Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders



Abstract
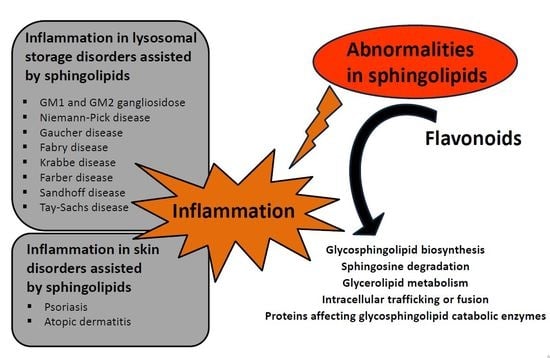
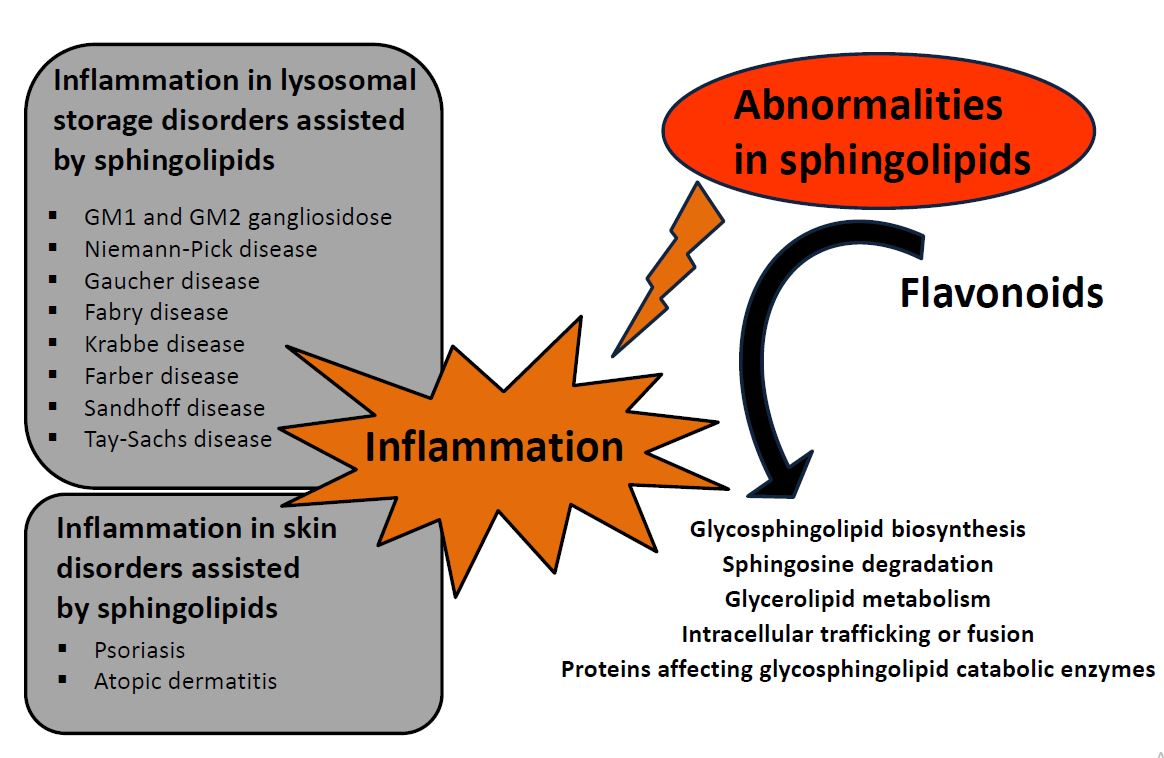{kind=link}
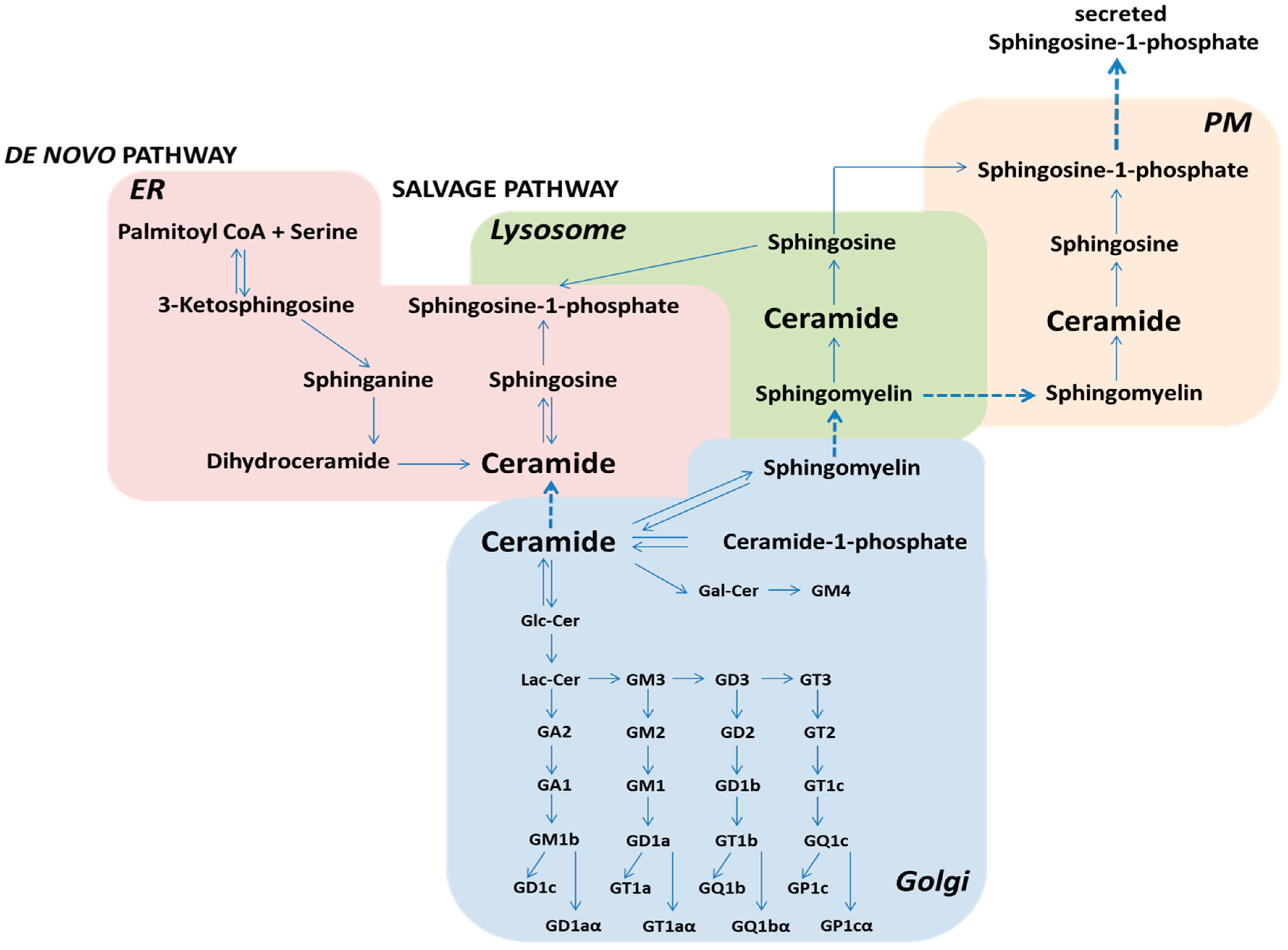{kind=link}
{kind=link}
1. Introduction to Sphingolipids
2. Sphingolipid Metabolism and Its Role in Cellular Processes
3. Diseases Associated with Defects in Sphingolipid Metabolism
3.1. Defect of Ganglioside Metabolism
3.1.1. GM1 Gangliosidosis
3.1.2. GM2 Gangliosidosis
3.1.3. Tay-Sachs Disease
3.1.4. Sandhoff Disease
3.1.5. GM2 Gangliosidosis AB Variant
3.1.6. Ganglioside GM3 Synthase Deficiency
3.2. Sialo-Oligosaccharides Accumulate in Sialidosis
3.3. Accumulation of Globosides
3.4. Accumulation of Glucocerebrosides
3.5. Accumulation of Galactocerebrosides
3.6. Storage of Ceramides
3.7. Lysosomal Storage of Sphingomyelin
4. Knowledge Implementation about the Metabolism of Sphingolipids in Learning the Mechanism of Other Diseases
4.1. Sphingolipids in Cell Signalling
4.2. Sphingolipids in Central Nervous System Inflammatory Disorders
4.3. SL Alterations in Metabolic Syndrome
4.4. SL Role in Skin Barrier Disorders
5. Flavonoids, Compounds Modulating Sphingolipid Metabolism Used in Lysosomal Storage Diseases and Skin Disorders
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)? Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, T.; Suzuki, K.; Popko, B. New perspectives on the function of myelin galactolipids. Trends Neurosci. 1998, 21, 126–130. [Google Scholar] [PubMed]
- Honke, K.; Hirahara, Y.; Dupree, J.; Suzuki, K.; Popko, B.; Fukushima, K.; Fukushima, J.; Nagasawa, T.; Yoshida, N.; Wada, Y.; et al. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc. Natl. Acad. Sci. USA 2002, 99, 4227–4232. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.C.; Jahng, A.; Maricic, I.; Kumar, V. Mini review: Immune response to myelin-derived sulfatide and CNS-demyelination. Neurochem. Res. 2007, 32, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, K.S.; Bremer, E.G.; Schwarting, G.A. P blood group regulation of glycosphingolipid levels in human erythrocytes. J. Biol. Chem. 1979, 254, 11196–11198. [Google Scholar] [PubMed]
- Sandhoff, K. My journey into the world of sphingolipids and sphingolipidoses. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 554–582. [Google Scholar] [CrossRef] [PubMed]
- De Chaves, E.P.; Sipione, S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010, 584, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 2009, 50, S440–S445. [Google Scholar] [CrossRef] [PubMed]
- Kotani, M.; Kawashima, I.; Ozawa, H.; Terashima, T.; Tai, T. Differential distribution of major gangliosides in rat central nervous system detected by specific monoclonal antibodies. Glycobiology 1993, 3, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Emoto, M.; Emoto, Y.; Yoshizawa, I.; Kita, E.; Shimizu, T.; Hurwitz, R.; Brinkmann, V.; Kaufmann, S.H. Alpha-GalCer ameliorates listeriosis by accelerating infiltration of Gr-1+ cells into the liver. Eur. J. Immunol. 2010, 40, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef] [PubMed]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Milhas, D.; Clarke, C.J.; Hannun, Y.A. Sphingomyelin metabolism at the plasma membrane: Implications for bioactive sphingolipids. FEBS Lett. 2010, 584, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Mandon, E.C.; Ehses, I.; Rother, J.; van Echten, G.; Sandhoff, K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 1992, 267, 11144–11148. [Google Scholar] [PubMed]
- Lannert, H.; Gorgas, K.; Meissner, I.; Wieland, F.T.; Jeckel, D. Functional organization of the Golgi apparatus in glycosphingolipid biosynthesis. Lactosylceramide and subsequent glycosphingolipids are formed in the lumen of the late Golgi. J. Biol. Chem. 1998, 273, 2939–2946. [Google Scholar] [CrossRef] [PubMed]
- Lannert, H.; Bunning, C.; Jeckel, D.; Wieland, F.T. Lactosylceramide is synthesized in the lumen of the Golgi apparatus. FEBS Lett. 1994, 342, 91–96. [Google Scholar] [CrossRef]
- Varki, A. Evolutionary forces shaping the Golgi glycosylation machinery: Why cell surface glycans are universal to living cells. Cold Spring Harb. Perspect. Biol. 2011, 3, a005462. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Weissman, J.S. Membranes in balance: Mechanisms of sphingolipid homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Gillard, B.K.; Clement, R.G.; Marcus, D.M. Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology 1998, 8, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signalling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Riboni, L.; Bassi, R.; Conti, M.; Tettamanti, G. Metabolism of exogenous ganglioside GM1 in cultured cerebellar granule cells. The fatty acid and sphingosine moieties formed during degradation are re-used for lipid biosynthesis. FEBS Lett. 1993, 322, 257–260. [Google Scholar] [CrossRef]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Scaglia, F. GM1 gangliosidosis: Review of clinical, molecular and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Oshima, A.; Sakuraba, H.; Nakano, T.; Yanagisawa, N.; Inui, K.; Okada, S.; Uyama, E.; Namba, R.; Kondo, K.; et al. GM1 gangliosidosis in adults: Clinical and molecular analysis of 16 Japanese patients. Ann. Neurol. 1992, 31, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C. Gangliosidoses. Handb. Clin. Neurol. 2013, 113, 1707–1708. [Google Scholar] [PubMed]
- Vallance, H.; Ford, J. Carrier testing for autosomal-recessive disorders. Crit. Rev. Clin. Lab. Sci. 2003, 40, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Kaback, M.M.; Desnick, R.J. Hexosaminidase a Deficiency; University of Washington: Seattle, WA, USA, 1993–2018; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1218/ (accessed on 15 October 2017).
- Sandhoff, K.; Andreae, U.; Jatzkewitz, H. Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Pathol. Eur. 1968, 3, 278–285. [Google Scholar] [CrossRef]
- Sandhoff Disease. Available online: http://ghr.nlm.nih.gov/condition/sandhoff-disease (accessed on 15 October 2017).
- Sheth, J.; Mistri, M.; Sheth, F.; Shah, R.; Bavdekar, A.; Godbole, K.; Nanavaty, N.; Datar, C.; Kamate, M.; Oza, N.; et al. Burden of lysosomal storage disorders in India: Experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014, 12, 51–63. [Google Scholar] [PubMed]
- Yun, Y.M.; Lee, S.N. A case refort of sandhoff disease. Korean J. Ophthalmol. 2005, 19, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Brunel-Guitton, C.; Lortie, A.; Gauvin, F.; Morales, C.R.; Mitchell, G.A.; Pshezhetsky, A.V. Atypical juvenile presentation of GM2 gangliosidosis AB in a patient compound-heterozygote for c.259G > T and c.164C > T mutations in the GM2A gene. Mol. Genet. Metab. Rep. 2017, 11, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Sheth, J.; Datar, C.; Mistri, M.; Bhavsar, R.; Sheth, F.; Shah, K. GM2 gangliosidosis AB variant: Novel mutation from India—A case report with a review. BMC Pediatr. 2016, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Fragaki, K.; Ait-El-Mkadem, S.; Chaussenot, A.; Gire, C.; Mengual, R.; Bonesso, L.; Beneteau, M.; Ricci, J.E.; Desquiret-Dumas, V.; Procaccio, V.; et al. Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency. Eur. J. Hum. Genet. 2013, 21, 528–534. [Google Scholar] [CrossRef] [PubMed]
- GM3 Synthase Deficiency. Available online: https://ghr.nlm.nih.gov/condition/gm3-synthase-deficiency (accessed on 15 October 2017).
- Boccuto, L.; Aoki, K.; Flanagan-Steet, H.; Chen, C.F.; Fan, X.; Bartel, F.; Petukh, M.; Pittman, A.; Saul, R.; Chaubey, A.; et al. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum. Mol. Genet. 2014, 23, 418–433. [Google Scholar] [PubMed]
- Seyrantepe, V.; Poupetova, H.; Froissart, R.; Zabot, M.T.; Maire, I.; Pshezhetsky, A.V. Molecular pathology of NEU1 gene in sialidosis. Hum. Mutat. 2003, 22, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Canafoglia, L. Sialidoses. Epileptic Disord. 2016, 18, 89–93. [Google Scholar] [PubMed]
- Caciotti, A.; Di Rocco, M.; Filocamo, M.; Grossi, S.; Traverso, F.; d’Azzo, A.; Cavicchi, C.; Messeri, A.; Guerrini, R.; Zammarchi, E.; et al. Type II sialidosis: Review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. J. Neurol. 2009, 256, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.R.; Niu, D.M. Fabry disease: Review and experience during newborn screening. Trends Cardiovasc. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R. Fabry disease. Handb. Clin. Neurol. 2015, 132, 231–248. [Google Scholar] [PubMed]
- Cox-Brinkman, J.; Vedder, A.; Hollak, C.; Richfield, L.; Mehta, A.; Orteu, K.; Wijburg, F.; Hammond, P. Three-dimensional face shape in fabry disease. Eur. J. Hum. Genet. 2007, 15, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Baker, R.; Pasquale, F.; Quarta, G.; Ebrahim, H.; Mehta, A.B.; Hughes, D.A. Prevalence of anderson-fabry disease in patients with hypertrophic cardiomyopathy: The European anderson-fabry disease survey. Heart 2011, 97, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Warnock, D.G.; Banikazemi, M.; Bultas, J.; Linthorst, G.E.; Packman, S.; Sorensen, S.A.; Wilcox, W.R.; Desnick, R.J. Fabry disease: Progression of nephropathy and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol. Dial. Transplant. 2009, 24, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Vedder, A.C.; Linthorst, G.E.; van Breemen, M.J.; Groener, J.E.; Bemelman, F.J.; Strijland, A.; Mannens, M.M.; Aerts, J.M.; Hollak, C.E. The dutch fabry cohort: Diversity of clinical manifestations and GB3 levels. J. Inherit. Metab. Dis. 2007, 30, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Gaucher disease: Gene frequencies and genotype/phenotype correlations. Genet. Test. 1997, 1, 5–12. [Google Scholar] [PubMed]
- Motta, M.; Camerini, S.; Tatti, M.; Casella, M.; Torreri, P.; Crescenzi, M.; Tartaglia, M.; Salvioli, R. Gaucher disease due to saposin C deficiency is an inherited lysosomal disease caused by rapidly degraded mutant proteins. Hum. Mol. Genet. 2014, 23, 5814–5826. [Google Scholar] [CrossRef] [PubMed]
- Jmoudiak, M.; Futerman, A.H. Gaucher disease: Pathological mechanisms and modern management. Br. J. Haematol. 2005, 129, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Zaizov, R.; Zlotogora, J. large scale screening for gaucher’s disease in Israel—A position paper by the national gaucher committee of the ministry of health. Harefuah 1997, 133, 107–108. [Google Scholar] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A review of gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis and treatment of gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Wenger, D.A.; Luzi, P.; Rafi, M.A. Krabbe disease: Are certain mutations disease-causing only when specific polymorphisms are present or when inherited in trans with specific second mutations? Mol. Genet. Metab. 2014, 111, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Vanier, M.T.; Mansson, J.E. Krabbe disease: A galactosylsphingosine (psychosine) lipidosis. J. Lipid Res. 1980, 21, 53–64. [Google Scholar] [PubMed]
- Bongarzone, E.R.; Escolar, M.L.; Gray, S.J.; Kafri, T.; Vite, C.H.; Sands, M.S. Insights into the pathogenesis and treatment of Krabbe disease. Pediatr. Endocr. Rev. 2016, 13, 689–696. [Google Scholar]
- Suzuki, K. Globoid cell leukodystrophy (Krabbe’s disease): Update. J. Child Neurol. 2003, 18, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Gelinas, J.; Sirrs, S. Phenotypic variability of Krabbe disease across the lifespan. Can. J. Neurol. Sci. 2014, 41, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Debs, R.; Froissart, R.; Aubourg, P.; Papeix, C.; Douillard, C.; Degos, B.; Fontaine, B.; Audoin, B.; Lacour, A.; Said, G.; et al. Krabbe disease in adults: Phenotypic and genotypic update from a series of 11 cases and a review. J. Inherit. Metab. Dis. 2013, 36, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Frohbergh, M.; He, X.; Schuchman, E.H. The molecular medicine of acid ceramidase. Biol. Chem. 2015, 396, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Stern, G. Niemann-Pick’s and Gaucher’s diseases. Parkinsonism Relat. Disord. 2014, 20 (Suppl. 1), S143–S146. [Google Scholar] [CrossRef]
- Brady, R.O.; Kanfer, J.N.; Mock, M.B.; Fredrickson, D.S. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc. Natl. Acad. Sci. USA 1966, 55, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.; Ziobro, R.; Becker, K.A.; Kolesnick, R.; Gulbins, E. Acid sphingomyelinase. In Handbook Experimental Pharmacology; Springer: Vienna, Austria, 2013; pp. 77–88. [Google Scholar]
- Wang, R.Y.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [PubMed]
- Borodzicz, S.; Rudnicka, L.; Mirowska-Guzel, D.; Cudnoch-Jedrzejewska, A. The role of epidermal sphingolipids in dermatologic diseases. Lipids Health Dis. 2016, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 1984, 220, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Suh, P.G.; Ryu, S.H.; Lee, S.Y. Studies of inositol phospholipid-specific phospholipase C. Science 1989, 244, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Loomis, C.R.; Merrill, A.H., Jr.; Bell, R.M. Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J. Biol. Chem. 1986, 261, 12604–12609. [Google Scholar] [PubMed]
- Dressler, K.A.; Mathias, S.; Kolesnick, R.N. Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science 1992, 255, 1715–1718. [Google Scholar] [CrossRef] [PubMed]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Hannun, Y.A. The acid sphingomyelinase/ceramide pathway: Biomedical significance and mechanisms of regulation. Curr. Mol. Med. 2010, 10, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Fernandez-Checa, J.C. Pharmacological modulation of sphingolipids and role in disease and cancer cell biology. Mini Rev. Med. Chem. 2007, 7, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Cremesti, A.; Paris, F.; Grassme, H.; Holler, N.; Tschopp, J.; Fuks, Z.; Gulbins, E.; Kolesnick, R. Ceramide enables fas to cap and kill. J. Biol. Chem. 2001, 276, 23954–23961. [Google Scholar] [CrossRef] [PubMed]
- Grassme, R.; Stegeman, D.F.; Drost, G.; Schumann, N.P.; Scholle, H. Selective spatial information from surface EMG after temporal filtering: The application to interference EMG using cross-covariance analysis. Clin. Neurophysiol. 2003, 114, 2338–2346. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Luberto, C.; Argraves, K.M. Enzymes of sphingolipid metabolism: From modular to integrative signaling. Biochemistry 2001, 40, 4893–4903. [Google Scholar] [CrossRef] [PubMed]
- Memon, R.A.; Holleran, W.M.; Uchida, Y.; Moser, A.H.; Ichikawa, S.; Hirabayashi, Y.; Grunfeld, C.; Feingold, K.R. Regulation of glycosphingolipid metabolism in liver during the acute phase response. J. Biol. Chem. 1999, 274, 19707–19713. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Deevska, G.M.; Nikolova-Karakashian, M.N. The twists and turns of sphingolipid pathway in glucose regulation. Biochimie 2011, 93, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Kikuta, J.; Shimazu, Y.; Meier-Schellersheim, M.; Germain, R.N. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J. Exp. Med. 2010, 207, 2793–2798. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Venable, M.E.; Lee, J.Y.; Smyth, M.J.; Bielawska, A.; Obeid, L.M. Role of ceramide in cellular senescence. J. Biol. Chem. 1995, 270, 30701–30708. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Pettus, B.J.; Rossi, M.J.; Wood, R.; Usta, J.; Szulc, Z.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A. Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J. Biol. Chem. 2002, 277, 12960–12969. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 2010, 24, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.; Lucks, J.; Fuchs, S.; Schiffmann, S.; Schreiber, Y.; Ferreiros, N.; Merkens, J.; Marschalek, R.; Geisslinger, G.; Grosch, S. Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int. J. Biochem. Cell Biol. 2012, 44, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, N.; Osta, W.; Bielawski, J.; Luberto, C.; Obeid, L.M.; Hannun, Y.A. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J. Biol. Chem. 2004, 279, 25101–25111. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar] [CrossRef]
- Brown, M.D.; Sacks, D.B. Protein scaffolds in map kinase signalling. Cell. Signal. 2009, 21, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.; Ayoub, M.; Storz, P.; Rennecke, J.; Fabbro, D.; Pfizenmaier, K. PKC zeta is a molecular switch in signal-transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic-acid. EMBO J. 1995, 14, 1961–1969. [Google Scholar] [PubMed]
- Zhang, Y.; Yao, B.; Delikat, S.; Bayoumy, S.; Lin, X.H.; Basu, S.; McGinley, M.; Chan-Hui, P.Y.; Lichenstein, H.; Kolesnick, R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 1997, 89, 63–72. [Google Scholar] [CrossRef]
- Verheij, M.; Bose, R.; Lin, X.H.; Yao, B.; Jarvis, W.D.; Grant, S.; Birrer, M.J.; Szabo, E.; Zon, L.I.; Kyriakis, J.M.; et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 1996, 380, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Brunner, J.; Hummel, R.; Vervoordeldonk, M.; Stabel, S.; van den Bosch, H.; Pfeilschifter, J. Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Clark, W.; Mumby, M.; Gao, F.; May, W.S. A functional role for the B56 alpha-subunit of protein phosphatase 2A in ceramide-mediated regulation of Bcl2 phosphorylation status and function. J. Biol. Chem. 2002, 277, 22847–22852. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Petronilli, V.; Di Lisa, F.; Bernardi, P. Commitment to apoptosis by GD3 ganglioside depends on opening of the mitochondrial permeability transition pore. J. Biol. Chem. 1999, 274, 22581–22585. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Colell, A.; Paris, R.; Fernandez-Checa, J.C. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition, cytochrome c release and caspase activation. FASEB J. 2000, 14, 847–858. [Google Scholar] [PubMed]
- Garcia-Ruiz, C.; Colell, A.; Morales, A.; Calvo, M.; Enrich, C.; Fernandez-Checa, J.C. Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J. Biol. Chem. 2002, 277, 36443–36448. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Garcia-Ruiz, C.; Roman, J.; Ballesta, A.; Fernandez-Checa, J.C. Ganglioside GD3 enhances apoptosis by suppressing the nuclear factor-kappa B-dependent survival pathway. FASEB J. 2001, 15, 1068–1070. [Google Scholar] [PubMed]
- Uzzo, R.G.; Rayman, P.; Kolenko, V.; Clark, P.E.; Cathcart, M.K.; Bloom, T.; Novick, A.C.; Bukowski, R.M.; Hamilton, T.; Finke, J.H. Renal cell carcinoma-derived gangliosides suppress nuclear factor-kappaB activation in T cells. J. Clin. Investig. 1999, 104, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Lee, H.J.; Lee, W.H.; Suk, K. Nf-kappaB as a common signaling pathway in ganglioside-induced autophagic cell death and activation of astrocytes. J. Neuroimmunol. 2010, 226, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, M.R.; Li, Z.; Jiang, X.C. Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim. Biophys. Acta 2005, 1737, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Wada, R.; Sasaki, T.; Deng, C.; Bierfreund, U.; Sandhoff, K.; Proia, R.L. A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Bosio, A.; Binczek, E.; Stoffel, W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 13280–13285. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, T.; Fujita, N.; Dupree, J.; Shi, R.; Blight, A.; Suzuki, K.; Popko, B. Myelination in the absence of galactocerebroside and sulfatide: Normal structure with abnormal function and regional instability. Cell 1996, 86, 209–219. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Go, S.; Takasaki, K.; Kakazu, Y.; Ohashi, M.; Nagafuku, M.; Kabayama, K.; Sekimoto, J.; Suzuki, S.; Takaiwa, K.; et al. Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc. Natl. Acad. Sci. USA 2009, 106, 9483–9488. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, K.; Burns, S.; Shrestha, S.; Chi, H. The S1P1-mTOR axis directs the reciprocal differentiation of TH1 and Treg cells. Nat. Immunol. 2010, 11, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Ridet, J.L.; Malhotra, S.K.; Privat, A.; Gage, F.H. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997, 20, 570–577. [Google Scholar] [CrossRef]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar] [PubMed]
- Boot, R.G.; Verhoek, M.; de Fost, M.; Hollak, C.E.; Maas, M.; Bleijlevens, B.; van Breemen, M.J.; van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2004, 103, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Boven, L.A.; van Meurs, M.; Boot, R.G.; Mehta, A.; Boon, L.; Aerts, J.M.; Laman, J.D. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 2004, 122, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; Evers, L.; Aerts, J.M.; van Oers, M.H. Elevated levels of M-CSF, sCD14 and IL8 in type 1 gaucher disease. Blood Cells Mol. Dis. 1997, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Frings, W.; Dreier, J.; Sorg, C. Only the soluble form of the scavenger receptor CD163 acts inhibitory on phorbol ester-activated T-lymphocytes, whereas membrane-bound protein has no effect. FEBS Lett. 2002, 526, 93–96. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Gratchev, A.; Guillot, P.; Hakiy, N.; Politz, O.; Orfanos, C.E.; Schledzewski, K.; Goerdt, S. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein betaiG-H3. Scand. J. Immunol. 2001, 53, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Pyo, H.; Joe, E.; Jung, S.; Lee, S.H.; Jou, I. Gangliosides activate cultured rat brain microglia. J. Biol. Chem. 1999, 274, 34584–34589. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Stone, K.; Jales, A.; Leitenberg, D.; Ladisch, S. Inhibition of TLR activation and up-regulation of IL-1R-associated kinase-M expression by exogenous gangliosides. J. Immunol. 2008, 180, 4425–4432. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial activation precedes acute neurodegeneration in sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Smith, D.A.; Williams, I.M.; Borja, M.C.; Neville, D.C.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Nsaids increase survival in the sandhoff disease mouse: Synergy with N-butyldeoxynojirimycin. Ann. Neurol. 2004, 56, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.P.; Proia, R.L. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in sandhoff disease mice. Proc. Natl. Acad. Sci. USA 2004, 101, 8425–8430. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Wallom, K.L.; Williams, I.M.; Jeyakumar, M.; Platt, F.M. Beneficial effects of anti-inflammatory therapy in a mouse model of niemann-pick disease type C1. Neurobiol. Dis. 2009, 36, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.B.; Yoon, H.J.; Park, S.H.; Kim, I.H.; Park, E.J. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J. Immunol. 2008, 181, 8077–8087. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Cheng, H.; Fryer, J.D.; Fagan, A.M.; Holtzman, D.M. Novel role for apolipoprotein e in the central nervous system. Modulation of sulfatide content. J. Biol. Chem. 2003, 278, 8043–8051. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, A.A.; Chen, Z.W.; Cook, S.D. Antibodies to sulfatide in cerebrospinal fluid of patients with multiple sclerosis. J. Neuroimmunol. 2003, 139, 76–80. [Google Scholar] [CrossRef]
- Han, X.; Fagan, A.M.; Cheng, H.; Morris, J.C.; Xiong, C.; Holtzman, D.M. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann. Neurol. 2003, 54, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Xu, J.; McKeel, D.W., Jr.; Han, X. Specificity and potential mechanism of sulfatide deficiency in Alzheimer’s disease: An electrospray ionization mass spectrometric study. Cell. Mol. Biol. 2003, 49, 809–818. [Google Scholar] [PubMed]
- Fabelo, N.; Martin, V.; Santpere, G.; Marin, R.; Torrent, L.; Ferrer, I.; Diaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from parkinson’s disease and incidental parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Holland, W.L.; Bar, J.; Sandhoff, K.; Summers, S.A. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J. Biol. Chem. 2005, 280, 20148–20153. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Boot, R.G.; van Eijk, M.; Groener, J.; Bijl, N.; Lombardo, E.; Bietrix, F.M.; Dekker, N.; Groen, A.K.; Ottenhoff, R.; et al. Glycosphingolipids and insulin resistance. Adv. Exp. Med. Biol. 2011, 721, 99–119. [Google Scholar] [PubMed]
- Inokuchi, J. Membrane microdomains and insulin resistance. FEBS Lett. 2010, 584, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Degroote, S.; Wolthoorn, J.; van Meer, G. The cell biology of glycosphingolipids. Semin. Cell Dev. Biol. 2004, 15, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Thematic review series: Skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis. J. Lipid Res. 2007, 48, 2531–2546. [Google Scholar] [CrossRef] [PubMed]
- Downing, D.T. Lipid and protein structures in the permeability barrier of mammalian epidermis. J. Lipid Res. 1992, 33, 301–313. [Google Scholar] [PubMed]
- Wertz, P.W.; Swartzendruber, D.C.; Madison, K.C.; Downing, D.T. Composition and morphology of epidermal cyst lipids. J. Investig. Dermatol. 1987, 89, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Landmann, L. The epidermal permeability barrier. Anat. Embryol. 1988, 178, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Holleran, W.M.; Ginns, E.I.; Menon, G.K.; Grundmann, J.U.; Fartasch, M.; McKinney, C.E.; Elias, P.M.; Sidransky, E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in gaucher disease. J. Clin. Investig. 1994, 93, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Sherer, D.M.; Ginns, E.I. Gaucher disease in the neonate: A distinct gaucher phenotype is analogous to a mouse model created by targeted disruption of the glucocerebrosidase gene. Pediatr. Res. 1992, 32, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.; Proia, R.L.; Sandhoff, K. Accumulation of protein-bound epidermal glucosylceramides in beta-glucocerebrosidase deficient type 2 gaucher mice. FEBS Lett. 1999, 447, 167–170. [Google Scholar] [CrossRef]
- Yamamoto, A.; Serizawa, S.; Ito, M.; Sato, Y. Effect of aging on sebaceous gland activity and on the fatty acid composition of wax esters. J. Investig. Dermatol. 1987, 89, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Wu, K.; Paul, P.; Marks, D.L.; Kobayashi, T.; Pittelkow, M.R.; Pagano, R.E. Up-regulation of glucosylceramide synthase expression and activity during human keratinocyte differentiation. J. Biol. Chem. 1998, 273, 9651–9655. [Google Scholar] [CrossRef] [PubMed]
- Marekov, L.N.; Steinert, P.M. Ceramides are bound to structural proteins of the human foreskin epidermal cornified cell envelope. J. Biol. Chem. 1998, 273, 17763–17770. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jia, Y.; Cheng, Z.W.; Gao, Y.; Zhang, G.L.; Li, J.Y.; He, C.F. Advancements in the maintenance of skin barrier/skin lipid composition and the involvement of metabolic enzymes. J. Cosmet. Dermatol. 2016, 15, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kanitakis, J. Anatomy, histology and immunohistochemistry of normal human skin. Eur. J. Dermatol. 2002, 12, 390–399. [Google Scholar] [PubMed]
- Breiden, B.; Sandhoff, K. The role of sphingolipid metabolism in cutaneous permeability barrier formation. Biochim. Biophys. Acta 2014, 1841, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Sahle, F.F.; Gebre-Mariam, T.; Dobner, B.; Wohlrab, J.; Neubert, R.H. Skin diseases associated with the depletion of stratum corneum lipids and stratum corneum lipid substitution therapy. Skin Pharmacol. Physiol. 2015, 28, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Van Smeden, J.; Bouwstra, J.A. Stratum corneum lipids: Their role for the skin barrier function in healthy subjects and atopic dermatitis patients. Curr. Probl. Dermatol. 2016, 49, 8–26. [Google Scholar] [PubMed]
- Van Smeden, J.; Janssens, M.; Gooris, G.S.; Bouwstra, J.A. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim. Biophys. Acta 2014, 1841, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Khnykin, D. Fatty acid transporters in skin development, function and disease. Biochim. Biophys. Acta 2014, 1841, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, M.; Gorgas, K.; Sandhoff, R. Ceramide synthesis in the epidermis. Biochim. Biophys. Acta 2014, 1841, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Holleran, W.M.; Takagi, Y.; Uchida, Y. Epidermal sphingolipids: Metabolism, function and roles in skin disorders. FEBS Lett. 2006, 580, 5456–5466. [Google Scholar] [CrossRef] [PubMed]
- Motta, S.; Monti, M.; Sesana, S.; Caputo, R.; Carelli, S.; Ghidoni, R. Ceramide composition of the psoriatic scale. Biochim. Biophys. Acta 1993, 1182, 147–151. [Google Scholar] [CrossRef]
- Lew, B.L.; Cho, Y.; Kim, J.; Sim, W.Y.; Kim, N.I. Ceramides and cell signaling molecules in psoriatic epidermis: Reduced levels of ceramides, PKC-alpha and JNK. J. Korean Med. Sci. 2006, 21, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kim, S.Y.; Kleuser, B.; Schafer-Korting, M.; Kim, K.H.; Park, K.C. Sphingosine-1-phosphate inhibits human keratinocyte proliferation via Akt/protein kinase B inactivation. Cell. Signal. 2004, 16, 89–95. [Google Scholar] [CrossRef]
- Vogler, R.; Sauer, B.; Kim, D.S.; Schafer-Korting, M.; Kleuser, B. Sphingosine-1-phosphate and its potentially paradoxical effects on critical parameters of cutaneous wound healing. J. Investig. Dermatol. 2003, 120, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Japtok, L.; Baumer, W.; Kleuser, B. Sphingosine-1-phosphate as signaling molecule in the skin: Relevance in atopic dermatitis. Allergo J. Int. 2014, 23, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Oizumi, A.; Nakayama, H.; Okino, N.; Iwahara, C.; Kina, K.; Matsumoto, R.; Ogawa, H.; Takamori, K.; Ito, M.; Suga, Y.; et al. Pseudomonas-derived ceramidase induces production of inflammatory mediators from human keratinocytes via sphingosine-1-phosphate. PLoS ONE 2014, 9, e89402. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Gruber, R.; Crumrine, D.; Menon, G.; Williams, M.L.; Wakefield, J.S.; Holleran, W.M.; Uchida, Y. Formation and functions of the corneocyte lipid envelope (CLE). Biochim. Biophys. Acta 2014, 1841, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Hara, M.; Nishio, H.; Sidransky, E.; Inoue, S.; Otsuka, F.; Suzuki, A.; Elias, P.M.; Holleran, W.M.; Hamanaka, S. Epidermal sphingomyelins are precursors for selected stratum corneum ceramides. J. Lipid Res. 2000, 41, 2071–2082. [Google Scholar] [PubMed]
- Ayala-Fontanez, N.; Soler, D.C.; McCormick, T.S. Current knowledge on psoriasis and autoimmune diseases. Psoriasis 2016, 6, 7–32. [Google Scholar]
- Gupta, R.; Debbaneh, M.G.; Liao, W. Genetic epidemiology of psoriasis. Curr. Dermatol. Rep. 2014, 3, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Suarez-Farinas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Barker, J.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar] [CrossRef]
- Boehncke, W.H.; Schon, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Grozdev, I.; Korman, N.; Tsankov, N. Psoriasis as a systemic disease. Clin. Dermatol. 2014, 32, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Grine, L.; Lambert, J. Psoriasis: Burning down the host. J. Dermatol. Treat. 2016, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Luger, T.A.; Loser, K. Novel insights into the pathogenesis of psoriasis. Clin. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Bochenska, K.; Smolinska, E.; Moskot, M.; Jakobkiewicz-Banecka, J.; Gabig-Ciminska, M. Models in the research process of psoriasis. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lew, B.L.; Seong, K.; Kim, N.I. An inverse relationship between ceramide synthesis and clinical severity in patients with psoriasis. J. Korean Med. Sci. 2004, 19, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Terao, M.; Takaishi, M.; Kataoka, S.; Goto-Inoue, N.; Setou, M.; Horie, K.; Sakamoto, F.; Ito, M.; Azukizawa, H.; et al. Barrier abnormality due to ceramide deficiency leads to psoriasiform inflammation in a mouse model. J. Investig. Dermatol. 2013, 133, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Pfister, S.; Kremmer, E.; Gerber, J.K.; Ring, J.; Behrendt, H. Alterations of glucosylceramide-beta-glucosidase levels in the skin of patients with psoriasis vulgaris. J. Investig. Dermatol. 2004, 123, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Stachowitz, S.; Ring, J.; Behrendt, H. The level of prosaposin is decreased in the skin of patients with psoriasis vulgaris. J. Investig. Dermatol. 2001, 116, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, A.; Fang, V.; Chen, C.; Schwab, S.R. Exit strategies: S1P signaling and T cell migration. Trends Immunol. 2015, 36, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Checa, A.; Xu, N.; Sar, D.G.; Haeggstrom, J.Z.; Stahle, M.; Wheelock, C.E. Circulating levels of sphingosine-1-phosphate are elevated in severe but not mild psoriasis and are unresponsive to anti-TNF-alpha treatment. Sci. Rep. 2015, 5, 12017. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Kim, J.Y.; Song, E.H.; Shin, M.K.; Cho, Y.H.; Kim, N.I. Altered levels of sphingosine and sphinganine in psoriatic epidermis. Ann. Dermatol. 2013, 25, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Kim, Y.I.; Shin, K.O.; Kim, B.W.; Lee, S.H.; Jeon, J.E.; Kim, H.J.; Lee, Y.M.; Mauro, T.M.; Elias, P.M.; et al. Sphingosine kinase 1 activation enhances epidermal innate immunity through sphingosine-1-phosphate stimulation of cathelicidin production. J. Dermatol. Sci. 2015, 79, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yamasaki, K.; Saito, R.; Fukushi-Takahashi, S.; Shimada-Omori, R.; Asano, M.; Aiba, S. Alarmin function of cathelicidin antimicrobial peptide LL37 through IL-36gamma induction in human epidermal keratinocytes. J. Immunol. 2014, 193, 5140–5148. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Elias, P.M.; Shin, K.O.; Lee, Y.M.; Hupe, M.; Borkowski, A.W.; Gallo, R.L.; Saba, J.; Holleran, W.M.; Uchida, Y. A novel role of a lipid species, sphingosine-1-phosphate, in epithelial innate immunity. Mol. Cell. Biol. 2013, 33, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Miyoshi, N. Electrophiles in foods: The current status of isothiocyanates and their chemical biology. Biosci. Biotechnol. Biochem. 2010, 74, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, R.J.; Lee, S.T.; Gardner, D.R.; Panter, K.E.; James, L.F. Phytochemicals: The good, the bad and the ugly? Phytochemistry 2007, 68, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [PubMed]
- Cushnie, T.P.; Lamb, A.J. Recent advances in understanding the antibacterial properties of flavonoids. Int. J. Antimicrob. Agents 2011, 38, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Ferrazzano, G.F.; Amato, I.; Ingenito, A.; Zarrelli, A.; Pinto, G.; Pollio, A. Plant polyphenols and their anti-cariogenic properties: A review. Molecules 2011, 16, 1486–1507. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, K.H.; Hong, I.; Hwang, J.S.; Rho, H.S.; Kim, D.H.; Chang, I.; Lee, M.O.; Lee, Y.S.; Hwang, J.S. Genistein stimulates differentiation in epidermal keratinocyte through activation of PPARs. FASEB J. 2006, 20, A612. [Google Scholar]
- Polkowski, K.; Mazurek, A.P. Biological properties of genistein. A review of in vitro and in vivo data. Acta Pol. Pharm. 2000, 57, 135–155. [Google Scholar] [PubMed]
- Pazyar, N.; Yaghoobi, R. Soybean: A potential antipsoriasis agent. Jundishapur J. Nat. Pharm. Prod. 2015, 10, e20924. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, G.; Jakobkiewicz-Banecka, J.; Gabig-Ciminska, M.; Piotrowska, E.; Narajczyk, M.; Kloska, A.; Malinowska, M.; Dziedzic, D.; Golebiewska, I.; Moskot, M.; et al. Genistein: A natural isoflavone with a potential for treatment of genetic diseases. Biochem. Soc. Trans. 2010, 38, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, A. Gene expression-targeted isoflavone therapy. IUBMB Life 2012, 64, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Montefusco, S.; Jakobkiewicz-Banecka, J.; Mozolewski, P.; Wegrzyn, A.; Di Bernardo, D.; Wegrzyn, G.; Medina, D.L.; Ballabio, A.; Gabig-Ciminska, M. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J. Biol. Chem. 2014, 289, 17054–17069. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Jakobkiewicz-Banecka, J.; Smolinska, E.; Banecki, B.; Wegrzyn, G.; Gabig-Ciminska, M. Activities of genes controlling sphingolipid metabolism in human fibroblasts treated with flavonoids. Metab. Brain Dis. 2015, 30, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Jakobkiewicz-Banecka, J.; Kloska, A.; Smolinska, E.; Mozolewski, P.; Malinowska, M.; Rychlowski, M.; Banecki, B.; Wegrzyn, G.; Gabig-Ciminska, M. Modulation of expression of genes involved in glycosaminoglycan metabolism and lysosome biogenesis by flavonoids. Sci. Rep. 2015, 5, 9378. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Jakobkiewicz-Banecka, J.; Smolinska, E.; Piotrowska, E.; Wegrzyn, G.; Gabig-Ciminska, M. Effects of flavonoids on expression of genes involved in cell cycle regulation and DNA replication in human fibroblasts. Mol. Cell. Biochem. 2015, 407, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Namikawa, S.; Takeuchi, K. Effect of the dry distillation tar of delipidated soybean (Glyteer) on a psoriasic model in the mouse (4). Nihon Yakurigaku Zasshi 1992, 99, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Shyong, E.Q.; Lu, Y.; Lazinsky, A.; Saladi, R.N.; Phelps, R.G.; Austin, L.M.; Lebwohl, M.; Wei, H. Effects of the isoflavone 4′,5,7-trihydroxyisoflavone (genistein) on psoralen plus ultraviolet A radiation (PUVA)-induced photodamage. Carcinogenesis 2002, 23, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Kim, Y.I.; Park, K. Signaling roles of ceramide and its metabolites in cutaneous antimicrobial defense. Dermatol. Sin. 2015, 33, 78–83. [Google Scholar] [CrossRef]
- Park, K.; Kim, Y.I.; Shin, K.O.; Seo, H.S.; Kim, J.Y.; Mann, T.; Oda, Y.; Lee, Y.M.; Holleran, W.M.; Elias, P.M.; et al. The dietary ingredient, genistein, stimulates cathelicidin antimicrobial peptide expression through a novel S1P-dependent mechanism. J. Nutr. Biochem. 2014, 25, 734–740. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moskot, M.; Bocheńska, K.; Jakóbkiewicz-Banecka, J.; Banecki, B.; Gabig-Cimińska, M. Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders. Int. J. Mol. Sci. 2018, 19, 247. https://doi.org/10.3390/ijms19010247
Moskot M, Bocheńska K, Jakóbkiewicz-Banecka J, Banecki B, Gabig-Cimińska M. Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders. International Journal of Molecular Sciences. 2018; 19(1):247. https://doi.org/10.3390/ijms19010247
Chicago/Turabian StyleMoskot, Marta, Katarzyna Bocheńska, Joanna Jakóbkiewicz-Banecka, Bogdan Banecki, and Magdalena Gabig-Cimińska. 2018. "Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders" International Journal of Molecular Sciences 19, no. 1: 247. https://doi.org/10.3390/ijms19010247
APA StyleMoskot, M., Bocheńska, K., Jakóbkiewicz-Banecka, J., Banecki, B., & Gabig-Cimińska, M. (2018). Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders. International Journal of Molecular Sciences, 19(1), 247. https://doi.org/10.3390/ijms19010247