Oliveira, J.P.; Pinho, A.; Moreira, L.; Rocha, L.; Santos, J.; Pinheiro, J.; Costa, P.P.; Lobato, L. Unrecognized fibrinogen Aα-chain amyloidosis: Results from targeted genetic testing. Am. J. Kidney Dis. 2017, 70, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Tavares, I.; Lobato, L.; Matos, C.; Santos, J.; Moreira, P.; Saraiva, M.J.; Henriques, A.C. Homozygosity for the E526V mutation in fibrinogen A alpha-chain amyloidosis: The first report. Case Rep. Nephrol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, S.X.; Zhang, Y.K. Hereditary fibrinogen Aα-chain amyloidosis caused by the E526V mutation: A case report and literature review. Beijing Da Xue Xue Bao Yi Xue Ban 2014, 46, 802–804. (In Chinese) [Google Scholar] [PubMed]
- Yazaki, M.; Fushimi, T.; Tokuda, T.; Kametani, F.; Yamamoto, K.; Matsuda, M.; Shimojo, H.; Hoshii, Y.; Higuchi, K.; Ikeda, S. A patient with severe renal amyloidosis associated with an immunoglobulin γ-heavy chain fragment. Am. J. Kidney Dis. 2004, 43, E23–E28. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299. [Google Scholar] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Mechanism of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Kant, J.A.; Fornace, A.J., Jr.; Saxe, D.; Simon, M.I.; McBride, O.W.; Crabtree, G.R. Evolution and organization of the fibrinogen locus on chromosome 4: Gene duplication accompanied by transposition and inversion. Proc. Natl. Acad. Sci. USA 1985, 82, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Koopman, J.; Haverkate, F.; Grimbergen, J.; Lord, S.T.; Mosesson, M.W.; DiOrio, J.P.; Siebenlist, K.R.; Legrand, C.; Soria, J.; Soria, C.; et al. The molecular basis for fibrinogen Dusart (Aa554 Arg->Cys) and its association with abnormal fibrinogen polymerization and thrombophilia. J. Clin. Investig. 1993, 91, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Margaglione, M.; Vecchione, G.; Santacroce, R.; D’Angelo, F.; Casetta, B.; Papa, M.L.; Grandone, E.; Di Minno, G. A frameshift mutation in the human fibrinogen α-chain gene (α (499) Ala frameshift stop) leading to dysfibrinogen San Giovanni Rotondo. Thromb. Haemost. 2001, 86, 1483–1488. [Google Scholar] [PubMed]
- Serpell, L.C.; Benson, M.D.; Liepnieks, J.J.; Fraser, P.E. Structural analyses of fibrinogen amyloid fibrils. Amyloid 2007, 14, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Ericzon, B.G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.; Pena, J.R.; Furtado, E.; Barroso, E.; Daniel, J.; Samuel, D.; et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Suhr, O.B.; Conceiçao, I.; Schmidt, H.H.; Trigo, P.; Kelly, J.W.; et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 2013, 260, 2802–2814. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kluve-Beckerman, B.; Zerdenrust, S.R.; Siesky, A.M.; Bodenmiller, D.M.; Showalter, A.D.; Sloop, K.W. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve 2006, 33, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Katoh, N.; Matsuda, M.; Tsuchiya-Suzuki, A.; Ikeda, S. Regression of gastroduodenal amyloid deposition in systemic AL amyloidosis after intensive chemotherapies. Br. J. Haematol. 2011, 153, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Katoh, N.; Matsushima, A.; Kurozumi, M.; Matsuda, M.; Ikeda, S. Marked and rapid regression of hepatic amyloid deposition in a patient with systemic light chain (AL) amyloidosis after high-dose melphalan therapy with stem cell transplantation. Intern. Med. 2014, 53, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Landau, H.; Hassoun, H.; Rosenzweig, M.A.; Maurer, M.; Liu, J.; Flombaum, C.; Bello, C.; Hoover, E.; Riedel, E.; Giralt, S.; et al. Bortezomib and dexamethasone consolidation following risk-adapted melphalan and stem cell transplantation for patients with newly diagnosed light-chain amyloidosis. Leukemia 2013, 27, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Takasugi, K. Successful use of a humanized anti-interleukin-6 receptor antibody, tocilizumab, to treat amyloid a amyloidosis complicating juvenileidiopathic arthritis. Arthritis Rheum. 2006, 54, 2997–3000. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Ohnishi, M.; Matoba, K.; Jouyama, K.; Yamada, A.; Sawada, N.; Mokuda, S.; Murata, Y.; Takasugi, K. Comparison of the clinical utility of tocilizumab and anti-TNF therapy in AA amyloidosis complicating rheumatic diseases. Mod. Rheumatol. 2014, 24, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Stangou, A.J.; Lachmann, H.J.; Goodman, H.J.; Wechalekar, A.D.; Acheson, J.; Tennent, G.A.; Bybee, A.; Gilbertson, J.; Rowczenio, D.; et al. Organ transplantation in hereditary apolipoprotein AI amyloidosis. Am. J. Transplant. 2006, 6, 2342–2347. [Google Scholar] [CrossRef] [PubMed]
- Magy, N.; Liepnieks, J.J.; Yazaki, M.; Kluve-Beckerman, B.; Benson, M.D. Renal transplantation for apolipoprotein AII amyloidosis. Amyloid 2003, 10, 224–228. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| FGA Mutations | Protein Variants | Clinical Features | Geographic Origin/Ethnicity | References |
|---|---|---|---|---|
| Missense Mutations | ||||
| c.1627G > A | E524K | Nephropathy | Scottish | Rowczenio et al. [15] |
| c.1633G > A | E526K | Nephropathy | Russian | Rowczenio et al. [15] |
| c.1634A > T | E526V | Nephropathy | US, Europe, Brazil, China | Uemichi et al. [9] |
| c.1670C > A | T538K | Nephropathy | Chinese | Gillmore et al. [13] |
| c.1676A > T | E540V | Nephropathy | German | Gillmore et al. [13] |
| c.1712C > A | P552H | Nephropathy | Afro-Caribbean | Gillmore et al. [13] |
| c.1718G > T | R554L | Nephropathy | Mexico, Europe, US (Afro-American) | Benson et al. [4] |
| Deletion Mutations | ||||
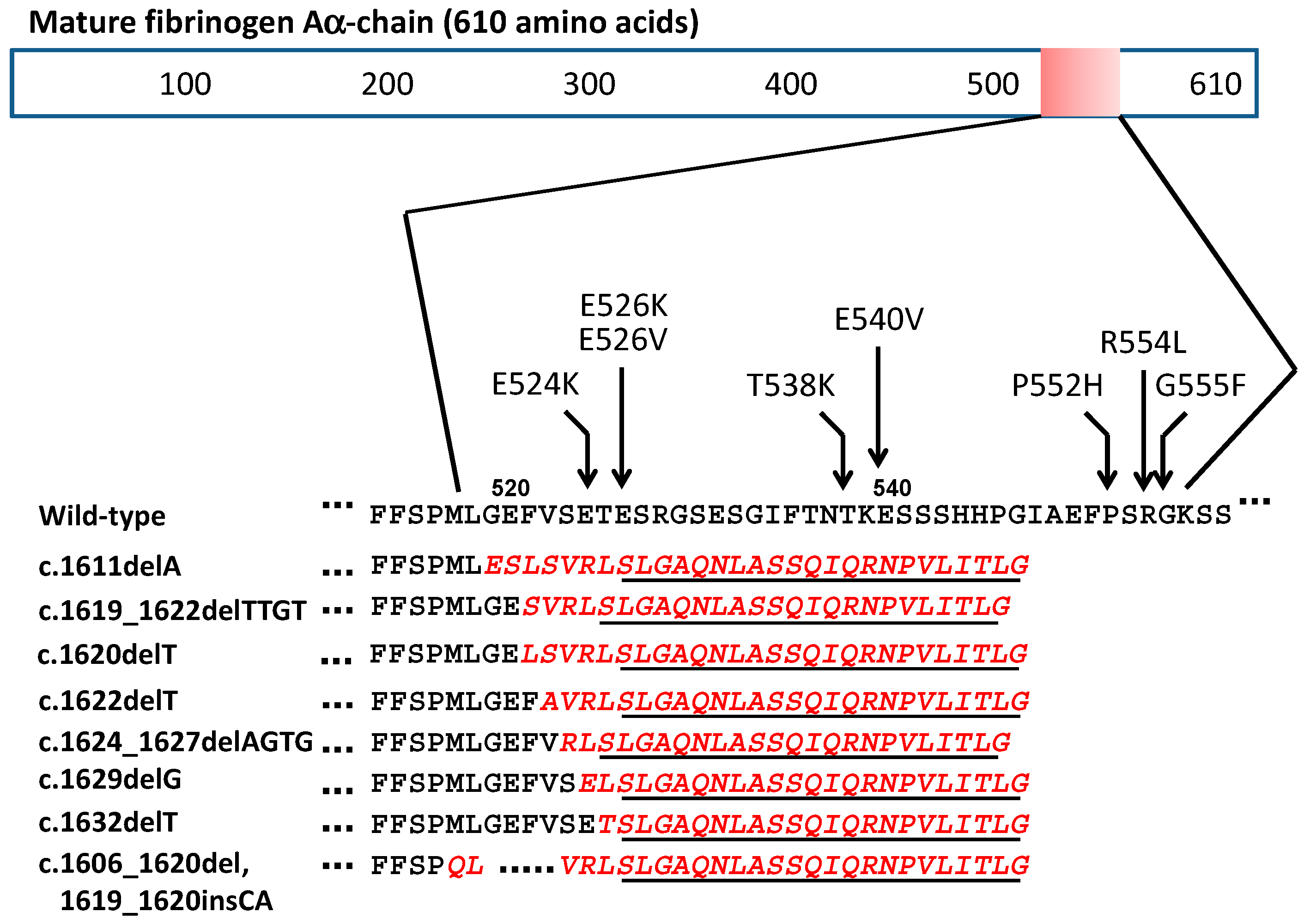
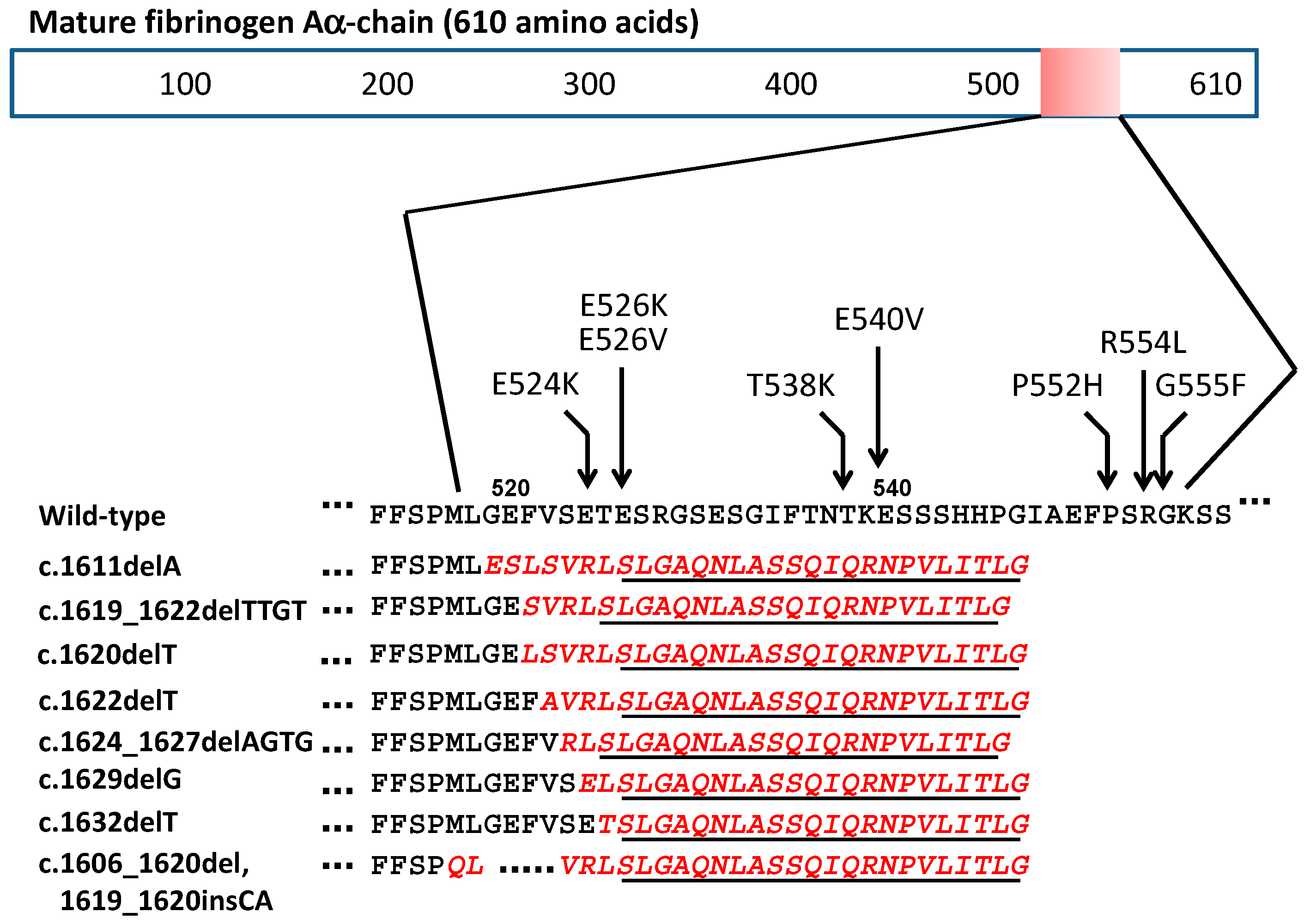
| c.1611delA (4886delA) | G519EfsX548 | Nephropathy | French | Rowczenio et al. [15] |
| c.1619_1622delTTGT (4894_4897delTTGT) | F521SfsX547 | Nephropathy | North African | Rowczenio et al. [15] |
| c.1620delT (4895delT) | F521LfsX548 | Nephropathy | French | Garnie et al. [16] |
| c.1622delT (4897delT) | V522AfsX548 | Nephropathy | French | Hamidi et al. [11] |
| c.1624_1627delAGTG (4899_4902delAGTG ) | S523RfsX547 | Nephropathy | Japanese | Yazaki et al. [14] |
| c.1629delG (4904delG) | E524EfsX548 | Nephropathy | US | Uemichi et al. [10] |
| c.1632delT (4907delT) | T525TfsX548 | Nephropathy | Chinese | Gillmore et al. [13] |
| Insertion-Deletion | ||||
| c.1606_1620del, 1619_1620insCA (4881_4895del, 4894_4895insCA) | M517_F521del insQSfsX548 | Nephropathy | Korean | Kang et al. [12] |
| c.1720_1721delGGinsTT (5445_5446delGGinsTT) | G555F | Nephropathy | Norwegian | Rowczenio et al. [15] |
| Types of Systemic Amyloidosis | Precursor Proteins | Therapeutic Options | References |
|---|---|---|---|
| Hereditary ATTR amyloidosis | Transthyretin | Liver transplant, tafamidis, diflunisal, gene therapies | [31,32,33,34,35] |
| Primary AL amyloidosis | Immunoglobulin light chain | Chemotherapies, stem cell transplantation | [36,37,38] |
| Reactive AA amyloidosis | Serum amyloid A (SAA) | Control of underlying diseases, biologic agents | [39,40] |
| Fibrinogen Aα-chain amyloidosis | Fibrinogen Aα-chain | Renal transplant, liver and renal transplant | [13,17] |
| Apolipoprotein A-I amyloidosis | Apolipoprotein A-I | Renal transplant, liver and renal transplant | [41] |
| Apolipoprotein A-II amyloidosis | Apolipoprotein A-II | Renal transplant | [42] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yazaki, M.; Yoshinaga, T.; Sekijima, Y.; Kametani, F.; Okumura, N. Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics. Int. J. Mol. Sci. 2018, 19, 320. https://doi.org/10.3390/ijms19010320
Yazaki M, Yoshinaga T, Sekijima Y, Kametani F, Okumura N. Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics. International Journal of Molecular Sciences. 2018; 19(1):320. https://doi.org/10.3390/ijms19010320
Chicago/Turabian StyleYazaki, Masahide, Tsuneaki Yoshinaga, Yoshiki Sekijima, Fuyuki Kametani, and Nobuo Okumura. 2018. "Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics" International Journal of Molecular Sciences 19, no. 1: 320. https://doi.org/10.3390/ijms19010320
APA StyleYazaki, M., Yoshinaga, T., Sekijima, Y., Kametani, F., & Okumura, N. (2018). Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics. International Journal of Molecular Sciences, 19(1), 320. https://doi.org/10.3390/ijms19010320