Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: A Case Report in a Southern Italian Family

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
Acknowledgments
Author Contributions
Conflicts of interest
References
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Petruzella, S.; Matloff, E.; Carter, D. Prevalence of BRCA1 and BRCA2 mutations in women diagnosed with ductal carcinoma in situ. JAMA 2005, 293, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Schildkraut, J.M.; Thompson, W.D.; Risch, N.J. The genetic attributable risk of breast and ovarian cancer. Cancer 1996, 77, 2318–2324. [Google Scholar] [CrossRef]
- Lavie, O.; Narod, S.; Lejbkowicz, F.; Dishon, S.; Goldberg, Y.; Gemer, O.; Rennert, G. Double heterozygosity in the BRCA1 and BRCA2 genes in the Jewish population. Ann. Oncol. 2011, 22, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Friedman, L.S.; Gayther, S.A.; Ponder, B.A.; Bobrow, L.; van der Looji, M.; Papp, J.; Olah, E. A breast/ovarian cancer patient with germline mutations in both BRCA1 and BRCA2. Nat. Genet. 1997, 15, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Baruch, R.; Dagan, E.; Kepten, I.; Freid, G. Co-segregation of BRCA1 185delAG mutation and BRCA2 6174delT in one single family. Eur. J. Cancer 1997, 33, 2283–2284. [Google Scholar] [CrossRef]
- Randall, T.C.; Bell, K.A.; Rebane, B.A.; Rubin, S.C.; Boyd, J. Germline mutations of the BRCA1 and BRCA2 genes in a breast and ovarian cancer patient. Gynecol. Oncol. 1998, 70, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Meynard, G.; Mansi, L.; Lebahar, P.; Villanueva, C.; Klajer, E.; Calcagno, F.; Vivalta, A.; Chaix, M.; Collonge-Rame, M.A.; Populaire, C.; et al. First description of a double heterozygosity for BRCA1 and BRCA2 pathogenic variants in a French metastatic breast cancer patient: A case report. Oncol. Rep. 2017, 37, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Leegte, B.; van der Hout, A.H.; Deffenbaugh, A.M.; Bakker, M.K.; Mulder, I.M.; ten Berge, A.; Leenders, E.P.; Wesseling, J.; de Hullu, J.; Hoogerbrugge, N.; et al. Phenotypic expression of double heterozygosity for BRCA1 and BRCA2 germline mutations. J. Med. Genet. 2005, 42, e20. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.R.; Gorringe, K.L.; Rowley, S.M.; Li, N.; McInerny, S.; Wong-Brown, M.W.; Devereux, L.; Li, J.; Lifepool, I.; Trainer, A.H.; et al. Reevaluation of the BRCA2 truncating allele c.9976A > T (p.Lys3326Ter) in a familial breast cancer context. Sci. Rep. 2015, 5, 14800. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, R.; Engstrom, P.G.; Helgadottir, H.; Eriksson, H.; Unneberg, P.; Kjellqvist, S.; Yang, M.; Linden, D.; Edsgard, D.; Hansson, J.; et al. The role of germline alterations in the DNA damage response genes BRIP1 and BRCA2 in melanoma susceptibility. Genes Chromosom. Cancer 2016, 55, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Meeks, H.D.; Song, H.; Michailidou, K.; Bolla, M.K.; Dennis, J.; Wang, Q.; Barrowdale, D.; Frost, D.; Embrace; McGuffog, L.; et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Wang, Y.; Shao, W.; Jin, J.; Du, M.; Ma, G.; Chu, H.; Wang, M.; Zhang, Z. Rare variants in BRCA2 and CHEK2 are associated with the risk of urinary tract cancers. Sci. Rep. 2016, 6, 33542. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.T.; Matsubayashi, H.; Rogers, C.D.; Philips, J.; Couch, F.J.; Brune, K.; Yeo, C.J.; Kern, S.E.; Hruban, R.H.; Goggins, M. Increased prevalence of the BRCA2 polymorphic stop codon K3326X among individuals with familial pancreatic cancer. Oncogene 2005, 24, 3652–3656. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.L.; Dyson, G.; Levin, N.K.; Chaudhry, S.; Rosati, R.; Kalpage, H.; Wernette, C.; Petrucelli, N.; Simon, M.S.; Tainsky, M.A. Reanalysis of BRCA1/2 negative high risk ovarian cancer patients reveals novel germline risk loci and insights into missing heritability. PLoS ONE 2017, 12, e0178450. [Google Scholar] [CrossRef] [PubMed]
- Higgs, J.E.; Harkness, E.F.; Bowers, N.L.; Howard, E.; Wallace, A.J.; Lalloo, F.; Newman, W.G.; Evans, D.G. The BRCA2 polymorphic stop codon: Stuff or nonsense? J. Med. Genet. 2015, 52, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Lee, M.H.; Bale, A.E.; Carter, D.; Haffty, B.G. Incidence of BRCA1 and BRCA2 mutations in young Korean breast cancer patients. J. Clin. Oncol. 2004, 22, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Lee, M.H.; Haffty, B.G. Double heterozygotes for non-Caucasian families with mutations in BRCA1 and BRCA2 genes. Breast J. 2006, 12, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.M.; Choi, D.H.; Nam, S.J.; Lee, J.E.; Kim, J.W.; Kim, S.W.; Kang, E.; Lee, M.H.; Ahn, S.H.; Kim, K.S.; et al. Characteristics of double heterozygosity for BRCA1 and BRCA2 germline mutations in Korean breast cancer patients. Breast Cancer Res. Treat. 2012, 131, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Liede, A.; Rehal, P.; Vesprini, D.; Jack, E.; Abrahamson, J.; Narod, S.A. A breast cancer patient of Scottish descent with germ-line mutations in BRCA1 and BRCA2. Am. J. Hum. Genet. 1998, 62, 1543–1544. [Google Scholar] [CrossRef] [PubMed]
- Loader, S.; Rowley, P.T. Deleterious mutations of both BRCA1 and BRCA2 in three siblings. Genet. Test. 1998, 2, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Tesoriero, A.; Andersen, C.; Southey, M.; Somers, G.; McKay, M.; Armes, J.; McCredie, M.; Giles, G.; Hopper, J.L.; Venter, D. De novo BRCA1 mutation in a patient with breast cancer and an inherited BRCA2 mutation. Am. J. Hum. Genet. 1999, 65, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Caldes, T.; de la Hoya, M.; Tosar, A.; Sulleiro, S.; Godino, J.; Ibanez, D.; Martin, M.; Perez-Segura, P.; Diaz-Rubio, E. A breast cancer family from Spain with germline mutations in both the BRCA1 and BRCA2 genes. J. Med. Genet. 2002, 39, e44. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Michiara, M.; Bella, M.A.; Zanelli, P.; Bortesi, B.; Capelletti, M.; Savi, M.; Neri, T.M.; Ardizzoni, A. A breast cancer patient from Italy with germline mutations in both the BRCA1 and BRCA2 genes. Breast Cancer Res. Treat. 2005, 91, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Fawcett, S.; Sigalas, E.; Bell, R.; Devery, S.; Andrieska, N.; Winship, I. Familial breast cancer: Double heterozygosity for BRCA1 and BRCA2 mutations with differing phenotypes. Fam. Cancer 2008, 7, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Pilato, B.; de Summa, S.; Danza, K.; Lambo, R.; Paradiso, A.; Tommasi, S. Maternal and paternal lineage double heterozygosity alteration in familial breast cancer: A first case report. Breast Cancer Res. Treat. 2010, 124, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Zuradelli, M.; Peissel, B.; Manoukian, S.; Zaffaroni, D.; Barile, M.; Pensotti, V.; Cavallari, U.; Masci, G.; Mariette, F.; Benski, A.C.; et al. Four new cases of double heterozygosity for BRCA1 and BRCA2 gene mutations: Clinical, pathological, and family characteristics. Breast Cancer Res. Treat. 2010, 124, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, A.Y.; Jonson, L.; Ejlertsen, B.; Gerdes, A.M.; Nielsen, F.C.; Hansen, T.V. Identification of a Danish breast/ovarian cancer family double heterozygote for BRCA1 and BRCA2 mutations. Fam. Cancer 2010, 9, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Augustyn, A.M.; Agostino, N.M.; Namey, T.L.; Nair, S.; Martino, M.A. Two patients with germline mutations in both BRCA1 and BRCA2 discovered unintentionally: A case series and discussion of BRCA testing modalities. Breast Cancer Res. Treat. 2011, 129, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Nomizu, T.; Matsuzaki, M.; Katagata, N.; Kobayashi, Y.; Sakuma, T.; Monma, T.; Saito, M.; Watanabe, F.; Midorikawa, S.; Yamaguchi, Y. A case of familial breast cancer with double heterozygosity for BRCA1 and BRCA2 genes. Breast Cancer 2015, 22, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Loubser, F.; de Villiers, J.N.; van der Merwe, N.C. Two double heterozygotes in a South African Afrikaner family: Implications for BRCA1 and BRCA2 predictive testing. Clin. Genet. 2012, 82, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Heidemann, S.; Fischer, C.; Engel, C.; Fischer, B.; Harder, L.; Schlegelberger, B.; Niederacher, D.; Goecke, T.O.; Doelken, S.C.; Dikow, N.; et al. Double heterozygosity for mutations in BRCA1 and BRCA2 in German breast cancer patients: Implications on test strategies and clinical management. Breast Cancer Res. Treat. 2012, 134, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Molinari, A.M.; Caliendo, G.; De Paola, M.L.; Giovanna, D.; Gambardella, A.L.; Petronella, P.; Cioffi, M. Double heterozygosity in the BRCA1 and BRCA2 genes in Italian family. Clin. Chem. Lab. Med. 2013, 51, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Janavicius, R. Founder BRCA1/2 mutations in the Europe: Implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010, 1, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Krajc, M.; Teugels, E.; Zgajnar, J.; Goelen, G.; Besic, N.; Novakovic, S.; Hocevar, M.; De Greve, J. Five recurrent BRCA1/2 mutations are responsible for cancer predisposition in the majority of Slovenian breast cancer families. BMC Med. Genet. 2008, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Cruz-Hernandez, J.J.; Sanchez-Valdivieso, E.; Rodriguez, C.A.; Gomez-Bernal, A.; Barco, E.; Fonseca, E.; Portugal, T.; Gonzalez-Sarmiento, R. BRCA1-2 mutations in breast cancer: Identification of nine new variants of BRCA1-2 genes in a population from central Western Spain. Cancer Lett. 2006, 233, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Kluska, A.; Balabas, A.; Paziewska, A.; Kulecka, M.; Nowakowska, D.; Mikula, M.; Ostrowski, J. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med. Genom. 2015, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Johannsson, O.T.; Staff, S.; Vallon-Christersson, J.; Kytola, S.; Gudjonsson, T.; Rennstam, K.; Hedenfalk, I.A.; Adeyinka, A.; Kjellen, E.; Wennerberg, J.; et al. Characterization of a novel breast carcinoma xenograft and cell line derived from a BRCA1 germ-line mutation carrier. Lab. Investig. 2003, 83, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Mazoyer, S.; Dunning, A.M.; Serova, O.; Dearden, J.; Puget, N.; Healey, C.S.; Gayther, S.A.; Mangion, J.; Stratton, M.R.; Lynch, H.T.; et al. A polymorphic stop codon in BRCA2. Nat. Genet. 1996, 14, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.G.; Liu, P.; Sharan, S.K. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat. Med. 2008, 14, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Hinson, S.R.; Ohashi, A.; Farrugia, D.; Wendt, P.; Tavtigian, S.V.; Deffenbaugh, A.; Goldgar, D.; Couch, F.J. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005, 65, 417–426. [Google Scholar] [PubMed]
- Morimatsu, M.; Donoho, G.; Hasty, P. Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to gamma-radiation and premature senescence. Cancer Res. 1998, 58, 3441–3447. [Google Scholar] [PubMed]
- Friedman, E.; Bar-Sade Bruchim, R.; Kruglikova, A.; Risel, S.; Levy-Lahad, E.; Halle, D.; Bar-On, E.; Gershoni-Baruch, R.; Dagan, E.; Kepten, I.; et al. Double heterozygotes for the Ashkenazi founder mutations in BRCA1 and BRCA2 genes. Am. J. Hum. Genet. 1998, 63, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Frank, T.S.; Deffenbaugh, A.M.; Reid, J.E.; Hulick, M.; Ward, B.E.; Lingenfelter, B.; Gumpper, K.L.; Scholl, T.; Tavtigian, S.V.; Pruss, D.R.; et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. J. Clin. Oncol. 2002, 20, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Peto, J.; Collins, N.; Barfoot, R.; Seal, S.; Warren, W.; Rahman, N.; Easton, D.F.; Evans, C.; Deacon, J.; Stratton, M.R. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J. Natl. Cancer Inst. 1999, 91, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Wu, S. Do mutations in BRCA1/BRCA2 confer a higher risk of skin cancer? Br. J. Dermatol. 2015, 172, 1473. [Google Scholar] [CrossRef] [PubMed]
- Gumaste, P.V.; Penn, L.A.; Cymerman, R.M.; Kirchhoff, T.; Polsky, D.; McLellan, B. Skin cancer risk in BRCA1/2 mutation carriers. Br. J. Dermatol. 2015, 172, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Leachman, S.A.; Lucero, O.M.; Sampson, J.E.; Cassidy, P.; Bruno, W.; Queirolo, P.; Ghiorzo, P. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.J.; Mitra, N.; Goldstein, A.M.; Tucker, M.A.; Avril, M.F.; Azizi, E.; Bergman, W.; Bishop, D.T.; Bressac-de Paillerets, B.; Bruno, W.; et al. Germline Variation at CDKN2A and Associations with Nevus Phenotypes among Members of Melanoma Families. J. Investig. Dermatol. 2017, 137, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
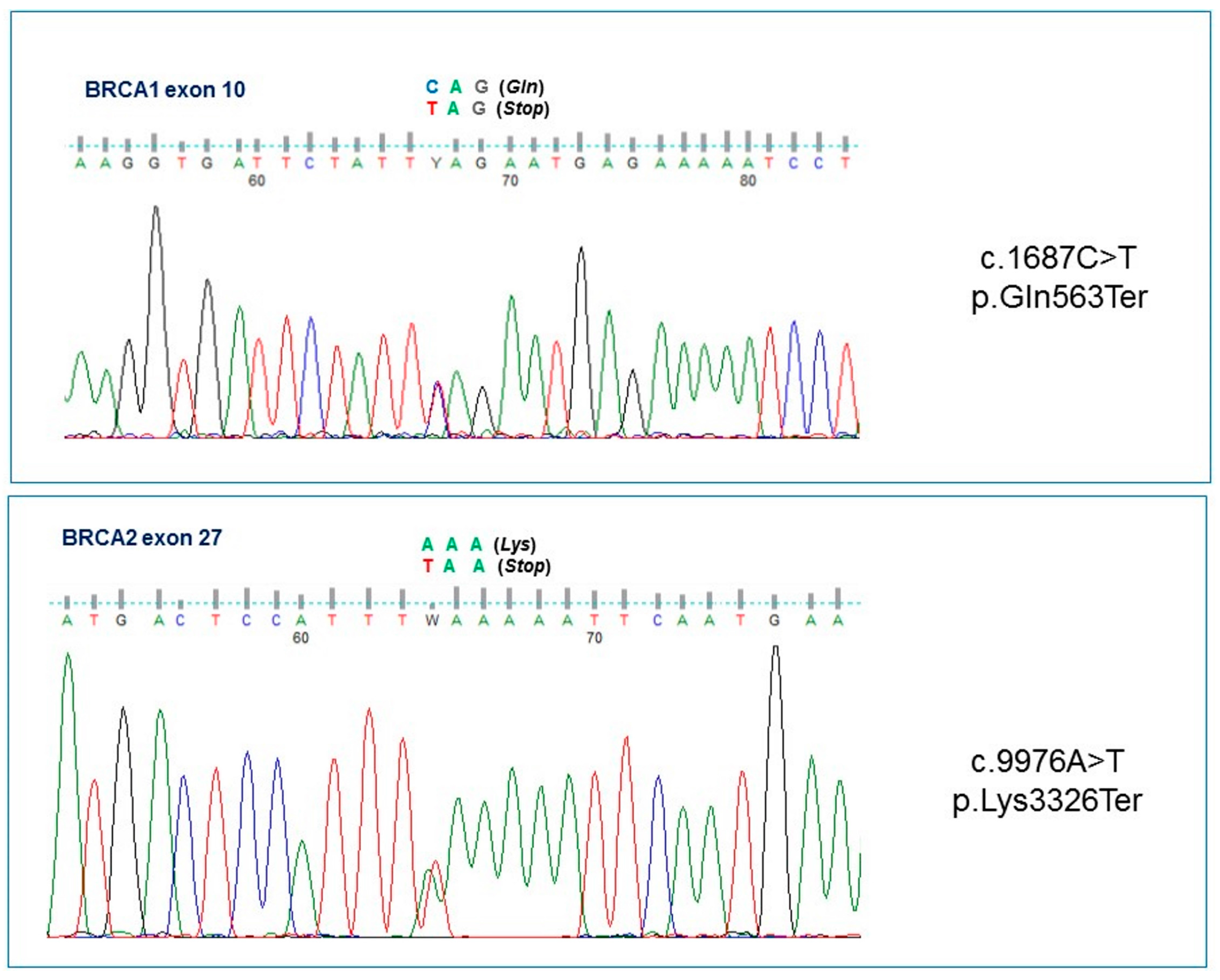

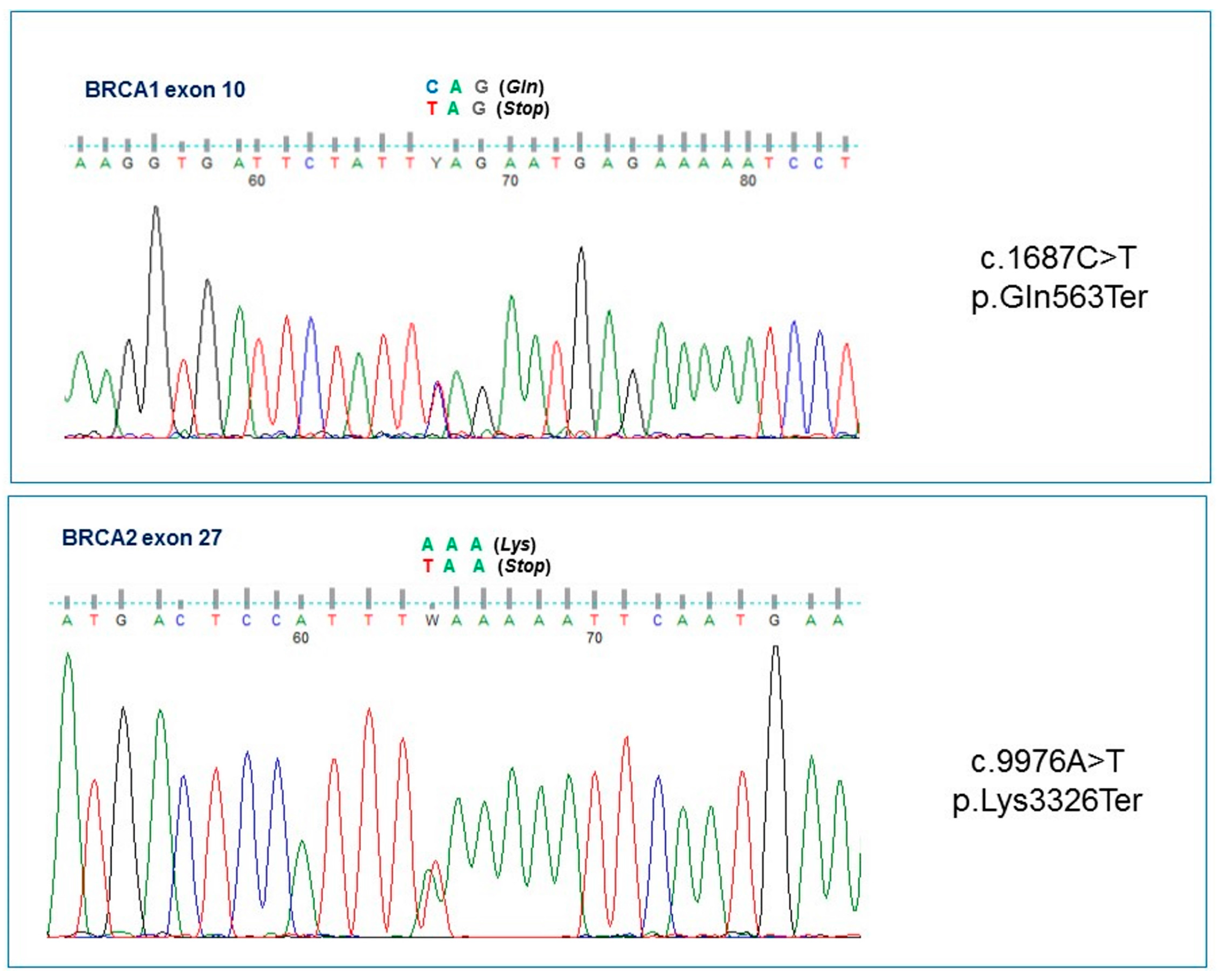{kind=link}
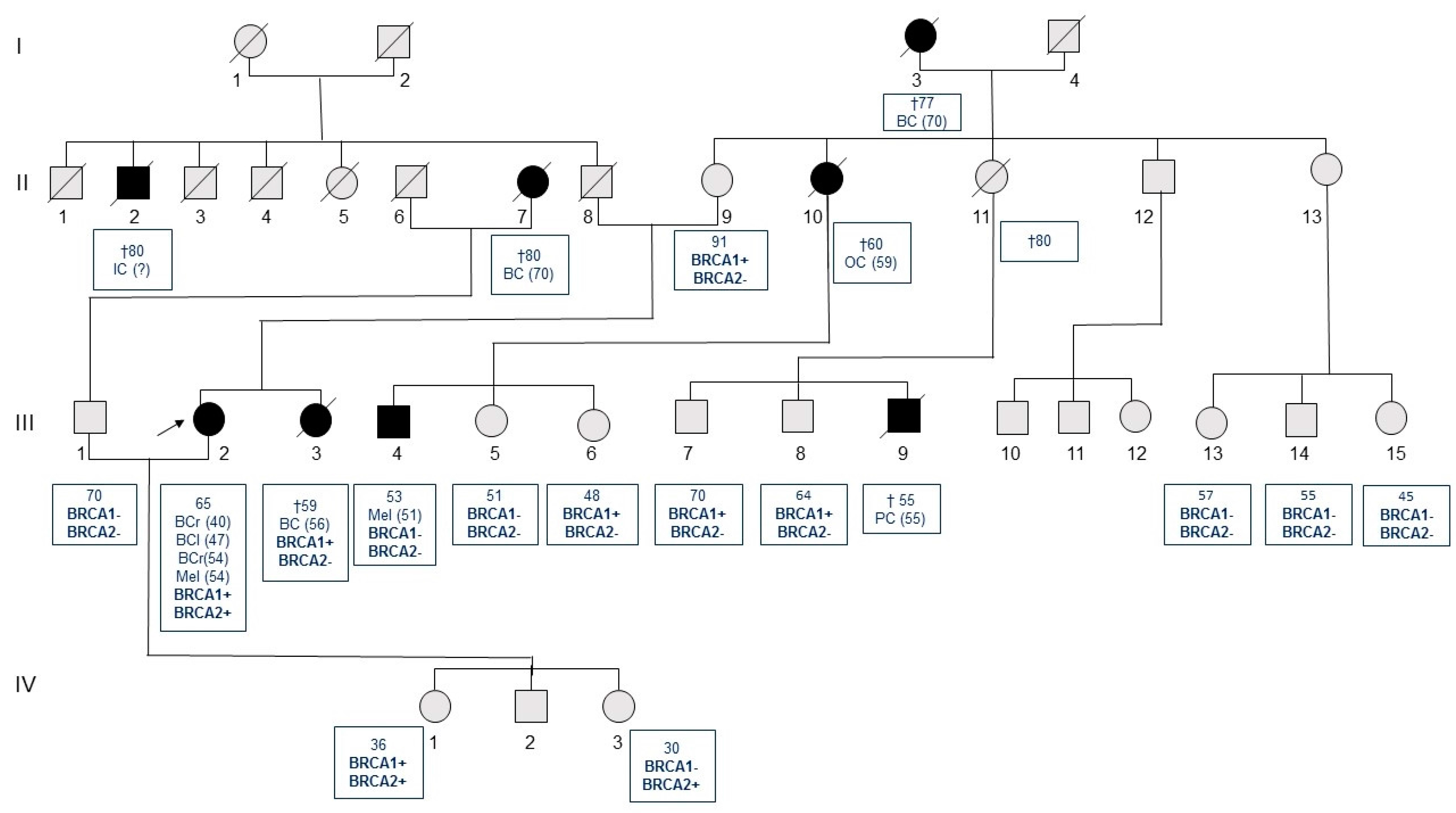
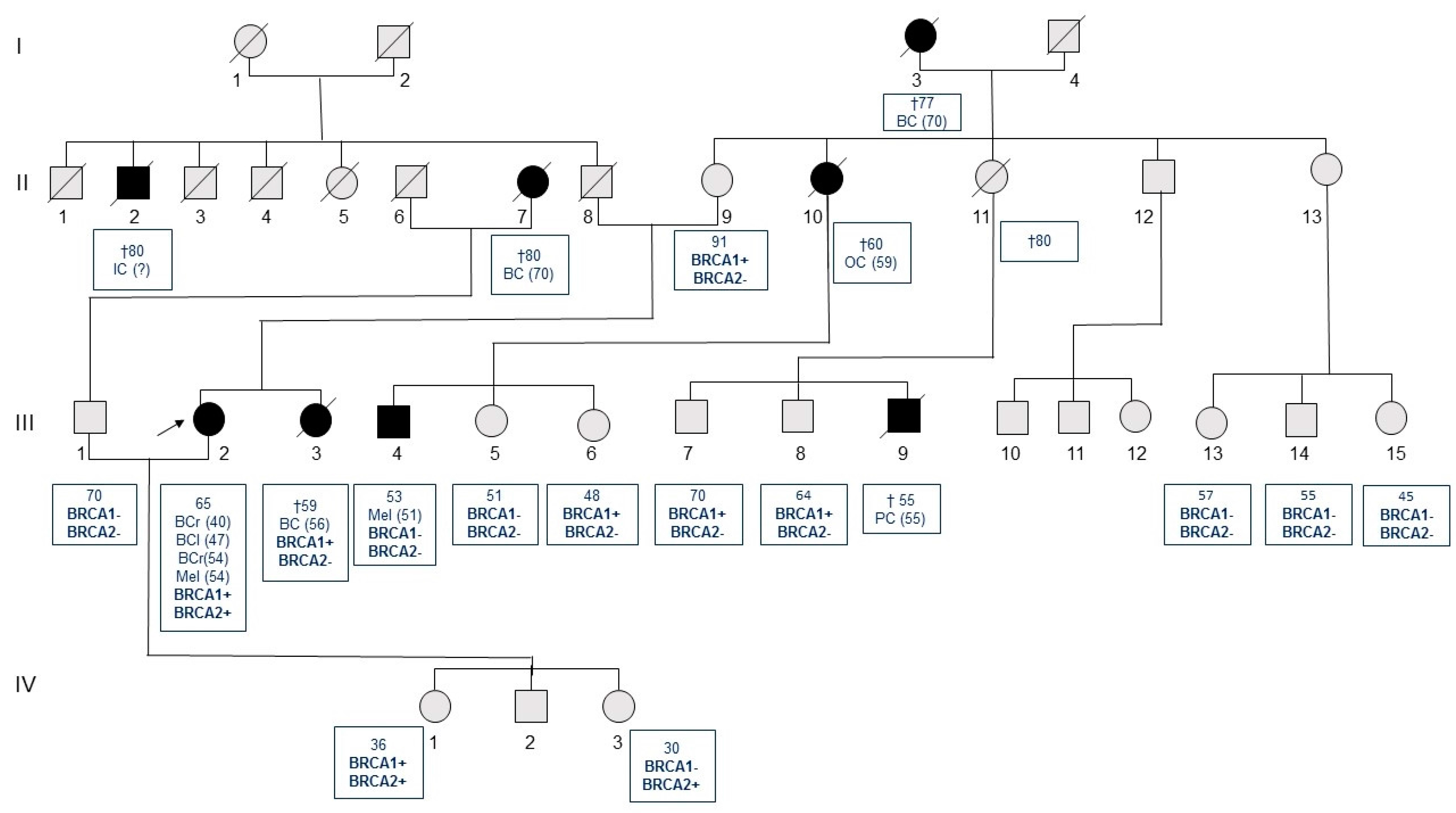{kind=link}
| Geographic Localization/Ethnic Group | BRCA1 Mutation | BRCA2 Mutation | Sex | Inheritance | Proband Cancer/Age of Onset (Years) [Relative with DH] | References | |
|---|---|---|---|---|---|---|---|
| Mother | Father | ||||||
| Scottish | c.2389G>T | c.3067_3068insA | F | WT | ND | Breast 35 | [20] |
| German descent | c.5080G>T | c.6405_6409delCTTAA. | M | BRCA1 | ND | Asymptomatic 36 [Sister asymptomatic 34] [Brother asymptomatic 30] | [21] |
| Australia (no Jewish ancestry) | c.3769_3770delGA | c.5946_5946delT | F | WT | BRCA2 | Breast < 40 | [22] |
| Spain | c.5123C>A | c.6275_6276delTT | F | BRCA1 BRCA2 | - | Breast 28 [Mother asymptomatic 70] [Sister asymptomatic 40] [Cousin asymptomatic 47] [Cousin asymptomatic 41] [Uncle prostate 66] [Aunt breast 70] [Aunt breast 66] | [23] |
| Korea | c.4981G>T | c.5946_5949delTGGA | F | BRCA1 BRCA2 | - | Breast 33 [Mother stomach 62] | [17] |
| Korea | c.1516_1520del5 | c.2798_2799delCA | F | ND | ND | Breast 26 | |
| Korea | c.1656_1656delT | c.4599A>C | F | ND | ND | Breast 37 | |
| Netherlands | c.2685_2686delAA | c.3487_3487delG | F | ND | ND | Ovarian 40, breast 45 | [9] |
| Netherlands | c.2685_2686delAA | c.4449_4449delA | F | ND | ND | Breast 28 | |
| European | c.962G>A | c.3170_3174delAGAAA | F | ND | ND | Breast 37 | [2] |
| Italy | c.4285_4286insG | c.7738C>T | F | ND | ND | Breast 37 | [24] |
| Australia | c.3331_3334delCAAG | c.631+2T>G | F | ND | ND | Breast 34, colon 35, breast 53 [Sister asymptomatic 65] | [25] |
| Italy | c.5263_5264insC | c.5796_5797delTA | F | BRCA1 | BRCA2 | Breast 38, ovarian 42 | [26] |
| Italy | c.835_835delC | c.8195T>G | F | ND | ND | Breast 43 | [27] |
| Italy | c.3916_3917delTT | c.5379_5379delG | F | WT | ND | Breast 30, ovarian 36 | |
| Italy | c.1687C>T | c.6469C>T | F | ND | ND | Breast 46, ovarian 58 | |
| Italy | c.2405_2406delTG | c.4284_4285insT | F | ND | ND | Breast and ovarian 52 | |
| Denmark | c.5096G>A | c.631+4A>G | F | - | BRCA1 BRCA2 | Breast 53, ovarian 59 [Father breast 76] [Son and daughter asymptomatic] | [28] |
| Caucasian | c.1961_1961delA | c.1444_1444delC | F | ND | ND | Ovarian b. 50 | [29] |
| Caucasian (maternal Ashkenazi ancestry) | c.5266_5267insC | c.4829_4830delTG | F | ND | ND | Breast u. 40 | |
| Korea | c.3627_3628insA | c.6724_6725delGA | F | ND | ND | Breast 26 | [19] |
| Korea | c.390C>A | c.3018_3018delA | F | ND | ND | Breast 45 | |
| Korea | c.5030_5033delCTAA | c.1399A>T | F | ND | ND | Breast 35 | |
| Japan | c.188T>A | c.5576_5579delTTAA | F | BRCA1 BRCA2 | Breast 55 [Father asymptomatic 51] [Cousin breast 41; Endometrial cancer 46] | [30] | |
| Afrikaners | c.2635G>T | c.7934_7934delG | F | BRCA1 | BRCA2 | Breast 42 [Healthy second cousin 49] | [31] |
| Germany | c.5263_5264insC | c.5645C>A | F | WT | BRCA1 BRCA2 | Breast b. 37; Ovarian b. 63 [Father prostate 68] | [32] |
| Germany | c.66_67delAG | c.5722_5723delCT | F | BRCA1 | BRCA2 | Breast u. 32 | |
| Germany | c.962G>A | c.2231C>G | F | BRCA1 BRCA2 | WT | Breast b. 31, 35 [Mother breast 40] | |
| Germany | c.3910_3910delG | c.2830A>T | F | BRCA1 BRCA2 | WT | Breast u. 39 [Mother breast 34; another cancer not reported 35] | |
| Germany | c.5193+1_5193+1delG | c.658_659delGT | F | ND | ND | Coecum 58, ovarian 61 | |
| Germany | c.3700_3704delGTAAA | c.1813_1814insA | F | ND | ND | Cervix 26, breast 40 | |
| Italy | c.547+2T>A | c.2830A>T c.426-57A>G | F | - | BRCA1 BRCA2 | Breast 35 [Father asymptomatic 72] | [33] |
| France | c.1016_1017insA | c.6814_6814delA | F | BRCA1 | WT | Breast 46 | [8] |
| Italy | c.1687C>T | c.9976A>T | F | BRCA1 | ND | Breast u (40), breast u (47), breast b (54), Mel (54) [Asymptomatic daughter (36)] | This report |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmirotta, R.; Lovero, D.; Stucci, L.S.; Silvestris, E.; Quaresmini, D.; Cardascia, A.; Silvestris, F. Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: A Case Report in a Southern Italian Family. Int. J. Mol. Sci. 2018, 19, 285. https://doi.org/10.3390/ijms19010285
Palmirotta R, Lovero D, Stucci LS, Silvestris E, Quaresmini D, Cardascia A, Silvestris F. Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: A Case Report in a Southern Italian Family. International Journal of Molecular Sciences. 2018; 19(1):285. https://doi.org/10.3390/ijms19010285
Chicago/Turabian StylePalmirotta, Raffaele, Domenica Lovero, Luigia Stefania Stucci, Erica Silvestris, Davide Quaresmini, Angela Cardascia, and Franco Silvestris. 2018. "Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: A Case Report in a Southern Italian Family" International Journal of Molecular Sciences 19, no. 1: 285. https://doi.org/10.3390/ijms19010285
APA StylePalmirotta, R., Lovero, D., Stucci, L. S., Silvestris, E., Quaresmini, D., Cardascia, A., & Silvestris, F. (2018). Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: A Case Report in a Southern Italian Family. International Journal of Molecular Sciences, 19(1), 285. https://doi.org/10.3390/ijms19010285