The Role of ERK Signaling in Experimental Autoimmune Encephalomyelitis


Abstract
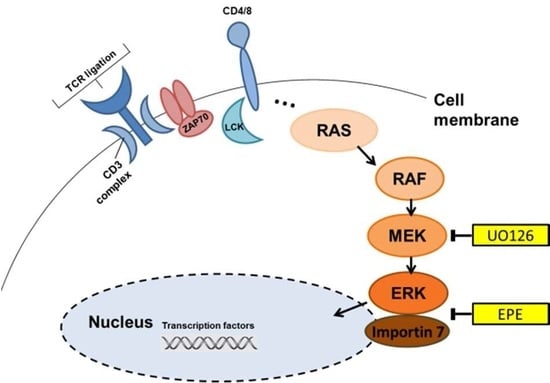
:
1. Introduction
2. Results
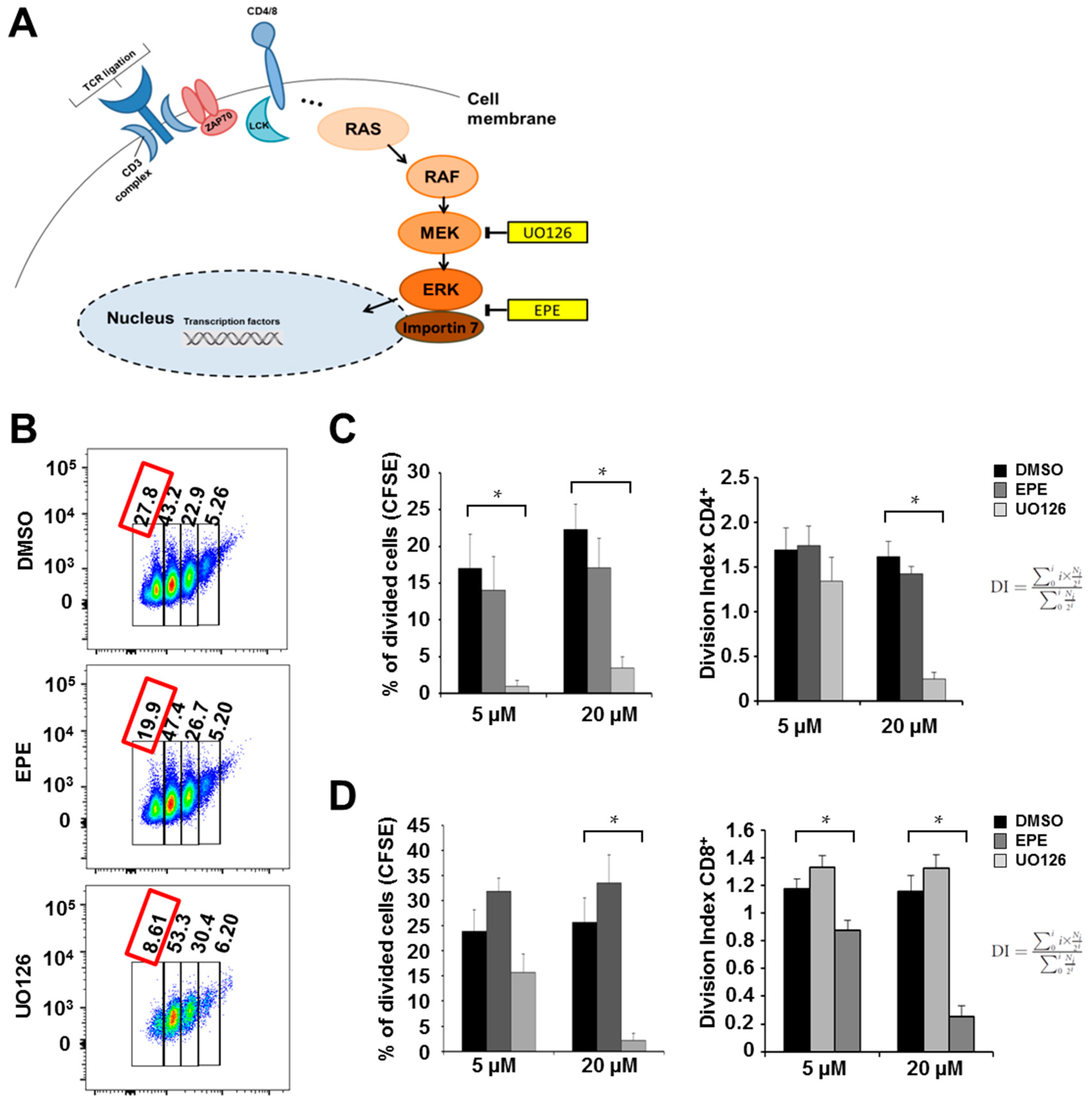
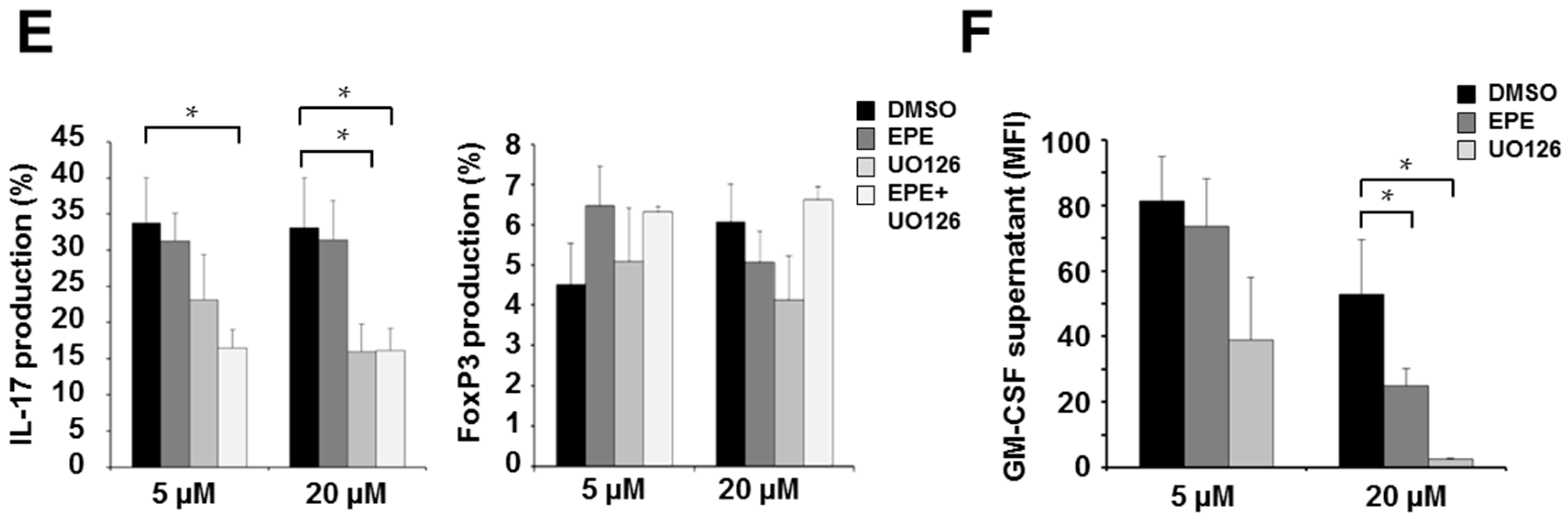
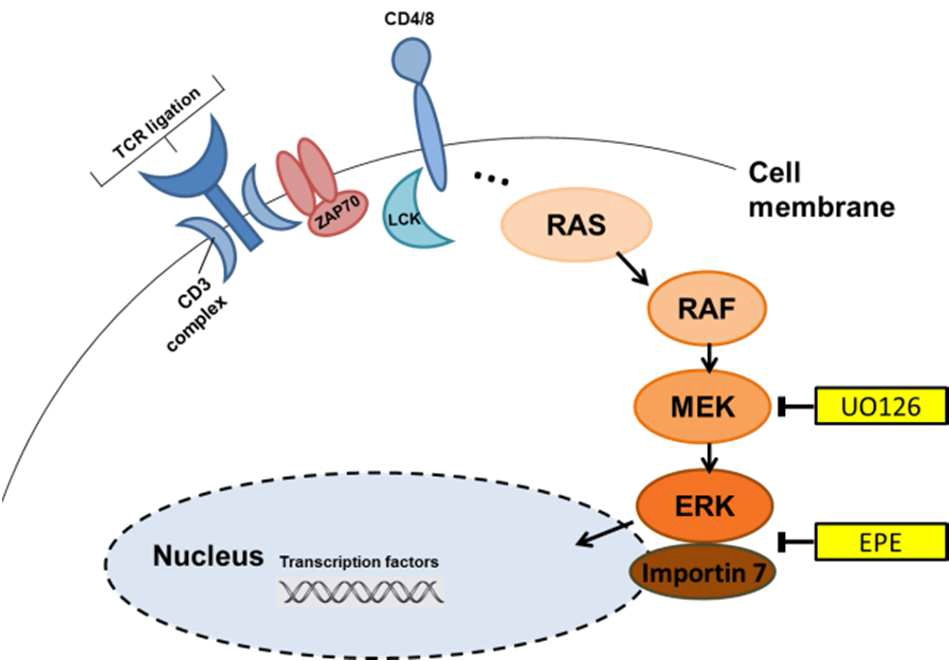
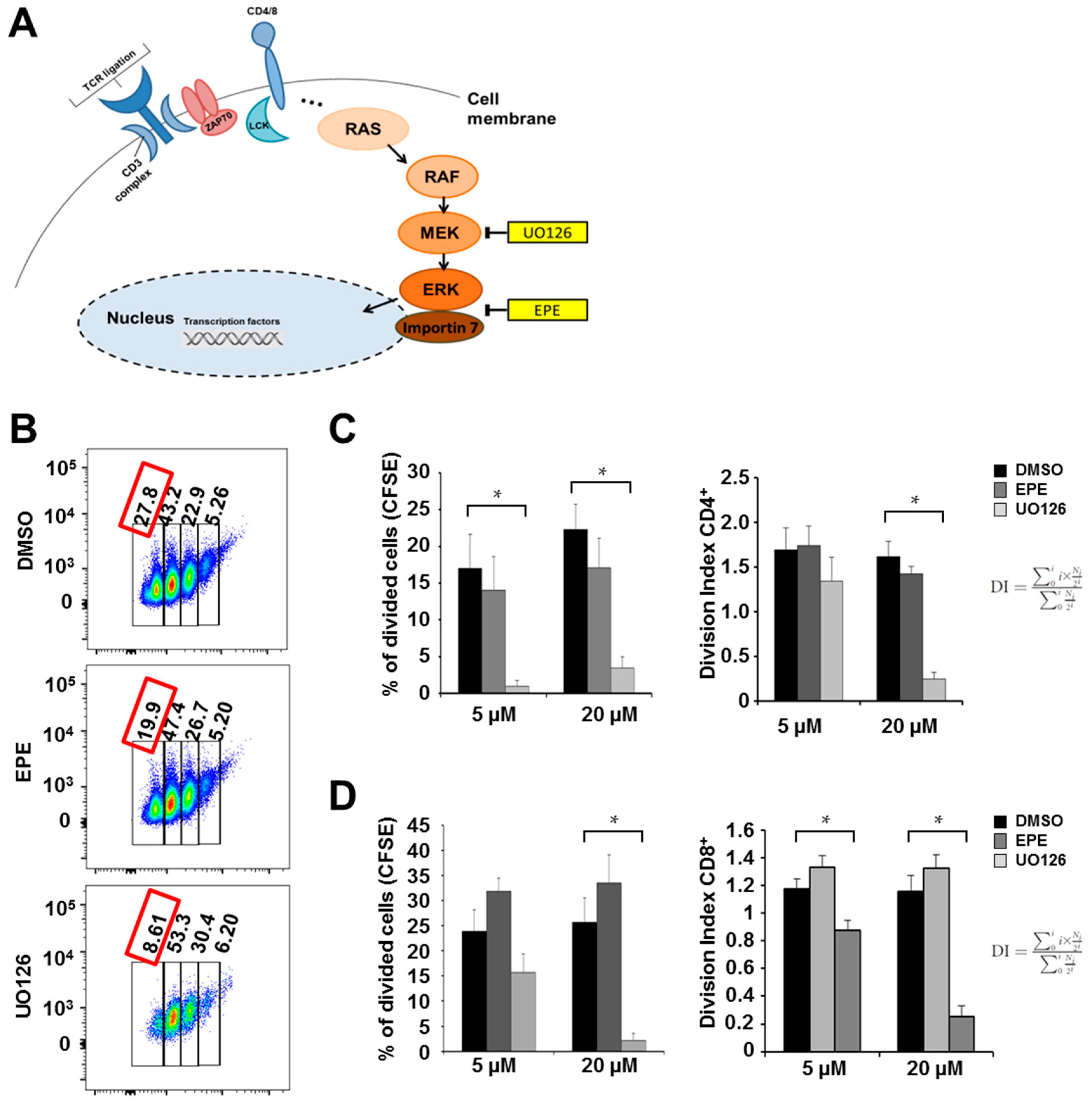
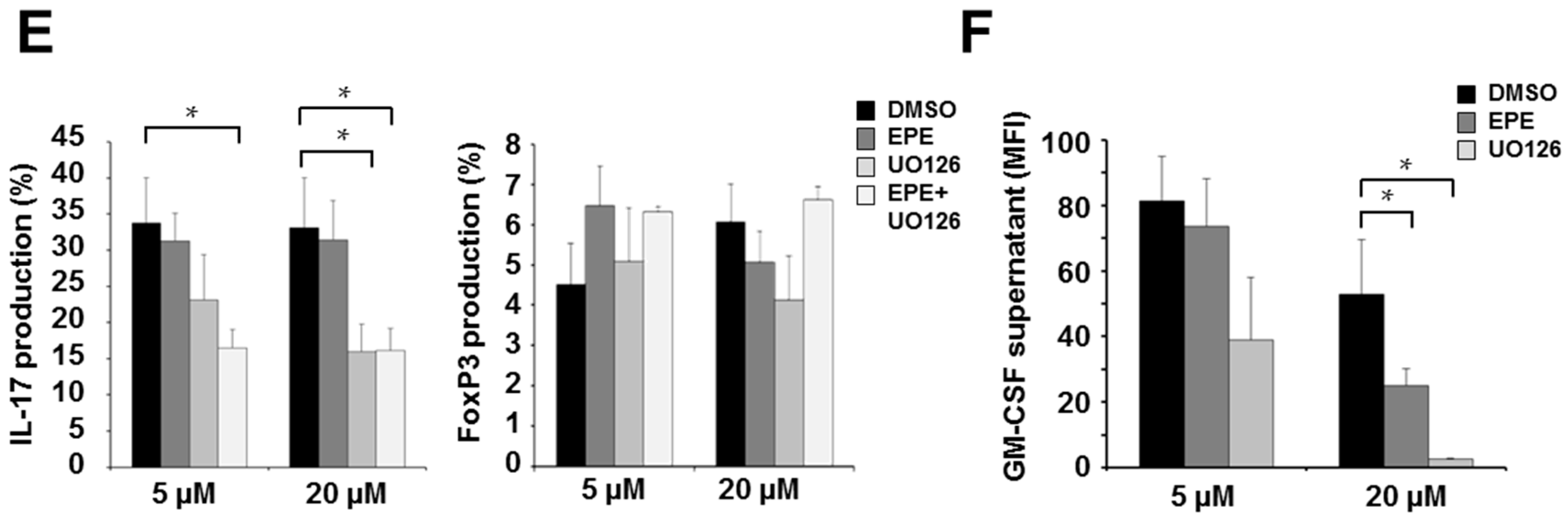
2.1. ERK Translocation Promotes Encephalitogenicity in T Cells by Facilitating GM-CSF Production
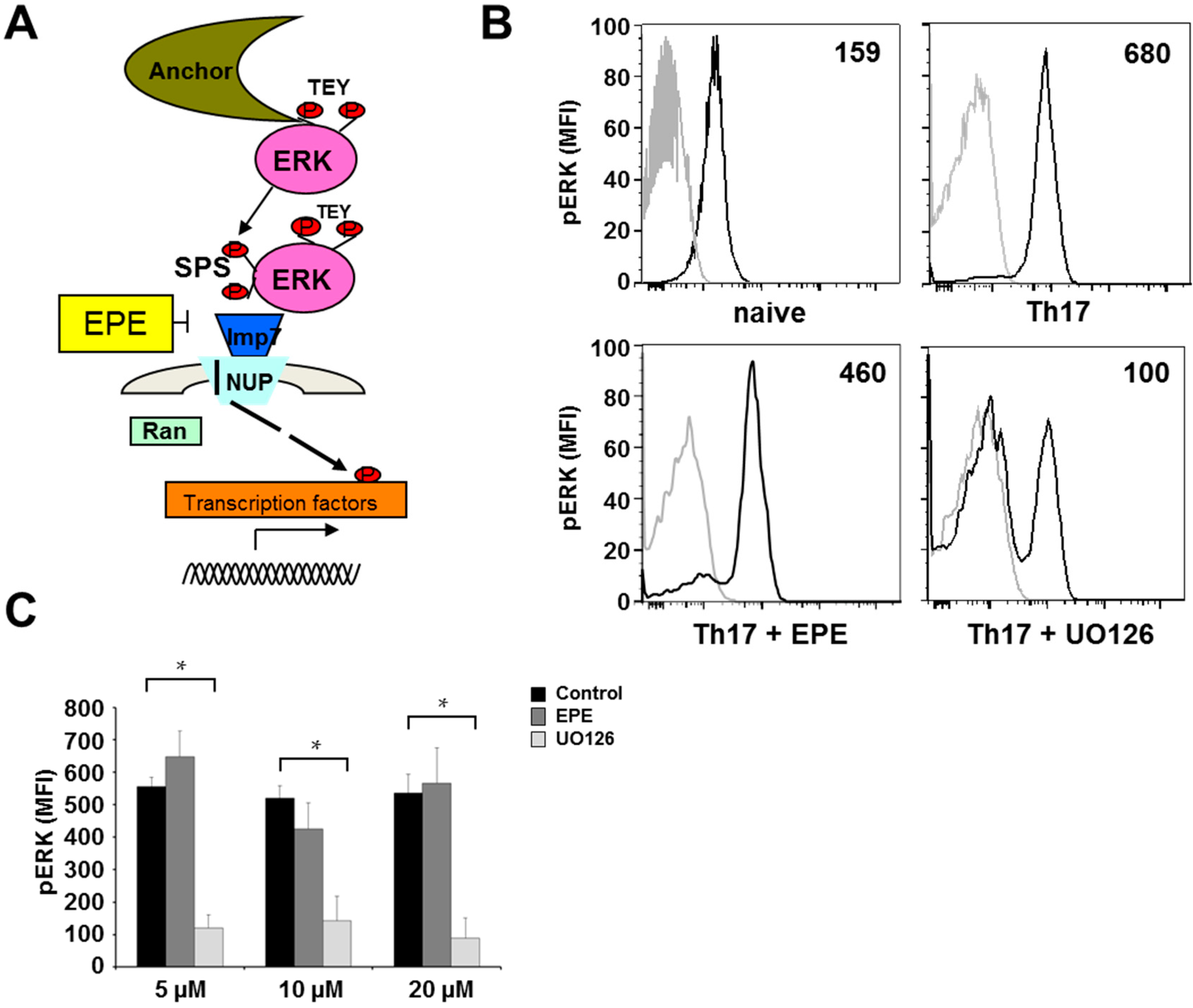
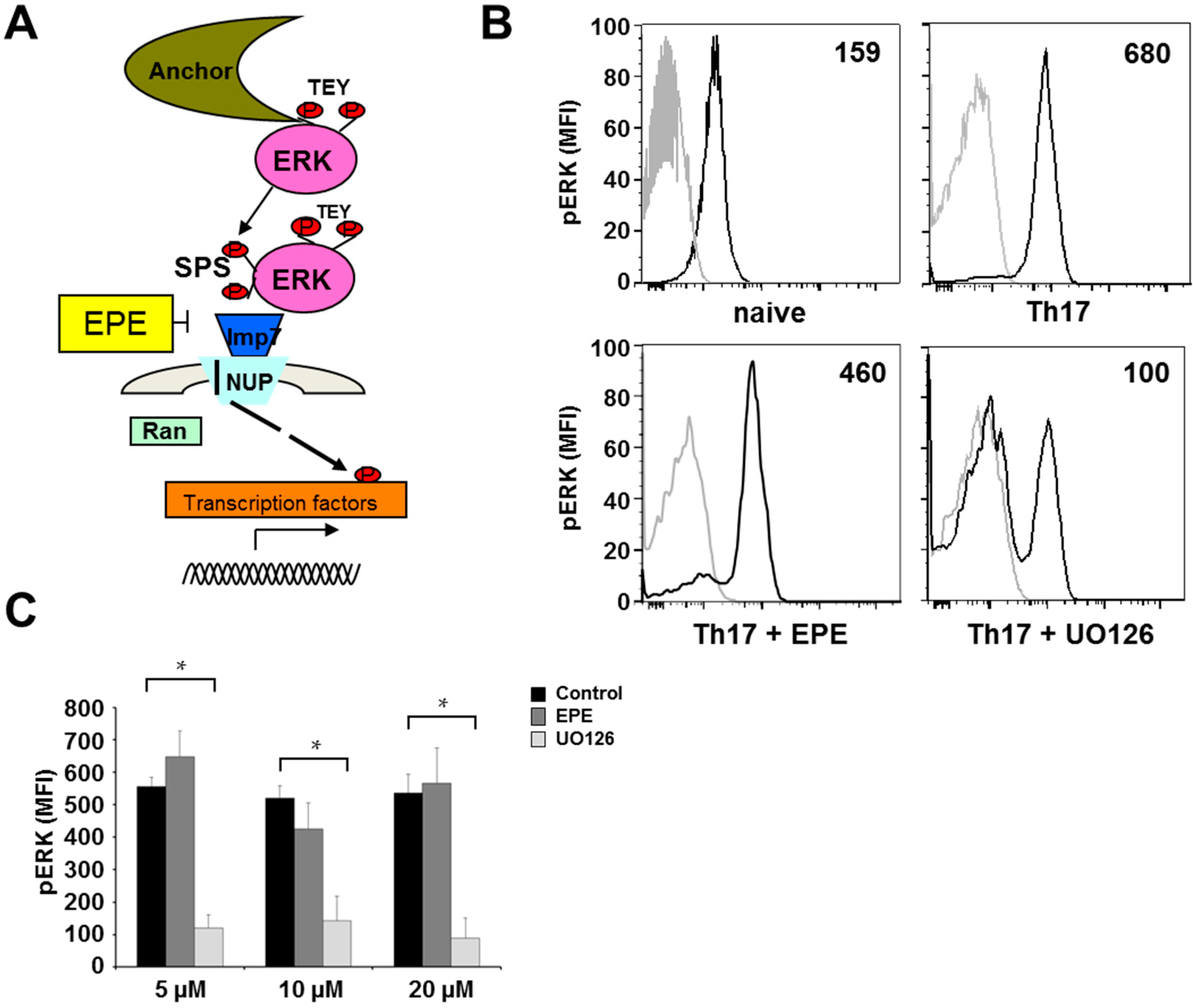
2.2. Differentiation Towards a Th17 Phenotype Increases ERK Phosphorylation with No Effect by EPE Peptide
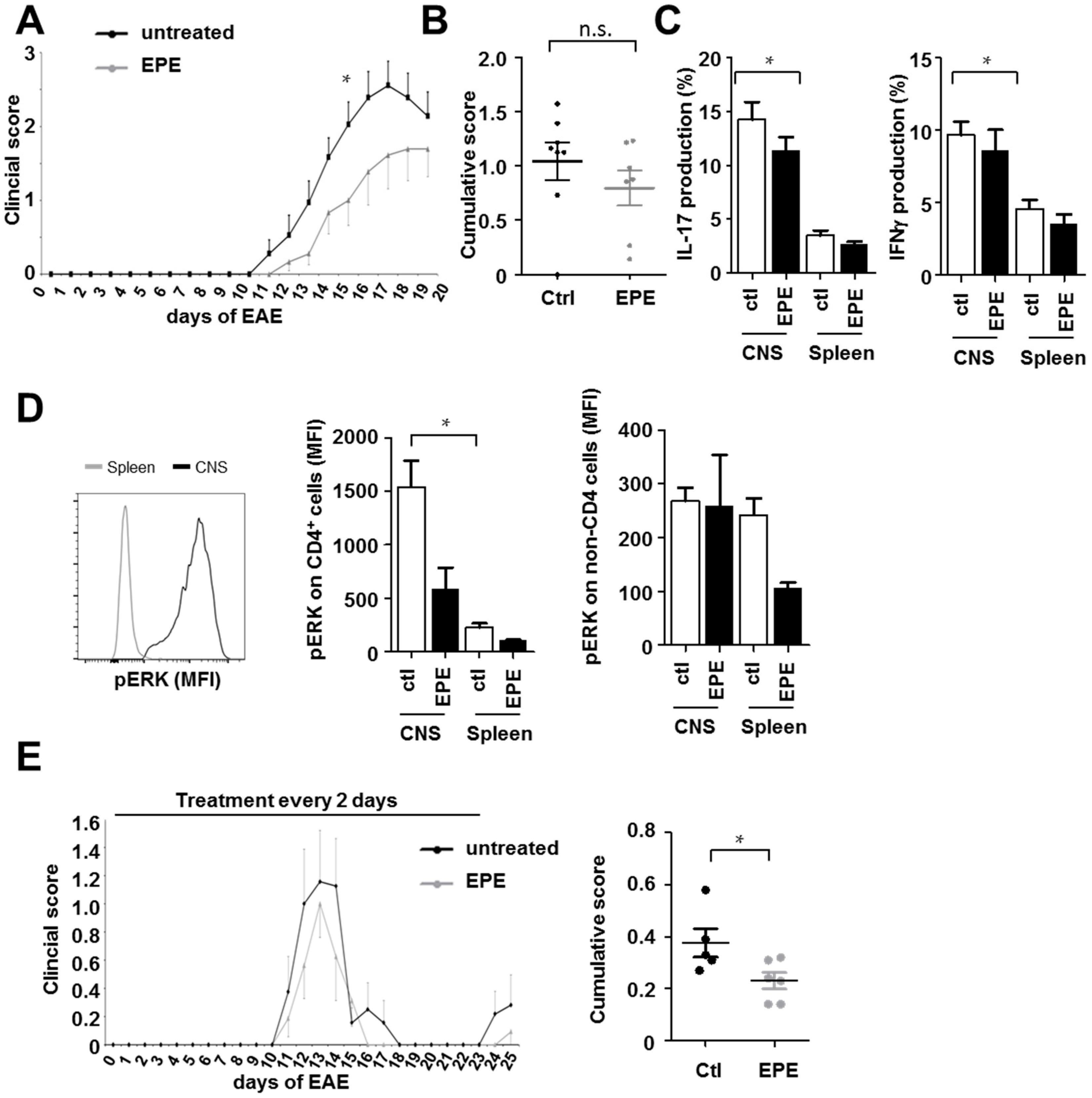
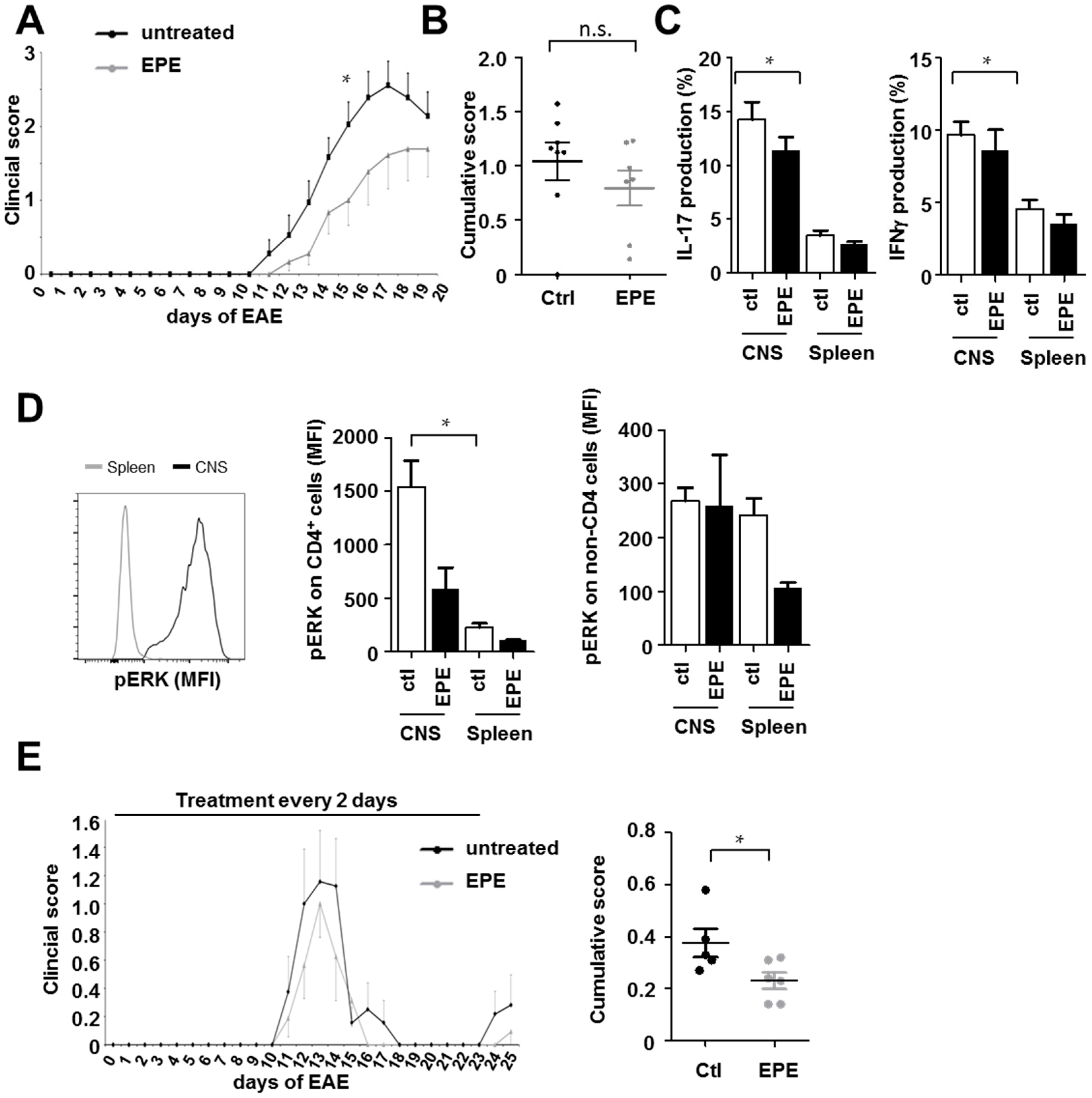
2.3. Inhibition with the EPE Peptide Has Only Discrete Influence on the Outcome in Two Different EAE Models
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Induction of Experimental Autoimmune Encephalomyelitis (EAE)
4.3. Synthesis of Peptide Constructs
4.4. Cell Isolation from Spleen and the Central Nervous System (CNS)
4.5. Th17 Cell Culture
4.6. Proliferation Assay
4.7. Antibodies
4.8. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BBB | Blood brain barrier |
| CFA | Complete Freund's adjuvant |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CNS | Central nervous system |
| DCs | Dendritic cells |
| DMSO | Dimethyl sulfoxide |
| EAE | Experimental autoimmune encephalomyelitis |
| EPE | Glu-Pro-Glu |
| ERK | Extracellular signal-regulated kinase |
| FCS | Fetal calf serum |
| GM-CSF | Granulocyte-macrophage-colony-stimulating factor |
| MAP | Mitogen-activated protein |
| MFI | Mean fluorescence intensity |
| MOG | Myelin oligodendrocyte glycoprotein |
| MS | Multiple sclerosis |
| NTS | Nuclear translocation signal |
| PBS | Phosphate buffered saline |
| PLP | Proteolipid protein |
| PMA | Phorbol 12-myristate 13-acetate |
| STAT | Signal transducer and activator of transcription |
| TCR | T cell receptor |
| WT | Wildtype |
References
- Plotnikov, A.; Chuderland, D.; Karamansha, Y.; Livnah, O.; Seger, R. Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol. Cell Biol. 2011, 31, 3515–3530. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Settleman, J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.S.; Morrison, D.K. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012, 26, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Seger, R. The ERK cascade inhibitors: Towards overcoming resistance. Drug Resist. Update 2016, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.; Ahn, M.; Jung, K.; Heo, S.; Kim, D.; Jee, Y.; Lim, Y.K.; Yeo, E.J. Activation of mitogen-activated protein kinases in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 140, 118–125. [Google Scholar] [CrossRef]
- Brereton, C.F.; Sutton, C.E.; Lalor, S.J.; Lavelle, E.C.; Mills, K.H.G. Inhibition of ERK MAPK suppresses IL-23- and IL-1-driven IL-17 production and attenuates autoimmune disease. J. Immunol. 2009, 183, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Bendix, I.; Pfueller, C.F.; Leuenberger, T.; Glezeva, N.; Siffrin, V.; Müller, Y.; Prozorovski, T.; Hansen, W.; Topphoff, U.S.; Loddenkemper, C.; et al. MAPK3 deficiency drives autoimmunity via DC arming. Eur. J. Immunol. 2010, 40, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Roose, J.P.; Mollenauer, M.; Gupta, V.A.; Stone, J.; Weiss, A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol. Cell Biol. 2005, 25, 4426–4441. [Google Scholar] [CrossRef] [PubMed]
- Roose, J.P.; Mollenauer, M.; Ho, M.; Kurosaki, T.; Weiss, A. Unusual interplay of two types of Ras activators, RasGRP and SOS, establishes sensitive and robust Ras activation in lymphocytes. Mol. Cell Biol. 2007, 27, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Deshpande, P.; Pryshchep, S.; Colmegna, I.; Liarski, V.; Weyand, C.M.; Goronzy, J.J. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J. Immunol. 2009, 183, 8258–8267. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Morris, E.J.; Hruza, A.; Mansueto, M.S.; Schroeder, G.K.; Arbanas, J.; McMasters, D.; Restaino, C.R.; Dayananth, P.; Black, S. Dissecting Therapeutic Resistance to ERK Inhibition. Mol. Cancer Ther. 2016, 15, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Das, D.; Heiser, L.H.; Bhattacharya, S.; Siwak, D.; Gendelman, R.; Bayani, N.; Wang, N.J.; Neve, R.M. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009, 69, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Jagani, Z.; Xiang, K.X.; Loo, A.; Dorsch, M.; Yao, M.Y.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Seger, R. The ERK signaling cascade–Views from different subcellular compartments. Biofactors 2009, 35, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Chuderland, D.; Seger, R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol. Biotechnol. 2005, 29, 57–74. [Google Scholar] [CrossRef]
- Plotnikov, A.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Chuderland, D.; Konson, A.; Seger, R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol. Cell 2008, 31, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Michailovici, I.; Harrington, H.A.; Azogui, H.H.; Yahalom-Ronen, Y.; Plotnikov, A.; Ching, S.; Stumpf, M.P.H.; Klein, O.D.; Seger, R.; Tzahor, E. Nuclear to cytoplasmic shuttling of ERK promotes differentiation of muscle stem/progenitor cells. Development 2014, 141, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Roederer, M. Interpretation of cellular proliferation data: Avoid the panglossian. Cytom. Part A 2011, 79, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases--The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Dillon, S.; Denning, T.L.; Pulendran, B. ERK1-/- mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. J. Immunol. 2006, 176, 5788–5796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Blattman, J.N.; Kennedy, N.J.; Duong, J.; Nguyen, T.; Wang, Y.; Davis, R.J.; Greenberg, P.D.; Flavell, R.A.; Dong, C. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 2004, 430, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.; Agrawal, A.; Dyke, T.V.; Landreth, G.; McCauley, L.; Koh, A.; Maliszewski, C.; Akira, S.; Pulendran, B. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J. Immunol. 2004, 172, 4733–4743. [Google Scholar] [CrossRef] [PubMed]
- Yi, A.K.; Yoon, J.G.; Yeo, S.J.; Hong, S.C.; English, B.K.; Krieg, A.M. Role of mitogen-activated protein kinases in CpG DNA-mediated IL-10 and IL-12 production: Central role of extracellular signal-regulated kinase in the negative feedback loop of the CpG DNA-mediated Th1 response. J. Immunol. 2002, 168, 4711–4720. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Durell, B.G.; Noelle, R.J. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J. Clin. Investig. 2003, 112, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.K.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Crews, C.M.; Alessandrini, A.; Erikson, R.L. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 1992, 258, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Patel, H.; Hedley, D.W. Measurement of MAP kinase activation by flow cytometry using phospho-specific antibodies to MEK and ERK: Potential for pharmacodynamic monitoring of signal transduction inhibitors. Cytometry 2001, 46, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J.; Staruch, M.J.; Fischer, P.; DaSilva, C.; Camacho, R. Inhibition of T cell activation by pharmacologic disruption of the MEK1/ERK MAP kinase or calcineurin signaling pathways results in differential modulation of cytokine production. J. Immunol. 1998, 160, 2579–2589. [Google Scholar] [PubMed]
- Li, Y.Q.; Hii, C.S.T.; Costabile, M.; Goh, D.; Der, C.J.; Ferrante, A. Regulation of lymphotoxin production by the p21ras-raf-MEK-ERK cascade in PHA/PMA-stimulated Jurkat cells. J. Immunol. 1999, 162, 3316–3320. [Google Scholar] [PubMed]
- Ambrosini, E.; Remoli, M.E.; Giacomini, E.; Rosicarelli, B.; Serafini, B.; Lande, R.; Aloisi, F.; Coccia, E.M. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 2005, 64, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Sagar, D.; Lamontagne, A.; Foss, C.A.; Khan, Z.K.; Pomper, M.G.; Jain, P. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood-brain barrier through paracellular transmigration and ERK activation. J. Neuroinflamm. 2012, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9563. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An. in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lee, Y.S.; Yu, C.R.; Egwuagu, C.E. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J. Immunol. 2008, 180, 6070–6076. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.N.; Chang, H.C.; Zisoulis, D.G.; Stritesky, G.L.; Yu, Q.; O’Malley, M.J.; Kapur, R.; Levy, D.E.; Kansas, G.S.; Kaplan, M.H. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 2007, 178, 4901–4907. [Google Scholar] [CrossRef] [PubMed]
- Zehorai, E.; Yao, Z.; Plotnikov, A.; Seger, R. The subcellular localization of MEK and ERK--a novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol. Cell Endocrinol. 2010, 314, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Sharp, L.L.; Schwarz, D.A.; Bott, C.M.; Marshall, C.J.; Hedrick, S.M. The influence of the MAPK pathway on T cell lineage commitment. Immunity 1997, 7, 609–618. [Google Scholar] [CrossRef]
- Joo, F. The blood-brain barrier in vitro: Ten years of research on microvessels isolated from the brain. Neurochem. Int. 1985, 7, 1–25. [Google Scholar] [CrossRef]
- Bittner, S.; Ruck, T.; Schuhmann, M.K.; Herrmann, A.M.; Maati, H.M.; Bobak, N.; Göbel, K.; Langhauser, F.; Stegner, D.; Stegner, P. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat. Med. 2013, 19, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Ruck, T.; Bittner, S.; Meuth, S.G. Blood-brain barrier modeling: challenges and perspectives. Neural. Regen. Res. 2015, 10, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.; Kashaw, S.K.; Jain, S.; Sau, S.; Iyer, A.K. Assessment of penetration potential of pH responsive double walled biodegradable nanogels coated with eucalyptus oil for the controlled delivery of 5-fluorouracil: In vitro and ex vivo studies. J. Control. Release 2017, 253, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.; Agarwalla, P.; Mukherjee, S.; Bag, I.; Sreedhar, B.; Pal-Bhadra, M.; Patra, C.P.; Banerjee, R. Cancer cell-selective promoter recognition accompanies antitumor effect by glucocorticoid receptor-targeted gold nanoparticle. Nanoscale 2014, 6, 6745–6754. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | (A) Wildtype (WT) (B) WT + EPE | (A) Wildtype (WT) (B) WT + EPE |
|---|---|---|
| n | 9 per group | 8 per group |
| Incidence | (A) 8/9 (B) 7/9 | (A) 5/8 (B) 6/8 |
| Mean day of onset (days ± SEM) | (A) 12.5 ± 0.4 (B) 14.3 ± 0.9 | (A) 11.8 ± 0.4 (B) 12 ± 0.4 |
| Mean day of disease maximum (days ± SEM) | (A) 2.9 ± 0.4 (B) 2.6 ± 0.4 | (A) 1.8 ± 0.3 (B) 1.3 ± 0.3 |
| Mean maximal score (± SEM) | (A) 2.9 ± 0.1 (B) 2.4 ± 0.3 | (A) 2 ± 0.1 (B) 1.7 ± 0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birkner, K.; Wasser, B.; Loos, J.; Plotnikov, A.; Seger, R.; Zipp, F.; Witsch, E.; Bittner, S. The Role of ERK Signaling in Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2017, 18, 1990. https://doi.org/10.3390/ijms18091990
Birkner K, Wasser B, Loos J, Plotnikov A, Seger R, Zipp F, Witsch E, Bittner S. The Role of ERK Signaling in Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2017; 18(9):1990. https://doi.org/10.3390/ijms18091990
Chicago/Turabian StyleBirkner, Katharina, Beatrice Wasser, Julia Loos, Alexander Plotnikov, Rony Seger, Frauke Zipp, Esther Witsch, and Stefan Bittner. 2017. "The Role of ERK Signaling in Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 18, no. 9: 1990. https://doi.org/10.3390/ijms18091990
APA StyleBirkner, K., Wasser, B., Loos, J., Plotnikov, A., Seger, R., Zipp, F., Witsch, E., & Bittner, S. (2017). The Role of ERK Signaling in Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 18(9), 1990. https://doi.org/10.3390/ijms18091990