Chaperones and the Proteasome System: Regulating the Construction and Demolition of Striated Muscle


Abstract
1. Introduction
2. The Chaperones
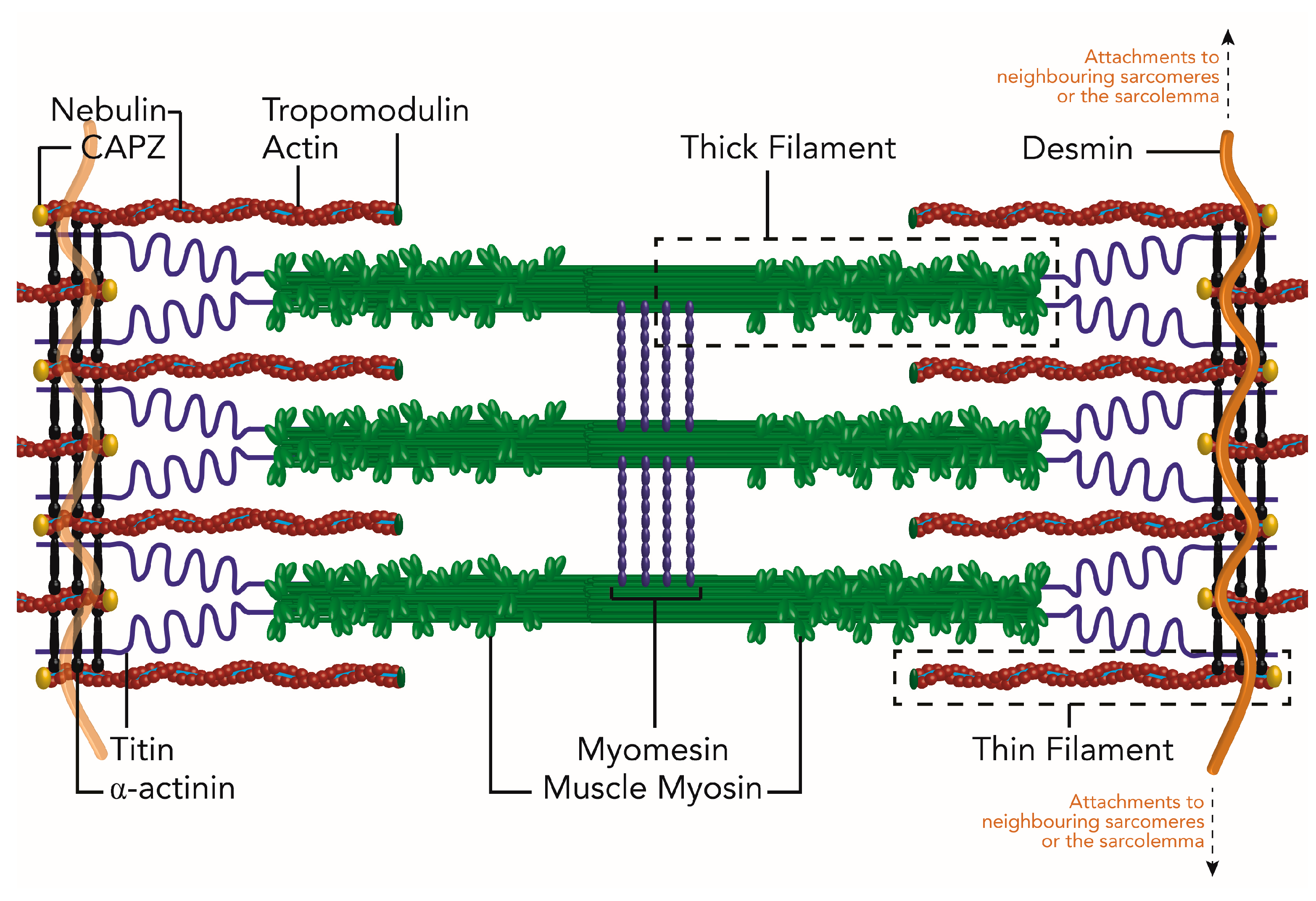
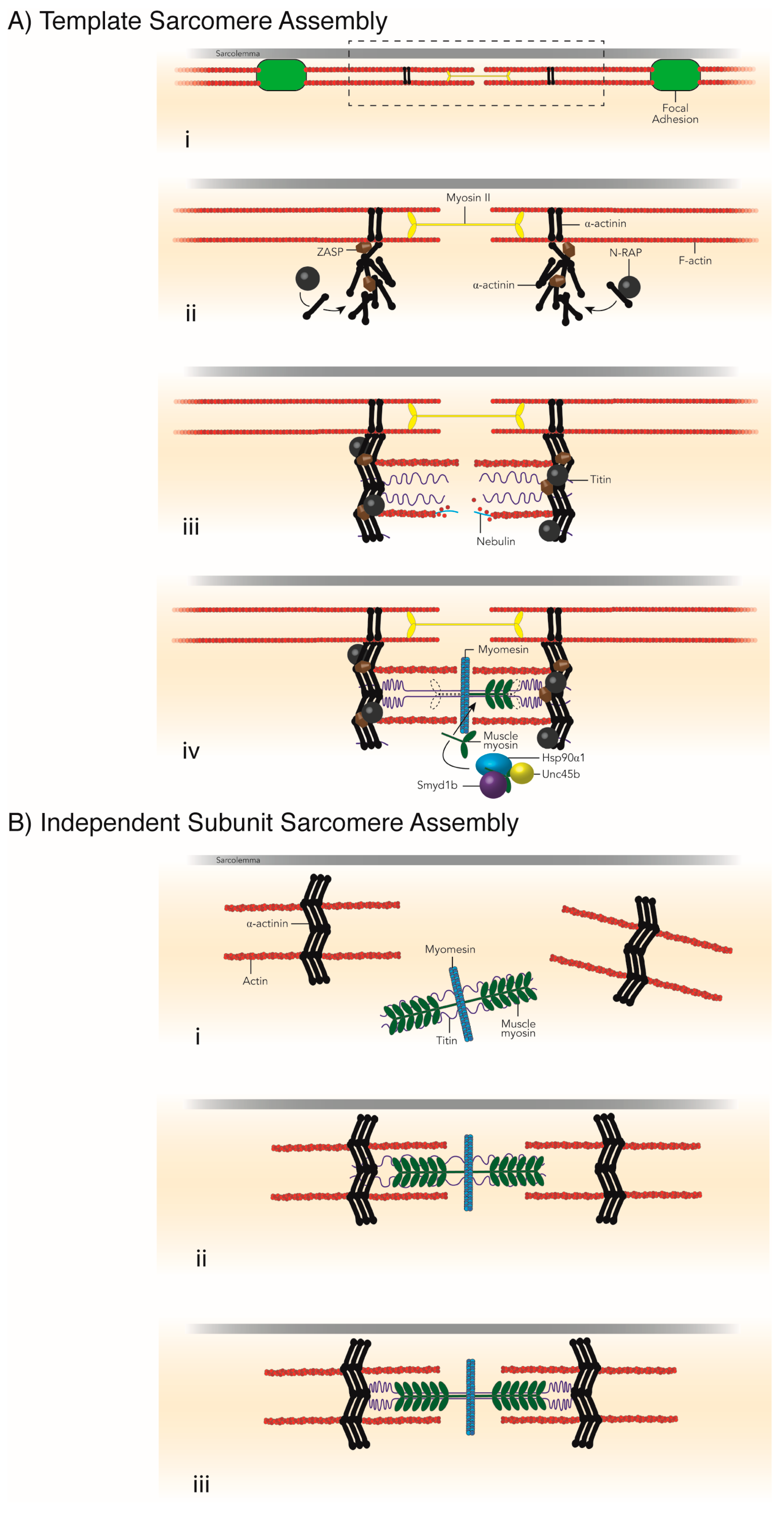
3. Necessity of Chaperones for Sarcomere Assembly
3.1. Z-Disc Assembly
3.2. Thin Filament Formation
3.3. Non-Muscle Myosin Filaments
3.4. Titin Folding and Incorporation
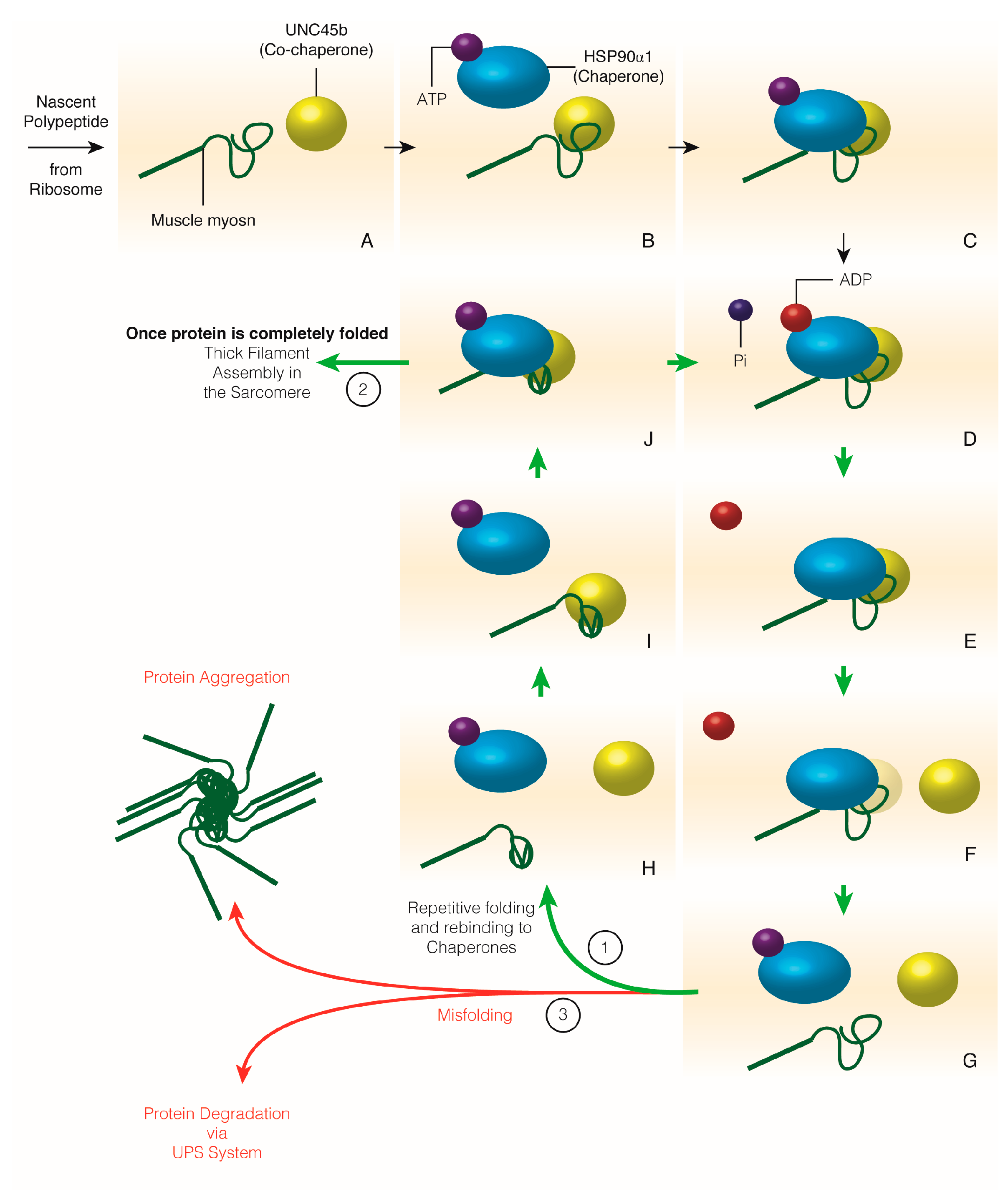
3.5. Muscle Myosin Folding and Thick Filament Assembly
3.6. M-Line Assembly
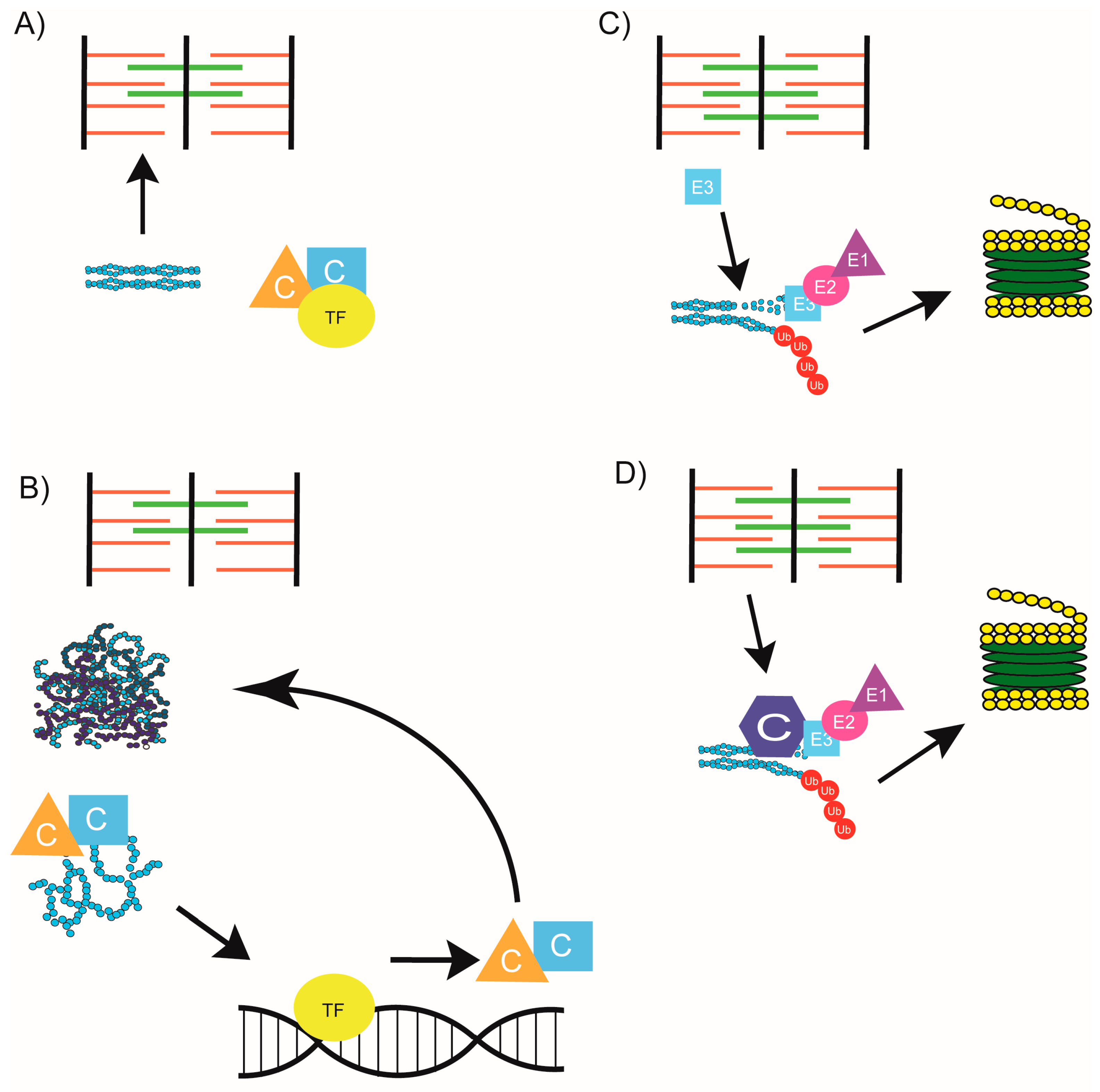
4. Chaperone Homeostasis
5. Chaperones beyond Assembly
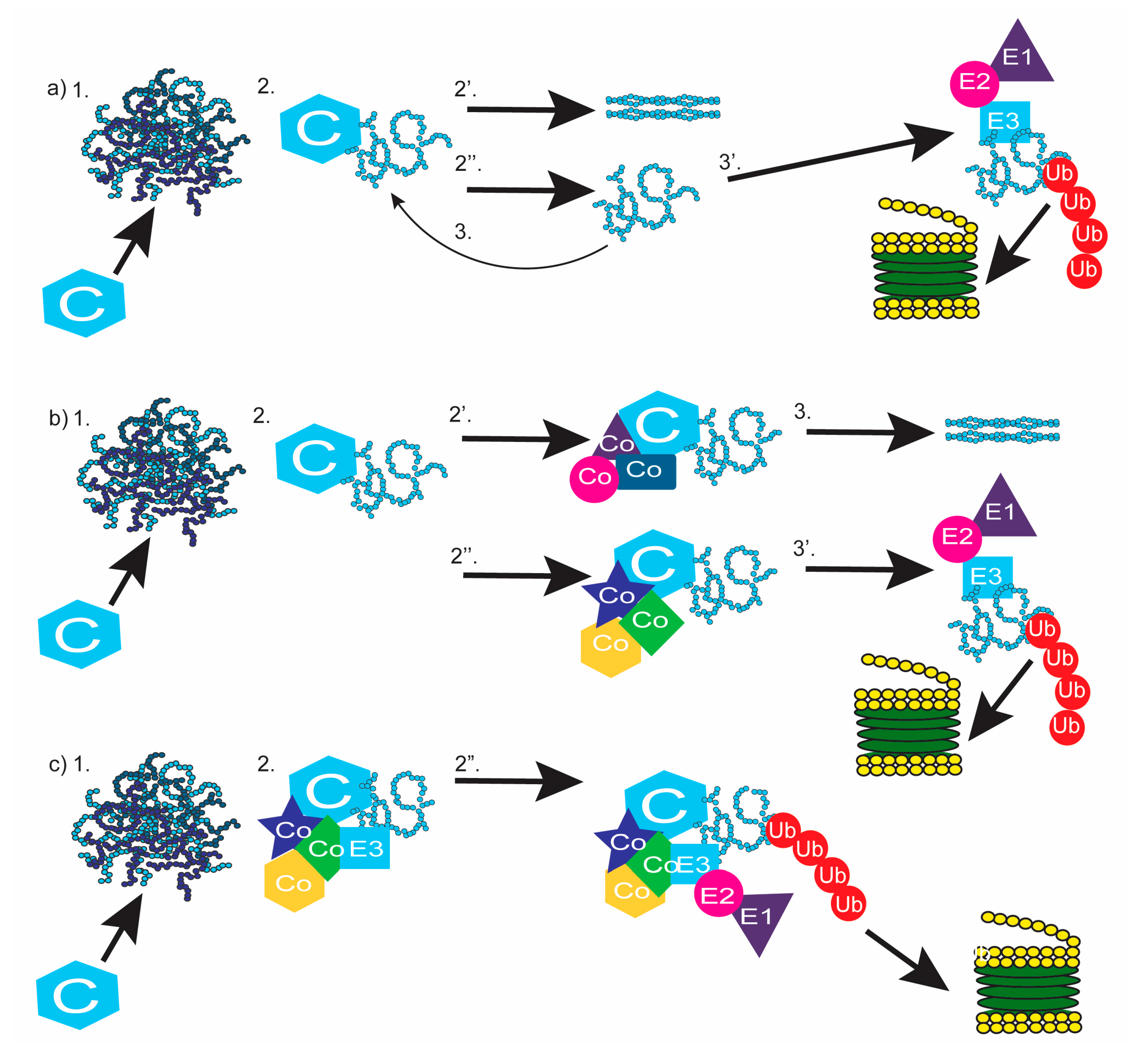
6. Completing the Chain: Communication between Chaperones and the Proteasome
7. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bianchi, T.; Gelosa, L. On the preventive detection of pathogenic staphylococci in the rhinopharynx of employees in the food industry in the province of Milan during the 5-year period of 1967–1971. Ann. Sclavo 1973, 15, 83–98. [Google Scholar] [PubMed]
- Van den Berg, B.; Wain, R.; Dobson, C.M.; Ellis, R.J. Macromolecular crowding perturbs protein refolding kinetics: Implications for folding inside the cell. EMBO J. 2000, 19, 3870–3875. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef] [PubMed]
- Kony, D.B.; Hunenberger, P.H.; van Gunsteren, W.F. Molecular dynamics simulations of the native and partially folded states of ubiquitin: Influence of methanol cosolvent, pH, and temperature on the protein structure and dynamics. Protein Sci. 2007, 16, 1101–1118. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.J.; Goldberger, R.F.; Anfinsen, C.B. The genetic control of tertiary protein structure: Studies with model systems. Cold Spring Harb. Symp. Quant. Biol. 1963, 28, 439–449. [Google Scholar] [CrossRef]
- Frydman, J.; Nimmesgern, E.; Ohtsuka, K.; Hartl, F.U. Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 1994, 370, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, J.P.; Hartl, F.U. Molecular chaperone functions of heat-shock proteins. Ann. Rev. Biochem. 1993, 62, 349–384. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Hemmingsen, S.M. Molecular chaperones: Proteins essential for the biogenesis of some macromolecular structures. Trends Biochem. Sci. 1989, 14, 339–342. [Google Scholar] [CrossRef]
- Malicdan, M.C.; Noguchi, S.; Nonaka, I.; Saftig, P.; Nishino, I. Lysosomal myopathies: An excessive build-up in autophagosomes is too much to handle. Neuromuscul. Disord. 2008, 18, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Sandri, M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy 2010, 6, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I. Autophagic vacuolar myopathy. Semin. Pediatr. Neurol. 2006, 13, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Paul, S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: Therapeutic approaches. BioEssays 2008, 30, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Janiesch, P.C.; Kim, J.; Mouysset, J.; Barikbin, R.; Lochmuller, H.; Cassata, G.; Krause, S.; Hoppe, T. The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat. Cell Biol. 2007, 9, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.C.; Goebel, H.H. Protein aggregate myopathies. Neurol. India 2005, 53, 273–279. [Google Scholar] [PubMed]
- Askanas, V.; Engel, W.K.; Nogalska, A. Inclusion body myositis: A degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol. 2009, 19, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Garlepp, M.J.; Mastaglia, F.L. Inclusion body myositis. J. Neurol. Neurosurg. Psychiatry 1996, 60, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Alberti, S.; Hohfeld, J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta 2004, 1695, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Nollen, E.A.; Kabakov, A.E.; Brunsting, J.F.; Kanon, B.; Hohfeld, J.; Kampinga, H.H. Modulation of in vivo HSP70 chaperone activity by Hip and Bag-1. J. Biol. Chem. 2001, 276, 4677–4682. [Google Scholar] [CrossRef] [PubMed]
- Abdulhaq, U.N.; Daana, M.; Dor, T.; Fellig, Y.; Eylon, S.; Schuelke, M.; Shaag, A.; Elpeleg, O.; Edvardson, S. Nemaline body myopathy caused by a novel mutation in troponin T1 (TNNT1). Muscle Nerve 2016, 53, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Simpson, K.; Tesi-Rocha, C.; Zhou, D.; Palmer, C.A.; Suchy, S.F. Novel large deletion in the ACTA1 gene in a child with autosomal recessive nemaline myopathy. Neuromuscul. Disord. 2014, 24, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Ilkovski, B.; Cooper, S.T.; Nowak, K.; Ryan, M.M.; Yang, N.; Schnell, C.; Durling, H.J.; Roddick, L.G.; Wilkinson, I.; Kornberg, A.J.; et al. Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene. Am. J. Hum. Genet. 2001, 68, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Wallefeld, W.; Krause, S.; Nowak, K.J.; Dye, D.; Horvath, R.; Molnar, Z.; Szabo, M.; Hashimoto, K.; Reina, C.; De Carlos, J.; et al. Severe nemaline myopathy caused by mutations of the stop codon of the skeletal muscle alpha actin gene (ACTA1). Neuromuscul. Disord. 2006, 16, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Md, E.P.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 Causes Severe Dominant Childhood Muscular Dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Keira, Y.; Noguchi, S.; Minami, N.; Hayashi, Y.K.; Nishino, I. Localization of Calpain 3 in Human Skeletal Muscle and Its Alteration in Limb-Girdle Muscular Dystrophy 2A Muscle. J. Biochem. 2003, 133, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Garvey, S.M.; Rajan, C.; Lerner, A.P.; Frankel, W.N.; Cox, G.A. The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics 2002, 79, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1–12. [Google Scholar] [PubMed]
- Hackman, P.; Marchand, S.; Sarparanta, J.; Vihola, A.; Penisson-Besnier, I.; Eymard, B.; Pardal-Fernandez, J.M.; Hammouda, E.-H.; Richard, I.; Illa, I.; et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul. Disord. 2008, 18, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Hackman, P.; Vihola, A.; Haravuori, H.; Marchand, S.; Sarparanta, J.; De Seze, J.; Labeit, S.; Witt, C.; Peltonen, L.; Richard, I.; et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002, 71, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, J.; Blandin, G.; Charton, K.; Vihola, A.; Marchand, S.; Milic, A.; Hackman, P.; Ehler, E.; Richard, I.; Udd, B. Interactions with M-band titin and calpain 3 link myospryn (CMYA5) to tibial and limb-girdle muscular dystrophies. J. Biol. Chem. 2010, 285, 30304–30315. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Thottakara, T.; Friedrich, F.W.; Reischmann, S.; Braumann, S.; Schlossarek, S.; Kramer, E.; Juhr, D.; Schluter, H.; van der Velden, J.; Munch, J.; et al. The E3 ubiquitin ligase Asb2beta is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation. J. Mol. Cell. Cardiol. 2015, 87, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Au, Y. The muscle ultrastructure: A structural perspective of the sarcomere. Cell. Mol. Life Sci. 2004, 61, 3016–3033. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Sykes, B.D. Targeting the sarcomere to correct muscle function. Nat. Rev. Drug Discov. 2015, 14, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Knoll, R.; Buyandelger, B.; Lab, M. The sarcomeric Z-disc and Z-discopathies. J. Biomed. Biotechnol. 2011, 2011, 569628. [Google Scholar] [CrossRef] [PubMed]
- Luther, P.K. The vertebrate muscle Z-disc: Sarcomere anchor for structure and signalling. J. Muscle Res. Cell Motil. 2009, 30, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.C.; Geeves, M.A. The structural basis of muscle contraction. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2000, 355, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Tskhovrebova, L.; Trinick, J. Roles of titin in the structure and elasticity of the sarcomere. J. Biomed. Biotechnol. 2010, 2010, 612482. [Google Scholar] [CrossRef] [PubMed]
- Myhre, J.L.; Pilgrim, D. A Titan but not necessarily a ruler: Assessing the role of titin during thick filament patterning and assembly. Anat. Rec. 2014, 297, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Carland, C.R.; Guo, Y.; Bernstein, S.I. Getting Folded: Chaperone Proteins in Muscle Development, Maintenance and Disease. Anat. Rec. 2014, 297, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhu, X. The molecular mechanisms of calpains action on skeletal muscle atrophy. Physiol. Res. 2016, 65, 547–560. [Google Scholar] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Chereau, D.; Boczkowska, M.; Skwarek-Maruszewska, A.; Fujiwara, I.; Hayes, D.B.; Rebowski, G.; Lappalainen, P.; Pollard, T.D.; Dominguez, R. Leiomodin is an actin filament nucleator in muscle cells. Science 2008, 320, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D. The premyofibril: Evidence for its role in myofibrillogenesis. Cell Motil. Cytoskelet. 1994, 28, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, S.; Bugg, C.E.; Wilkinson, K.D.; Vierstra, R.D.; Hatfield, P.M.; Cook, W.J. Comparison of the three-dimensional structures of human, yeast, and oat ubiquitin. J. Biol. Chem. 1987, 262, 6396–6399. [Google Scholar] [PubMed]
- Kriegenburg, F.; Ellgaard, L.; Hartmann-Petersen, R. Molecular chaperones in targeting misfolded proteins for ubiquitin-dependent degradation. FEBS J. 2012, 279, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef] [PubMed]
- Kettern, N.; Dreiseidler, M.; Tawo, R.; Hohfeld, J. Chaperone-assisted degradation: Multiple paths to destruction. Biol. Chem. 2010, 391, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. The molecular chaperone concept. Semin. Cell Biol. 1990, 1, 1–9. [Google Scholar] [PubMed]
- Caplan, A.J. What is a co-chaperone? Cell Stress Chaperones 2003, 8, 105–107. [Google Scholar] [CrossRef]
- Langer, T.; Lu, C.; Echols, H.; Flanagan, J.; Hayer, M.K.; Hartl, F.U. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 1992, 356, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Srivastava, P. Heat-shock proteins. Curr. Protoc. Immunol. 2014. Appendix 1, Appendix 1T. [Google Scholar]
- Craig, E.A.; Gambill, B.D.; Nelson, R.J. Heat shock proteins: Molecular chaperones of protein biogenesis. Microbiol. Rev. 1993, 57, 402–414. [Google Scholar] [PubMed]
- Etard, C.; Armant, O.; Roostalu, U.; Gourain, V.; Ferg, M.; Strähle, U. Loss of function of myosin chaperones triggers Hsf1-mediated transcriptional response in skeletal muscle cells. Genome Biol. 2015, 16, 267. [Google Scholar] [CrossRef] [PubMed]
- Siegert, R.; Leroux, M.R.; Scheufler, C.; Hartl, F.U.; Moarefi, I. Structure of the molecular chaperone prefoldin: Unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 2000, 103, 621–632. [Google Scholar] [CrossRef]
- Sanger, J.W.; Wang, J.; Holloway, B.; Du, A.; Sanger, J.M. Myofibrillogenesis in skeletal muscle cells in zebrafish. Cell Motil. Cytoskelet. 2009, 66, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.W.; Kang, S.; Siebrands, C.C.; Freeman, N.; Du, A.; Wang, J.; Stout, A.L.; Sanger, J.M. How to build a myofibril. J. Muscle Res. Cell Motil. 2005, 26, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.W.; Wang, J.; Fan, Y.; White, J.; Sanger, J.M. Assembly and dynamics of myofibrils. J. Biomed. Biotechnol. 2010, 2010, 858606. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.C.; Schock, F. The initial steps of myofibril assembly: Integrins pave the way. Nat. Rev. Mol. Cell Biol. 2009, 10, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Dlugosz, A.A.; Antin, P.B.; Nachmias, V.T.; Holtzer, H. The relationship between stress fiber-like structures and nascent myofibrils in cultured cardiac myocytes. J. Cell Biol. 1984, 99, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.M.; Mittal, B.; Pochapin, M.B.; Sanger, J.W. Myofibrillogenesis in living cells microinjected with fluorescently labeled alpha-actinin. J. Cell Biol. 1986, 102, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.W.; Mittal, B.; Sanger, J.M. Analysis of myofibrillar structure and assembly using fluorescently labeled contractile proteins. J. Cell Biol. 1984, 98, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.H.; DiLullo, C.; Schultheiss, T.; Holtzer, S.; Murray, J.M.; Choi, J.; Fischman, D.A.; Holtzer, H. The vinculin/sarcomeric-alpha-actinin/alpha-actin nexus in cultured cardiac myocytes. J. Cell Biol. 1992, 117, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Holtzer, H.; Hijikata, T.; Lin, Z.X.; Zhang, Z.Q.; Holtzer, S.; Protasi, F.; Franzini-Armstrong, C.; Sweeney, H.L. Independent assembly of 1.6 microns long bipolar MHC filaments and I-Z-I bodies. Cell Struct. Funct. 1997, 22, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.L.; Herrera, A.H.; Horowits, R. Targeting and functional role of N-RAP, a nebulin-related LIM protein, during myofibril assembly in cultured chick cardiomyocytes. J. Cell Sci. 2001, 114, 4229–4238. [Google Scholar] [PubMed]
- Just, S.; Meder, B.; Berger, I.M.; Etard, C.; Trano, N.; Patzel, E.; Hassel, D.; Marquart, S.; Dahme, T.; Vogel, B.; et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J. Cell Sci. 2011, 124, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Jani, K.; Schock, F. Zasp is required for the assembly of functional integrin adhesion sites. J. Cell Biol. 2007, 179, 1583–1597. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Wang, P.; Streuli, C.; Geiger, B.; Humphries, M.J.; Ballestrem, C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 2007, 179, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Tokuyasu, K.T. Immunocytochemical studies of cardiac myofibrillogenesis in early chick embryos. III. Generation of fasciae adherentes and costameres. J. Cell Biol. 1989, 108, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Volk, T.; Fessler, L.I.; Fessler, J.H. A role for integrin in the formation of sarcomeric cytoarchitecture. Cell 1990, 63, 525–536. [Google Scholar] [CrossRef]
- Hilenski, L.L.; Ma, X.H.; Vinson, N.; Terracio, L.; Borg, T.K. The role of beta 1 integrin in spreading and myofibrillogenesis in neonatal rat cardiomyocytes in vitro. Cell Motil. Cytoskelet. 1992, 21, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Manisastry, S.M.; Zaal, K.J.; Horowits, R. Myofibril assembly visualized by imaging N-RAP, alpha-actinin, and actin in living cardiomyocytes. Exp. Cell Res. 2009, 315, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Littlefield, R.; Almenar-Queralt, A.; Fowler, V.M. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 2001, 3, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Mardahl-Dumesnil, M.; Fowler, V.M. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. J. Cell Biol. 2001, 155, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.B.; Ribbeck, K.; Hagler, D.J.; Spitzer, N.C. A calcium signaling cascade essential for myosin thick filament assembly in Xenopus myocytes. J. Cell Biol. 1998, 141, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Du, A.; Sanger, J.M.; Linask, K.K.; Sanger, J.W. Myofibrillogenesis in the first cardiomyocytes formed from isolated quail precardiac mesoderm. Dev. Biol. 2003, 257, 382–394. [Google Scholar] [CrossRef]
- Kachur, T.; Ao, W.; Berger, J.; Pilgrim, D. Maternal UNC-45 is involved in cytokinesis and colocalizes with non-muscle myosin in the early Caenorhabditis elegans embryo. J. Cell Sci. 2004, 117, 5313–5321. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, T.; Andresen, C.; Unger, A.; Just, S.; Rottbauer, W.; Linke, W.A. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim. Biophys. Acta 2013, 1833, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Donlin, L.T.; Andresen, C.; Just, S.; Rudensky, E.; Pappas, C.T.; Kruger, M.; Jacobs, E.Y.; Unger, A.; Zieseniss, A.; Dobenecker, M.W.; et al. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev. 2012, 26, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Linke, W.A. The giant protein titin: A regulatory node that integrates myocyte signaling pathways. J. Biol. Chem. 2011, 286, 9905–9912. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lowe, T.; Hoppe, T. Protein quality control gets muscle into shape. Trends Cell Biol. 2008, 18, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Etard, C.; Behra, M.; Fischer, N.; Hutcheson, D.; Geisler, R.; Strahle, U. The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis. Dev. Biol. 2007, 308, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, T.A.; Haramis, A.P.; Etard, C.; Prodromou, C.; Vaughan, C.K.; Ashworth, R.; Ray, S.; Behra, M.; Holder, N.; Talbot, W.S.; et al. The ATPase-dependent chaperoning activity of Hsp90a regulates thick filament formation and integration during skeletal muscle myofibrillogenesis. Development 2008, 135, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Prill, K.; Reid, P.W.; Wohlgemuth, S.L.; Pilgrim, D.B. Still Heart Encodes a Structural HMT, SMYD1b, with Chaperone-Like Function during Fast Muscle Sarcomere Assembly. PLoS ONE 2015, 10, e0142528. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Srikakulam, R.; Winkelmann, D.A. Unc45 activates Hsp90-dependent folding of the myosin motor domain. J. Biol. Chem. 2008, 283, 13185–13193. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Wittung-Stafshede, P.; Guidry, J.; Horne, B.E.; Landry, S.J. The J-domain of Hsp40 couples ATP hydrolysis to substrate capture in Hsp70. Biochemistry 2003, 42, 4937–4944. [Google Scholar] [CrossRef] [PubMed]
- Srikakulam, R.; Liu, L.; Winkelmann, D.A. Unc45b forms a cytosolic complex with Hsp90 and targets the unfolded myosin motor domain. PLoS ONE 2008, 3, e2137. [Google Scholar] [CrossRef] [PubMed]
- Bujalowski, P.J.; Nicholls, P.; Oberhauser, A.F. UNC-45B chaperone: The role of its domains in the interaction with the myosin motor domain. Biophys. J. 2014, 107, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, Y.; Wang, Z.; Gao, J.; Xu, J.; Chu, W.; Zhang, J.; Fang, S.; Du, S.J. Smyd1b is required for skeletal and cardiac muscle function in zebrafish. Mol. Biol. Cell 2013, 24, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- Du, S.J.; Li, H.; Bian, Y.; Zhong, Y. Heat-shock protein 90alpha1 is required for organized myofibril assembly in skeletal muscles of zebrafish embryos. Proc. Natl. Acad. Sci. USA 2008, 105, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Sass, J.B.; Martin, C.C.; Krone, P.H. Restricted expression of the zebrafish hsp90alpha gene in slow and fast muscle fiber lineages. Int. J. Dev. Biol. 1999, 43, 835–838. [Google Scholar] [PubMed]
- Agarkova, I.; Perriard, J.C. The M-band: An elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 2005, 15, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, A.; Lange, S.; Holt, M.; Vihola, A.; Carmignac, V.; Ferreiro, A.; Udd, B.; Gautel, M. Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band: Implications for hereditary myopathies. J. Cell Sci. 2008, 121, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Kontrogianni-Konstantopoulos, A.; Catino, D.H.; Strong, J.C.; Sutter, S.; Borisov, A.B.; Pumplin, D.W.; Russell, M.W.; Bloch, R.J. Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J. 2006, 20, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Agarkova, I.; Perriard, J.C.; Ehler, E. The sarcomeric M-band during development and in disease. J. Muscle Res. Cell Motil. 2005, 26, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.D.; Pierce, S.A.; Sims, R.J.; Yamagishi, H.; Weihe, E.K.; Harriss, J.V.; Maika, S.D.; Kuziel, W.A.; King, H.L.; Olson, E.N.; et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J.; Weihe, E.K.; Zhu, L.; O’Malley, S.; Harriss, J.V.; Gottlieb, P.D. m-Bop, a repressor protein essential for cardiogenesis, interacts with skNAC, a heart- and muscle-specific transcription factor. J. Biol. Chem. 2002, 277, 26524–26529. [Google Scholar] [CrossRef] [PubMed]
- Bonnemann, C.G.; Laing, N.G. Myopathies resulting from mutations in sarcomeric proteins. Curr. Opin. Neurol. 2004, 17, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Bernick, E.P.; Zhang, P.J.; Du, S. Knockdown and overexpression of Unc-45b result in defective myofibril organization in skeletal muscles of zebrafish embryos. BMC Cell Biol. 2010, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, T.; Cassata, G.; Barral, J.M.; Springer, W.; Hutagalung, A.H.; Epstein, H.F.; Baumeister, R. Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell 2004, 118, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Portbury, A.L.; Patterson, C. Build it up-Tear it down: Protein quality control in the cardiac sarcomere. Cardiovasc. Res. 2009, 81, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Barral, J.M.; Hutagalung, A.H.; Brinker, A.; Hartl, F.U.; Epstein, H.F. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science 2002, 295, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, P.C.; Briand, P.A.; Picard, D. A Remodeled Hsp90 Molecular Chaperone Ensemble with the Novel Cochaperone Aarsd1 Is Required for Muscle Differentiation. Mol. Cell. Biol. 2016, 36, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Laskey, R.A.; Honda, B.M.; Mills, A.D.; Finch, J.T. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 1978, 275, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Molecular chaperones: Assisting assembly in addition to folding. Trends Biochem. Sci. 2006, 31, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Furst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Bimston, D.N.; Matsuzawa, S.; Freeman, B.C.; Aime-Sempe, C.; Xie, Z.; Morimoto, R.I.; Reed, J.C. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 1997, 16, 4887–4896. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Rogon, C.; Hohfeld, J. To be, or not to be—Molecular chaperones in protein degradation. Cell. Mol. Life Sci. 2007, 64, 2525–2541. [Google Scholar] [CrossRef] [PubMed]
- Bercovich, B.; Stancovski, I.; Mayer, A.; Blumenfeld, N.; Laszlo, A.; Schwartz, A.L.; Ciechanover, A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem. 1997, 272, 9002–9010. [Google Scholar] [CrossRef] [PubMed]
- Luders, J.; Demand, J.; Hohfeld, J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 2000, 275, 4613–4617. [Google Scholar] [CrossRef] [PubMed]
- Demand, J.; Alberti, S.; Patterson, C.; Hohfeld, J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef]
- Dai, Q.; Zhang, C.; Wu, Y.; McDonough, H.; Whaley, R.A.; Godfrey, V.; Li, H.H.; Madamanchi, N.; Xu, W.; Neckers, L.; et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003, 22, 5446–5458. [Google Scholar] [CrossRef] [PubMed]
- Etard, C.; Behra, M.; Ertzer, R.; Fischer, N.; Jesuthasan, S.; Blader, P.; Geisler, R.; Strahle, U. Mutation in the delta-subunit of the nAChR suppresses the muscle defects caused by lack of Dystrophin. Dev. Dyn. 2005, 234, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Behra, M.; Cousin, X.; Bertrand, C.; Vonesch, J.L.; Biellmann, D.; Chatonnet, A.; Strahle, U. Acetylcholinesterase is required for neuronal and muscular development in the zebrafish embryo. Nat. Neurosci. 2002, 5, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Myhre, J.L.; Hills, J.A.; Prill, K.; Wohlgemuth, S.L.; Pilgrim, D.B. The titin A-band rod domain is dispensable for initial thick filament assembly in zebrafish. Dev. Biol. 2014, 387, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.Y.; Patterson, C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [PubMed]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Hohfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [PubMed]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [PubMed]
- Gottesman, S.; Wickner, S.; Maurizi, M.R. Protein quality control: Triage by chaperones and proteases. Genes Dev. 1997, 11, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational quality control: Folding, refolding, and degrading proteins. Science 1999, 286, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Odunuga, O.O.; Longshaw, V.M.; Blatch, G.L. Hop: More than an Hsp70/Hsp90 adaptor protein. BioEssays 2004, 26, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Minami, Y.; Minami, M.; Chiba, T.; Tanaka, K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001, 2, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, T.; Bukau, B.; Buchberger, A. Protein turnover: A CHIP programmed for proteolysis. Curr. Biol. 2002, 12, R26–R28. [Google Scholar] [CrossRef]
- Kanelakis, K.C.; Murphy, P.J.; Galigniana, M.D.; Morishima, Y.; Takayama, S.; Reed, J.C.; Toft, D.O.; Pratt, W.B. hsp70 interacting protein Hip does not affect glucocorticoid receptor folding by the hsp90-based chaperone machinery except to oppose the effect of BAG-1. Biochemistry 2000, 39, 14314–14321. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M. Cytoskeletal protein kinases: Titin and its relations in mechanosensing. Pflug. Arch. 2011, 462, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Mrosek, M.; Labeit, D.; Witt, S.; Heerklotz, H.; von Castelmur, E.; Labeit, S.; Mayans, O. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J. 2007, 21, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Xiang, F.; Yakovenko, A.; Vihola, A.; Hackman, P.; Rostkova, E.; Kristensen, J.; Brandmeier, B.; Franzen, G.; Hedberg, B.; et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005, 308, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.S.; Spencer, M. Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenance. Neuromuscul. Disord. 2008, 18, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Imajoh-Ohmi, S.; Emori, Y.; Kawasaki, H.; Ohno, S.; Minami, Y.; Suzuki, K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989, 264, 20106–20111. [Google Scholar] [PubMed]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Moriscot, A.S.; Baptista, I.L.; Bogomolovas, J.; Witt, C.; Hirner, S.; Granzier, H.; Labeit, S. MuRF1 is a muscle fiber-type II associated factor and together with MuRF2 regulates type-II fiber trophicity and maintenance. J. Struct. Biol. 2010, 170, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sarcomere Protein or Structure | Chaperones Required for Assembly | Chaperones and UPS Members Required for Maintenance | Muscle Disease |
|---|---|---|---|
| fast myosin | Hsp40 Hsp70 Hsp90α1 Unc45b Smyd1b | CHIP MURF2 | Inclusion Body Myopathy [17,18] |
| slow myosin | Hsp40 Hsp70 Hsp90α1 Unc45b | CHIP | Inclusion Body Myopathy [17,18,19,20] |
| actin | GimC TriC Leiomodin 2 Leiomodin 3 | αβ-crystallin | Nemaline Myopathy [21,22,23,24] |
| α-actinin | ZASP N-RAP | MURF1 (associates with N-RAP in Yeast-2-hybrid) | Muscular Dystrophy [25] Nemaline Myopathy [24] |
| titin | αβ-crystallin | Hsp90α1 Smyd2 αβ-crystallin MURF1 (associates with titin in Yeast-2-hybrid) Calpain3 | Limb Girdle Muscular Dystrophy [26,27,28] Tibial Muscular Dystrophy [29,30,31] Dilated Cardiomyopathy [32,33] |
| nebulin | Unknown | Unknown | Nemaline Myopathy |
| non-muscle myosin | Unc45 is a likely chaperone candidate | Unknown | Unknown |
| desmin | αβ-crystallin | Asb2β | Desmin Related Myopathies (DRM) Hypertrophic Cardiomyopathy [34] |
| troponin I | Unknown | MURF1 | Nemaline Myopathy [21] |
| Factor Name | Homologues Implicated in Muscle | Factor Type | Expression | Model Organism |
|---|---|---|---|---|
| UNC45 | (SM-Unc45) Unc45b | Chaperone/Co-chaperone | Cardiac, fast and slow skeletal muscle | Human, Mouse, Xenopus, Zebrafish, C. elegans |
| (GC-Unc45) Unc45a | Co-chaperone | Generally Expressed | Human, Mouse, Zebrafish | |
| Hsp90 | Hsp90α1 | Chaperone | Cardiac, Skeletal muscle and Neural Tissue | Zebrafish |
| Hsp90α2 | Proposed chaperone | Cardiac, Skeletal muscle and Neural Tissue | Zebrafish | |
| Hsp90αβ | Unknown | Generally Expressed | Zebrafish | |
| Smyd1 | m-Bop/Smyd1b | Chaperone/Co-chaperone | Cardiac, Fast skeletal muscle | Mouse/Zebrafish |
| Smyd1a | Proposed chaperone | Skeletal Muscle | Zebrafish | |
| Smyd2 | Smyd2a | Methyltransferase | Generally Expressed | Mouse, Zebrafish |
| αβ-crystallin | αβ-crystallin/CRYAB/Hspb1 | Chaperone | Cardiac and Skeletal muscle | Human, Mouse, Zebrafish |
| MuRF1/Trim63 | MuRF1/Trim63 | E3 enzyme | Cardiac and Skeletal muscle | Mouse, Zebrafish (Zebrafish have Trim63a and Trim63b) |
| MuRF2/Trim55 | MuRF2/Trim55 | E3 enzyme | Cardiac and Skeletal muscle | Mouse, Zebrafish (Zebrafish have Trim55a and Trim55b) |
| MuRF3/Trim54 | MuRF3/Trim54 | E3 enzyme | Cardiac and Skeletal muscle | Mouse, Zebrafish |
| MuRF4/Trim101 | MuRF4/Trim101 | E3 enzyme | Skeletal Muscle | Zebrafish |
| Bag-1 | Bag-1 | Co-chaperone | Generally Expressed | Human, Mouse, Zebrafish |
| Bag-3/Starvin | Bag-3/Starvin | Co-chaperone | Striated muscle Z-disc | Human, Mouse, Zebrafish, Drosophila |
| Stub1/CHIP | Stub1/CHIP | Co-chaperone/E3 enzyme | Cytosol, ER | Human, Zebrafish, Mouse, C. elegans |
| Hsf-1 | HSf-1 | Transcription Factor | Generally Expressed | Humans, Mice, Zebrafish, C. elegans |
| Hsc-70 | Hsp70 | Chaperone | Generally Expressed Under Heat Shock | Human, Mice, Zebrafish, E. coli |
| Hsp70-1 | Chaperone | Generally Expressed Under Heat Shock | Human, Mouse, Zebrafish | |
| Hsp70-3 | Chaperone | Unknown | Human, Mouse, Zebrafish | |
| ST13/Hip | Hip | Co-chaperone | Cytoplasm | Human, Mouse, Zebrafish, Drosophila |
| STIP1/Hop | STIP1/Hop | Co-chaperone | Nucleus, Cytoplasm | Human, Mouse, Zebrafish, C. elegans, Drosophila |
| Calpain-3 | Calpain-3 | Protease | Generally Expressed | Human, Mouse, Zebrafish |
| SRF | SRF | Transcription factor | Nucleus | Human, Mouse, Zebrafish |
| UFD2/Ube4b | UFD2/Ub24b | E3 enzyme | Nucleus, Cytoplasm | C. elegans, Human, Mouse, Zebrafish |
| CHN-1 | CHN-1 | GTPase activating protein | Cytosol | Human, C. elegans |
| p97/Vcp/CDC48 | p97/CDC48 | ATPase | Generally Expressed | Human, Mouse, Zebrafish, C. elegans |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlisle, C.; Prill, K.; Pilgrim, D. Chaperones and the Proteasome System: Regulating the Construction and Demolition of Striated Muscle. Int. J. Mol. Sci. 2018, 19, 32. https://doi.org/10.3390/ijms19010032
Carlisle C, Prill K, Pilgrim D. Chaperones and the Proteasome System: Regulating the Construction and Demolition of Striated Muscle. International Journal of Molecular Sciences. 2018; 19(1):32. https://doi.org/10.3390/ijms19010032
Chicago/Turabian StyleCarlisle, Casey, Kendal Prill, and Dave Pilgrim. 2018. "Chaperones and the Proteasome System: Regulating the Construction and Demolition of Striated Muscle" International Journal of Molecular Sciences 19, no. 1: 32. https://doi.org/10.3390/ijms19010032
APA StyleCarlisle, C., Prill, K., & Pilgrim, D. (2018). Chaperones and the Proteasome System: Regulating the Construction and Demolition of Striated Muscle. International Journal of Molecular Sciences, 19(1), 32. https://doi.org/10.3390/ijms19010032