Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia


Abstract
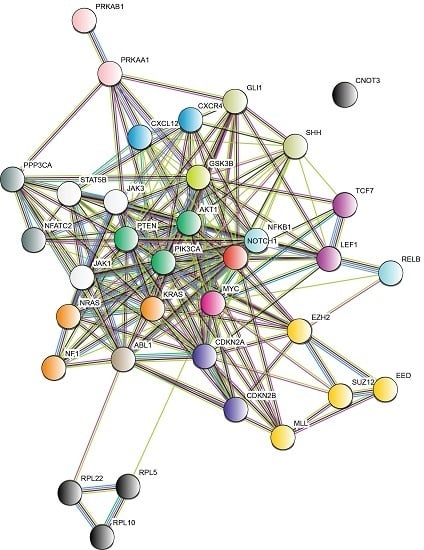
{kind=link}
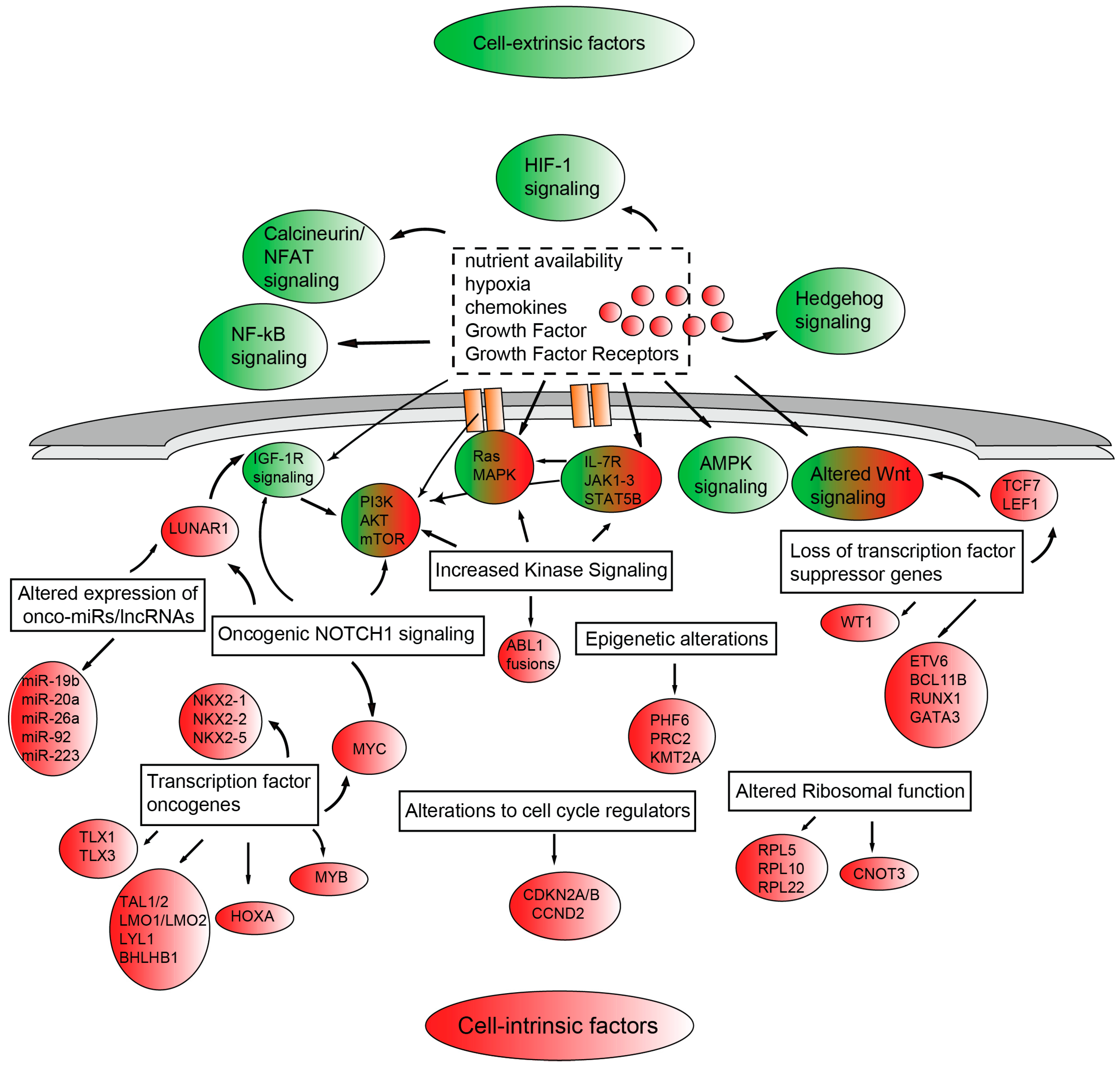{kind=link}
1. Introduction
2. Normal T Lymphocyte Development
3. Genetics of T-ALL
4. T-ALL Molecular Subgroups
5. Signaling Pathways Involved in the Development of T-ALL
5.1. Oncogenic NOTCH Signaling and NOTCH1-Myc Signaling Axis
5.2. PI3K/AKT/mTOR Pathway
5.3. IL-7R/JAK/STAT Pathway
5.4. RAS Pathway
5.5. ABL Kinase
5.6. NF-κB Pathway
5.7. Hedgehog Pathway
5.8. Calcineurin/NFAT Pathway
5.9. Wnt Signaling
5.10. Altered Metabolic Homeostasis
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef]
- Inukai, T.; Kiyokawa, N.; Campana, D.; Coustan-Smith, E.; Kikuchi, A.; Kobayashi, M.; Takahashi, H.; Koh, K.; Manabe, A.; Kumagai, M.; et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: Results of the Tokyo children’s cancer study group study L99-15. Br. J. Haematol. 2012, 156, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Quang, C.T.; Ghysdael, J. Microenvironmental cues for T-cell acute lymphoblastic leukemia development. Immunol. Rev. 2016, 271, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.; Cayuela, J.M.; Berkeley, J.; Sigaux, F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood 1994, 84, 4038–4044. [Google Scholar] [PubMed]
- Lind, E.F.; Prockop, S.E.; Porritt, H.E.; Petrie, H.T. Mapping precursor movement through the postnatal thymus reveals specific microenvironments supporting defined stages of early lymphoid development. J. Exp. Med. 2001, 194, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Grabstein, K.H.; Waldschmidt, T.J.; Finkelman, F.D.; Hess, B.W.; Alpert, A.R.; Boiani, N.E.; Namen, A.E.; Morrissey, P.J. Inhibition of murine B and T lymphopoiesis in vivo by an anti-interleukin 7 monoclonal antibody. J. Exp. Med. 1993, 178, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Nishikawa, S.; Ohno, N.; Akiyama, N.; Tamakoshi, M.; Yoshida, H. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 9125–9129. [Google Scholar] [CrossRef] [PubMed]
- Rodewald, H.R.; Kretzschmar, K.; Swat, W.; Takeda, S. Intrathymically expressed c-kit ligand (stem cell factor) is a major factor driving expansion of very immature thymocytes in vivo. Immunity 1995, 3, 313–319. [Google Scholar] [CrossRef]
- Koch, U.; Fiorini, E.; Benedito, R.; Besseyrias, V.; Schuster-Gossler, K.; Pierres, M.; Manley, N.R.; Duarte, A.; Macdonald, H.R.; Radtke, F. Delta-like 4 is the essential, nonredundant ligand for NOTCH1 during thymic T cell lineage commitment. J. Exp. Med. 2008, 205, 2515–2523. [Google Scholar] [CrossRef] [PubMed]
- Weerkamp, F.; Pike-Overzet, K.; Staal, F.J. T-sing progenitors to commit. Trends Immunol. 2006, 27, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Zuniga-Pflucker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Von Boehmer, H. Unique features of the pre-T-cell receptor α-chain: Not just a surrogate. Nat. Rev. Immunol. 2005, 5, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Ciofani, M.; Zuniga-Pflucker, J.C. NOTCH promotes survival of pre-T cells at the β-selection checkpoint by regulating cellular metabolism. Nat. Immunol. 2005, 6, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Aplan, P.D.; Davey, M.P.; Nakahara, K.; Tchorz, K.; Kurtzberg, J.; Hershfield, M.S.; Haynes, B.F.; Cohen, D.I.; Waldmann, T.A.; et al. Chromosomal translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor delta-chain diversity region and results in a previously unreported fusion transcript. Proc. Natl. Acad. Sci. USA 1989, 86, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Brown, L.; Yang, C.Y.; Tsan, J.T.; Siciliano, M.J.; Espinosa, R., 3rd; Le Beau, M.M.; Baer, R.J. TAL2, a helix-loop-helix gene activated by the (7;9)(q34;q32) translocation in human T-cell leukemia. Proc. Natl. Acad. Sci. USA 1991, 88, 11416–11420. [Google Scholar] [CrossRef] [PubMed]
- Mellentin, J.D.; Smith, S.D.; Cleary, M.L. lyl-1, a novel gene altered by chromosomal translocation in T cell leukemia, codes for a protein with a helix-loop-helix DNA binding motif. Cell 1989, 58, 77–83. [Google Scholar] [CrossRef]
- Wang, J.; Jani-Sait, S.N.; Escalon, E.A.; Carroll, A.J.; de Jong, P.J.; Kirsch, I.R.; Aplan, P.D. The t(14;21)(q11.2;q22) chromosomal translocation associated with T-cell acute lymphoblastic leukemia activates the BHLHB1 gene. Proc. Natl. Acad. Sci. USA 2000, 97, 3497–3502. [Google Scholar] [CrossRef] [PubMed]
- Royer-Pokora, B.; Loos, U.; Ludwig, W.D. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene 1991, 6, 1887–1893. [Google Scholar] [PubMed]
- McGuire, E.A.; Hockett, R.D.; Pollock, K.M.; Bartholdi, M.F.; O’Brien, S.J.; Korsmeyer, S.J. The t(11;14)(p15;q11) in a T-cell acute lymphoblastic leukemia cell line activates multiple transcripts, including Ttg-1, a gene encoding a potential zinc finger protein. Mol. Cell. Biol. 1989, 9, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Roberts, C.W.; Minden, M.; Crist, W.M.; Korsmeyer, S.J. Deregulation of a homeobox gene, HOX11, by the t(10;14) in T cell leukemia. Science 1991, 253, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Bernard, O.A.; Busson-LeConiat, M.; Ballerini, P.; Mauchauffe, M.; Della Valle, V.; Monni, R.; Nguyen Khac, F.; Mercher, T.; Penard-Lacronique, V.; Pasturaud, P.; et al. A new recurrent and specific cryptic translocation, t(5;14)(q35;q32), is associated with expression of the Hox11L2 gene in T acute lymphoblastic leukemia. Leukemia 2001, 15, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Homminga, I.; Pieters, R.; Langerak, A.W.; de Rooi, J.J.; Stubbs, A.; Verstegen, M.; Vuerhard, M.; Buijs-Gladdines, J.; Kooi, C.; Klous, P.; et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 2011, 19, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; Kaufmann, M.; Drexler, H.G.; MacLeod, R.A. The cardiac homeobox gene NKX2-5 is deregulated by juxtaposition with BCL11B in pediatric T-ALL cell lines via a novel t(5;14)(q35.1;q32.2). Cancer Res. 2003, 63, 5329–5334. [Google Scholar] [PubMed]
- Soulier, J.; Clappier, E.; Cayuela, J.M.; Regnault, A.; Garcia-Peydro, M.; Dombret, H.; Baruchel, A.; Toribio, M.L.; Sigaux, F. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005, 106, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Erikson, J.; Finger, L.; Sun, L.; ar-Rushdi, A.; Nishikura, K.; Minowada, J.; Finan, J.; Emanuel, B.S.; Nowell, P.C.; Croce, C.M. Deregulation of c-myc by translocation of the α-locus of the T-cell receptor in T-cell leukemias. Science 1986, 232, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Clappier, E.; Cuccuini, W.; Kalota, A.; Crinquette, A.; Cayuela, J.M.; Dik, W.A.; Langerak, A.W.; Montpellier, B.; Nadel, B.; Walrafen, P.; et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 2007, 110, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Graux, C.; Cools, J.; Melotte, C.; Quentmeier, H.; Ferrando, A.; Levine, R.; Vermeesch, J.R.; Stul, M.; Dutta, B.; Boeckx, N.; et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat. Genet. 2004, 36, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, H.; Beverloo, H.B.; van Drunen, E.; Janssens, A.; Hahlen, K.; Poppe, B.; Van Roy, N.; Marynen, P.; De Paepe, A.; Slater, R.; et al. Molecular cytogenetic and clinical findings in ETV6/ABL1-positive leukemia. Genes Chromosomes Cancer 2001, 30, 274–282. [Google Scholar] [CrossRef]
- Remke, M.; Pfister, S.; Kox, C.; Toedt, G.; Becker, N.; Benner, A.; Werft, W.; Breit, S.; Liu, S.; Engel, F.; et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-β and PI3K-AKT pathways and deletions at 6q15-16.1 as a genomic marker for unfavorable early treatment response. Blood 2009, 114, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Palomero, T.; Khiabanian, H.; Van der Meulen, J.; Castillo, M.; Van Roy, N.; De Moerloose, B.; Philippe, J.; Gonzalez-Garcia, S.; Toribio, M.L.; et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, J.; Sanghvi, V.; Mavrakis, K.; Durinck, K.; Fang, F.; Matthijssens, F.; Rondou, P.; Rosen, M.; Pieters, T.; Vandenberghe, P.; et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 2015, 125, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Thalhammer-Scherrer, R.; Mitterbauer, G.; Simonitsch, I.; Jaeger, U.; Lechner, K.; Schneider, B.; Fonatsch, C.; Schwarzinger, I. The immunophenotype of 325 adult acute leukemias: Relationship to morphologic and molecular classification and proposal for a minimal screening program highly predictive for lineage discrimination. Am. J. Clin. Pathol. 2002, 117, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Durinck, K.; Goossens, S.; Peirs, S.; Wallaert, A.; Van Loocke, W.; Matthijssens, F.; Pieters, T.; Milani, G.; Lammens, T.; Rondou, P.; et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp. Hematol. 2015, 43, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, K.; Atak, Z.K.; Li, N.; Vicente, C.; Patchett, S.; Girardi, T.; Gianfelici, V.; Geerdens, E.; Clappier, E.; Porcu, M.; et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Tosello, V.; Mansour, M.R.; Barnes, K.; Paganin, M.; Sulis, M.L.; Jenkinson, S.; Allen, C.G.; Gale, R.E.; Linch, D.C.; Palomero, T.; et al. WT1 mutations in T-ALL. Blood 2009, 114, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.L.; Akkapeddi, P.; Alcobia, I.; Almeida, A.R.; Cardoso, B.A.; Fragoso, R.; Serafim, T.L.; Barata, J.T. From the outside, from within: Biological and therapeutic relevance of signal transduction in T-cell acute lymphoblastic leukemia. Cell Signal. 2017, 38, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, J.; van Roy, N.; van Vlierberghe, P.; Speleman, F. The epigenetic landscape of T-cell acute lymphoblastic leukemia. Int. J. Biochem. Cell Biol. 2014, 53, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T cell fate specification in mice with an induced inactivation of NOTCH1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila NOTCH gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Buonamici, S.; Sulis, M.L.; Palomero, T.; Vilimas, T.; Basso, G.; Ferrando, A.; Aifantis, I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007, 204, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi-Elias, P.; Hu, T.; Jenkins, D.; Firestone, B.; Gans, S.; Kurth, E.; Capodieci, P.; Deplazes-Lauber, J.; Petropoulos, K.; Thiel, P.; et al. Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene 2016, 35, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Maraver, A.; Latkowski, J.A.; Henderson, T.; Schlessinger, K.; Ding, Y.; Shen, J.; Tadokoro, C.E.; Lafaille, J.J. Characterization of two distinct lymphoproliferative diseases caused by ectopic expression of the NOTCH ligand DLL4 on T cells. PLoS ONE 2013, 8, e84841. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of NOTCH1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navas, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to NOTCH inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [PubMed]
- Real, P.J.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; de Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. γ-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.O.; Zapata, M.C.; Singh, A.; Jo, W.S.; Spencer, N.; Choi, Y.S. γ secretase inhibitors enhance vincristine-induced apoptosis in T-ALL in a NOTCH-independent manner. Apoptosis Int. J. Program. Cell Death 2014, 19, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the γ-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2012, 11, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual NOTCH receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Agnusdei, V.; Minuzzo, S.; Frasson, C.; Grassi, A.; Axelrod, F.; Satyal, S.; Gurney, A.; Hoey, T.; Seganfreddo, E.; Basso, G.; et al. Therapeutic antibody targeting of NOTCH 1 in T-acute lymphoblastic leukemia xenografts. Leukemia 2014, 28, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by NOTCH signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Wendorff, A.A.; Koch, U.; Wunderlich, F.T.; Wirth, S.; Dubey, C.; Bruning, J.C.; MacDonald, H.R.; Radtke, F. Hes1 is a critical but context-dependent mediator of canonical NOTCH signaling in lymphocyte development and transformation. Immunity 2010, 33, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S.A.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Belver, L.; Xu, L.; Qin, Y.; Kageyama, R.; Ferrando, A.A. Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood 2015, 125, 2806–2814. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Dose, M.; Khan, I.; Guo, Z.; Kovalovsky, D.; Krueger, A.; von Boehmer, H.; Khazaie, K.; Gounari, F. c-Myc mediates pre-TCR-induced proliferation but not developmental progression. Blood 2006, 108, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. NOTCH1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [PubMed]
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Juntilla, M.M.; Koretzky, G.A. Critical roles of the PI3K/Akt signaling pathway in T cell development. Immunol. Lett. 2008, 116, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Juntilla, M.M.; Wofford, J.A.; Birnbaum, M.J.; Rathmell, J.C.; Koretzky, G.A. Akt1 and Akt2 are required for αβ thymocyte survival and differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 12105–12110. [Google Scholar] [CrossRef] [PubMed]
- Swat, W.; Montgrain, V.; Doggett, T.A.; Douangpanya, J.; Puri, K.; Vermi, W.; Diacovo, T.G. Essential role of PI3Kdelta and PI3Kγ in thymocyte survival. Blood 2006, 107, 2415–2422. [Google Scholar] [CrossRef] [PubMed]
- Buckler, J.L.; Walsh, P.T.; Porrett, P.M.; Choi, Y.; Turka, L.A. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J. Immunol. 2006, 177, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.P.; Andres, P.G.; Howland, K.C.; Abbas, A.K.; Weiss, A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-γ but not TH2 cytokines. Nat. Immunol. 2001, 2, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.; Neuberg, D.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.S.; Choudhury, B.; Campbell, P.J.; Feng, B.; Wong, K.-K.; Protopopov, A.; O’Neil, J.; Gutierrez, A.; Ivanova, E.; Perna, I.; et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007, 447, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Jotta, P.Y.; Ganazza, M.A.; Silva, A.; Viana, M.B.; da Silva, M.J.; Zambaldi, L.J.G.; Barata, J.T.; Brandalise, S.R.; Yunes, J.A. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia 2009, 24, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. The genomic landscape of acute lymphoblastic leukemia in children and young adults. ASH Educ. Program Book 2014, 2014, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.T.; Silva, A.; Brandao, J.G.; Nadler, L.M.; Cardoso, A.A.; Boussiotis, V.A. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J. Exp. Med. 2004, 200, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.E.; Shah, N.; Bajer, A.A.; LeBien, T.W. IL-7 activates the phosphatidylinositol 3-kinase/AKT pathway in normal human thymocytes but not normal human B cell precursors. J. Immunol. 2008, 180, 8109–8117. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, S.; Garcia-Peydro, M.; Martin-Gayo, E.; Ballestar, E.; Esteller, M.; Bornstein, R.; de la Pompa, J.L.; Ferrando, A.A.; Toribio, M.L. CSL-MAML-dependent NOTCH1 signaling controls T lineage-specific IL-7R{α} gene expression in early human thymopoiesis and leukemia. J. Exp. Med. 2009, 206, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Wolfe, A.L.; Oricchio, E.; Palomero, T.; de Keersmaecker, K.; McJunkin, K.; Zuber, J.; James, T.; Khan, A.A.; Leslie, C.S.; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in NOTCH-induced T-cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2010, 12, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Grebliunaite, R.; Feng, H.; Kozakewich, E.; Zhu, S.; Guo, F.; Payne, E.; Mansour, M.; Dahlberg, S.E.; Neuberg, D.S.; et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Hales, E.C.; Orr, S.M.; Larson Gedman, A.; Taub, J.W.; Matherly, L.H. NOTCH1 receptor regulates AKT protein activation loop (Thr308) dephosphorylation through modulation of the PP2A phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell acute lymphoblastic leukemia cells. J. Biol. Chem. 2013, 288, 22836–22848. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, A. Mechanisms by which IGF-I may promote cancer. Cancer Biol. Ther. 2003, 2, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Gusscott, S.; Kuchenbauer, F.; Humphries, R.K.; Weng, A.P. NOTCH-mediated repression of miR-223 contributes to IGF1R regulation in T-ALL. Leukemia Res. 2012, 36, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of NOTCH-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Hoshii, T.; Kasada, A.; Hatakeyama, T.; Ohtani, M.; Tadokoro, Y.; Naka, K.; Ikenoue, T.; Ikawa, T.; Kawamoto, H.; Fehling, H.J.; et al. Loss of mTOR complex 1 induces developmental blockage in early T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Antunes, I.L.; Chiarini, F.; Orsini, E.; Buontempo, F.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; Melchionda, F.; Pession, A.; et al. Activity of the pan-class I phosphoinositide 3-kinase inhibitor NVP-BKM120 in T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Cappellini, A.; Sparta, A.M.; Chiarini, F.; Buontempo, F.; Evangelisti, C.; Orsini, E.; McCubrey, J.A.; Martelli, A.M. PI3K pan-inhibition impairs more efficiently proliferation and survival of T-cell acute lymphoblastic leukemia cell lines when compared to isoform-selective PI3K inhibitors. Oncotarget 2015, 6, 10399–10414. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, P.S.; Whye, D.W.; Efimenko, E.; Chen, J.; Tosello, V.; De Keersmaecker, K.; Kashishian, A.; Thompson, M.A.; Castillo, M.; Cordon-Cardo, C.; et al. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell 2012, 21, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Simioni, C.; Neri, L.M.; Tabellini, G.; Ricci, F.; Bressanin, D.; Chiarini, F.; Evangelisti, C.; Cani, A.; Tazzari, P.L.; Melchionda, F.; et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 2012, 26, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; Da Silva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Fala, F.; Tazzari, P.L.; Ricci, F.; Astolfi, A.; Pession, A.; Pagliaro, P.; McCubrey, J.A.; Martelli, A.M. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009, 69, 3520–3528. [Google Scholar] [CrossRef] [PubMed]
- Fransecky, L.; Mochmann, L.H.; Baldus, C.D. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, W.Q.; Aiello, F.B.; Mazzucchelli, R.; Asefa, B.; Khaled, A.R.; Durum, S.K. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev. 2005, 16, 513–533. [Google Scholar] [CrossRef] [PubMed]
- ElKassar, N.; Gress, R.E. An overview of IL-7 biology and its use in immunotherapy. J. Immunotoxicol. 2010, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, R.; Durum, S.K. Interleukin-7 receptor expression: Intelligent design. Nat. Rev. Immunol. 2007, 7, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Von Freeden-Jeffry, U.; Vieira, P.; Lucian, L.A.; McNeil, T.; Burdach, S.E.; Murray, R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 1995, 181, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Ziegler, S.F.; Buckley, R.H.; Leonard, W.J. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat. Genet. 1998, 20, 394–397. [Google Scholar] [PubMed]
- Noguchi, M.; Yi, H.; Rosenblatt, H.M.; Filipovich, A.H.; Adelstein, S.; Modi, W.S.; McBride, O.W.; Leonard, W.J. Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell 1993, 73, 147–157. [Google Scholar] [CrossRef]
- Rich, B.E.; Campos-Torres, J.; Tepper, R.I.; Moreadith, R.W.; Leder, P. Cutaneous lymphoproliferation and lymphomas in interleukin 7 transgenic mice. J. Exp. Med. 1993, 177, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.; Ma, M.C.; Snow, J.W.; Miners, M.J.; Herndier, B.G.; Goldsmith, M.A. Haploinsufficiency identifies STAT5 as a modifier of IL-7-induced lymphomas. Oncogene 2005, 24, 5252–5257. [Google Scholar] [CrossRef] [PubMed]
- Dibirdik, I.; Langlie, M.C.; Ledbetter, J.A.; Tuel-Ahlgren, L.; Obuz, V.; Waddick, K.G.; Gajl-Peczalska, K.; Schieven, G.L.; Uckun, F.M. Engagement of interleukin-7 receptor stimulates tyrosine phosphorylation, phosphoinositide turnover, and clonal proliferation of human T-lineage acute lymphoblastic leukemia cells. Blood 1991, 78, 564–570. [Google Scholar] [PubMed]
- Touw, I.; Pouwels, K.; van Agthoven, T.; van Gurp, R.; Budel, L.; Hoogerbrugge, H.; Delwel, R.; Goodwin, R.; Namen, A.; Löwenberg, B. Interleukin-7 is a growth factor of precursor B and T acute lymphoblastic leukemia. Blood 1990, 75, 2097–2101. [Google Scholar] [PubMed]
- Digel, W.; Schmid, M.; Heil, G.; Conrad, P.; Gillis, S.; Porzsolt, F. Human interleukin-7 induces proliferation of neoplastic cells from chronic lymphocytic leukemia and acute leukemias. Blood 1991, 78, 753–759. [Google Scholar] [PubMed]
- Silva, A.; Laranjeira, A.B.A.; Martins, L.R.; Cardoso, B.A.; Demengeot, J.; Andrés Yunes, J.; Seddon, B.; Barata, J.T. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011, 71, 4780–4789. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.T.; Cardoso, A.A.; Nadler, L.M.; Boussiotis, V.A. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood 2001, 98, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Gírio, A.; Cebola, I.; Santos, C.I.; Antunes, F.; Barata, J.T. Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia 2011, 25, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Flex, E.; Petrangeli, V.; Stella, L.; Chiaretti, S.; Hornakova, T.; Knoops, L.; Ariola, C.; Fodale, V.; Clappier, E.; Paoloni, F.; et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 2008, 205, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Bains, T.; Heinrich, M.C.; Loriaux, M.M.; Beadling, C.; Nelson, D.; Warrick, A.; Neff, T.L.; Tyner, J.W.; Dunlap, J.; Corless, C.L.; et al. Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia 2012, 26, 2144–2146. [Google Scholar] [CrossRef] [PubMed]
- Kontro, M.; Kuusanmaki, H.; Eldfors, S.; Pemovska, T.; Rajala, H.L.M.; Edgren, H.; Ellonen, P.; Lagstrom, S.; Lundan, T.; Kallioniemi, O.; et al. Novel Activating STAT5B Mutations As Drivers Of T-ALL. Blood 2013, 122, 3863. [Google Scholar]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stutz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, e188–e192. [Google Scholar] [CrossRef] [PubMed]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffe, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Onnebo, S.M.; Rasighaemi, P.; Kumar, J.; Liongue, C.; Ward, A.C. Alternative TEL-JAK2 fusions associated with T-cell acute lymphoblastic leukemia and atypical chronic myelogenous leukemia dissected in zebrafish. Haematologica 2012, 97, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Lahortiga, I.; El Chaar, T.; De Keersmaecker, K.; Mentens, N.; Graux, C.; Van Roosbroeck, K.; Ferrando, A.A.; Langerak, A.W.; Meijerink, J.P.; et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Soulier, J.; Asnafi, V.; Mentens, N.; Hornakova, T.; Knoops, L.; Constantinescu, S.; Sigaux, F.; Meijerink, J.P.; Vandenberghe, P.; et al. PTPN2 negatively regulates oncogenic JAK1 in T-cell acute lymphoblastic leukemia. Blood 2011, 117, 7090–7098. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, C.S.; Brown, F.C.; Collett, M.; Saw, J.; Chiu, S.K.; Sonderegger, S.E.; Lucas, S.E.; Alserihi, R.; Chau, N.; Toribio, M.L.; et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia 2016, 30, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Radaelli, E.; Blanchet, O.; Durinck, K.; Van der Meulen, J.; Peirs, S.; Taghon, T.; Tremblay, C.S.; Costa, M.; Farhang Ghahremani, M.; et al. ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat. Commun. 2015, 6, 5794. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular pathways: Molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Vannucchi, A.M.; Cervantes, F.; Harrison, C.; Morra, E.; Kantarjian, H.; Verstovsek, S. Ruxolitinib and survival improvement in patients with myelofibrosis. Leukemia 2015, 29, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015, 125, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Buijs-Gladdines, J.G.; Cante-Barrett, K.; Stubbs, A.P.; Vroegindeweij, E.M.; Smits, W.K.; van Marion, R.; Dinjens, W.N.; Horstmann, M.; Kuiper, R.P.; et al. IL-7 Receptor Mutations and Steroid Resistance in Pediatric T cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study. PLoS Med. 2016, 13, e1002200. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
- Irving, J.; Matheson, E.; Minto, L.; Blair, H.; Case, M.; Halsey, C.; Swidenbank, I.; Ponthan, F.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 2014, 124, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Oshima, K.; Khiabanian, H.; da Silva-Almeida, A.C.; Tzoneva, G.; Abate, F.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Carpenter, Z.; Penson, A.; Perez-Garcia, A.; et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, 11306–11311. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Cornejo, M.G.; Scholl, C.; Liu, J.; Leeman, D.S.; Haydu, J.E.; Fröhling, S.; Lee, B.H.; Gilliland, D.G. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor NOTCH1 mutations and are sensitive to {γ}-secretase inhibitors. Blood 2008, 112, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Du, J.; Liu, Y.; Meline, B.; Chang, Y.I.; Ranheim, E.A.; Wang, J.; Zhang, J. NOTCH1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J. Biol. Chem. 2013, 288, 18219–18227. [Google Scholar] [CrossRef] [PubMed]
- Von Lintig, F.C.; Huvar, I.; Law, P.; Diccianni, M.B.; Yu, A.L.; Boss, G.R. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2000, 6, 1804–1810. [Google Scholar] [PubMed]
- Yokota, S.; Nakao, M.; Horiike, S.; Seriu, T.; Iwai, T.; Kaneko, H.; Azuma, H.; Oka, T.; Takeda, T.; Watanabe, A.; et al. Mutational analysis of the N-ras gene in acute lymphoblastic leukemia: A study of 125 Japanese pediatric cases. Int. J. Hematol. 1998, 67, 379–387. [Google Scholar] [CrossRef]
- Perentesis, J.P.; Bhatia, S.; Boyle, E.; Shao, Y.; Shu, X.O.; Steinbuch, M.; Sather, H.N.; Gaynon, P.; Kiffmeyer, W.; Envall-Fox, J.; et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia 2004, 18, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Zhang, Y.; Chang, J.; Zheng, S.; Metayer, C.; Zhang, L.; Smith, M.T.; Ma, X.; Selvin, S.; Buffler, P.A.; et al. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia 2005, 19, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Van Vlierberghe, P.; Van Den Ouweland, A.M.W.; Beverloo, H.B.; Terlouw-Kromosoeto, J.N.R.; Van Wering, E.R.; Reinhardt, D.; Horstmann, M.; Kaspers, G.J.L.; Pieters, R.; et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood 2008, 111, 4322–4328. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, B.A.; Lapinski, P.E.; Oliver, J.A.; Ksionda, O.; Parada, L.F.; Zhu, Y.; Maillard, I.; Chiang, M.; Roose, J.; King, P.D. Cutting Edge: Codeletion of the Ras GTPase-Activating Proteins (RasGAPs) Neurofibromin 1 and p120 RasGAP in T Cells Results in the Development of T Cell Acute Lymphoblastic Leukemia. J. Immunol. 2015, 195, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Dower, N.A.; Stang, S.L.; Bottorff, D.A.; Ebinu, J.O.; Dickie, P.; Ostergaard, H.L.; Stone, J.C. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol. 2000, 1, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Mues, M.; Roose, J.P. Distinct oncogenic Ras signals characterized by profound differences in flux through the RasGDP/RasGTP cycle. Small GTPases 2017, 8, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ksionda, O.; Melton, A.; Bache, J.; Tenhagen, M.; Bakker, J.; Harvey, R.; Winter, S.; Rubio, I.; Roose, J. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines HHS Public Access. Oncogene 2016, 14, 3658–3668. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liu, X.; Yu, W.M.; Meyerson, H.J.; Guo, C.; Gerson, S.L.; Qu, C.K. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J. Exp. Med. 2011, 208, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Hagemeijer, A.; Graux, C. ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 2010, 49, 299–308. [Google Scholar] [PubMed]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. ASH Educ. Program Book 2012, 2012, 389–396. [Google Scholar]
- Raanani, P.; Trakhtenbrot, L.; Rechavi, G.; Rosenthal, E.; Avigdor, A.; Brok-Simoni, F.; Leiba, M.; Amariglio, N.; Nagler, A.; Ben-Bassat, I. Philadelphia-chromosome-positive T-lymphoblastic leukemia: Acute leukemia or chronic myelogenous leukemia blastic crisis. Acta Haematol. 2005, 113, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Graux, C.; Stevens-Kroef, M.; Lafage, M.; Dastugue, N.; Harrison, C.J.; Mugneret, F.; Bahloula, K.; Struski, S.; Grégoire, M.J.; Nadal, N.; et al. Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 2009, 23, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, T.; Gökbuget, N.; Reinhardt, R.; Rieder, H.; Hoelzer, D.; Schwartz, S. NUP214-ABL1 in adult T-ALL: The GMALL study group experience. Blood 2006, 108, 3556–3559. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Moorman, A.V.; Cazzaniga, G.; Stanulla, M.; Harvey, R.C.; Roberts, K.G.; Heatley, S.L.; Loh, M.L.; Konopleva, M.; Chen, I.M.; et al. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica 2016, 101, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, K.; Graux, C.; Odero, M.D.; Mentens, N.; Somers, R.; Maertens, J.; Wlodarska, I.; Vandenberghe, P.; Hagemeijer, A.; Marynen, P.; et al. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32). Blood 2005, 105, 4849–4852. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Tong, W.; Manshouri, T.; Vega, F.; Lennon, P.A.; Cools, J.; Gilliland, D.G.; Lee, F.; Cortes, J.; Kantarjian, H.; et al. Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 2008, 22, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, K.; Versele, M.; Cools, J.; Superti-Furga, G.; Hantschel, O. Intrinsic differences between the catalytic properties of the oncogenic NUP214-ABL1 and BCR-ABL1 fusion protein kinases. Leukemia 2008, 22, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Eide, C.A.; Deininger, M.W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Fulford, T.S.; Messina, N.L.; Grumont, R.J. NF-κB control of T cell development. Nat. Immunol. 2013, 15, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Jimi, E.; Phillips, R.J.; Barber, D.F.; Rincon, M.; Hayday, A.C.; Flavell, R.A.; Ghosh, S. NF-Kappa B Activation by the pre-T Cell receptor serves as a selective survival signal in T lymphocyte development. Immunity 2000, 13, 677–689. [Google Scholar] [CrossRef]
- Jimi, E.; Strickland, I.; Voll, R.E.; Long, M.; Ghosh, S. Differential role of the transcription factor NF-kappaB in selection and survival of CD4+ and CD8+ thymocytes. Immunity 2008, 29, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-κB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-H.; Xiao, Y.; Hu, H.; Jin, J.; Yu, J.; Zhou, X.; Wu, X.; Johnson, H.M.; Akira, S.; Pasparakis, M.; et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat. Immunol. 2012, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, V.; Hammond, K.J. L.; Howells, N.; Pfeffer, K.; Weih, F. Differential requirement for Rel/nuclear factor kappa B family members in natural killer T cell development. J. Exp. Med. 2003, 197, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, S.; Gugasyan, R.; Kyparissoudis, K.; Grumont, R.; Banerjee, A.; Tsichlis, P.; Gerondakis, S.; Godfrey, D.I. Distinct roles in NKT cell maturation and function for the different transcription factors in the classical NF-κB pathway. Immunol. Cell Biol. 2011, 89, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.D.; Scheidereit, C. Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Poglio, S.; Cahu, X.; Uzan, B.; Besnard-Guerin, C.; Lapillonne, H.; Leblanc, T.; Baruchel, A.; Landman-Parker, J.; Petit, A.; Baleydier, F.; et al. Rapid childhood T-ALL growth in xenograft models correlates with mature phenotype and NF-kappaB pathway activation but not with poor prognosis. Leukemia 2015, 29, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.L.; et al. Targeting the NF-κB signaling pathway in NOTCH1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Ingles-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; et al. The NOTCH/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.R.; Williame, M.; Gachet, S.; Cormier, F.; Janin, A.; Weih, D.; Weih, F.; Ghysdael, J. RelB-dependent stromal cells promote T-cell leukemogenesis. PLoS ONE 2008, 3, e2555. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.; Ventura, J.-J.; Cusson, N.; Kelliher, M. NF-kappaB activation in premalignant mouse tal-1/scl thymocytes and tumors. Blood 2003, 102, 2593–2596. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Campese, A.F.; Alesse, E.; Vacca, A.; Felli, M.P.; Balestri, A.; Stoppacciaro, A.; Tiveron, C.; Tatangelo, L.; Giovarelli, M.; et al. Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in NOTCH3 transgenic mice. EMBO J. 2000, 19, 3337–3348. [Google Scholar] [CrossRef] [PubMed]
- Scupoli, M.T.; Donadelli, M.; Cioffi, F.; Rossi, M.; Perbellini, O.; Malpeli, G.; Corbioli, S.; Vinante, F.; Krampera, M.; Palmieri, M.; et al. Bone marrow stromal cells and the upregulation of interleukin-8 production in human T-cell acute lymphoblastic leukemia through the CXCL12/CXCR4 axis and the NF-kappaB and JNK/AP-1 pathways. Haematologica 2008, 93, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Bertaina, A.; Vinti, L.; Strocchio, L.; Gaspari, S.; Caruso, R.; Algeri, M.; Coletti, V.; Gurnari, C.; Romano, M.; Cefalo, M.G.; et al. The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br. J. Haematol. 2017, 176, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.; Segal, R.A. Hedgehog signal transduction: Key players, oncogenic drivers, and cancer therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Mar, B.G.; Amakye, D.; Aifantis, I.; Buonamici, S. The controversial role of the Hedgehog pathway in normal and malignant hematopoiesis. Leukemia 2011, 25, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Hager-Theodorides, A.L.; Outram, S.V.; Ross, S.E.; Varas, A.; Crompton, T. Reduced thymocyte development in sonic hedgehog knockout embryos. J. Immunol. 2004, 172, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Outram, S.V.; Varas, A.; Pepicelli, C.V.; Crompton, T. Hedgehog signaling regulates differentiation from double-negative to double-positive thymocyte. Immunity 2000, 13, 187–197. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Graves, S.; Meng, F.; Mandal, M.; Mashayekhi, M.; Aifantis, I. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat. Immunol. 2006, 7, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Crompton, T.; Outram, S.V.; Hager-Theodorides, A.L. Sonic hedgehog signalling in T-cell development and activation. Nat. Rev. Immunol. 2007, 7, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Drakopoulou, E.; Outram, S.V.; Rowbotham, N.J.; Ross, S.E.; Furmanski, A.L.; Saldana, J.I.; Hager-Theodorides, A.L.; Crompton, T. Non-redundant role for the transcription factor Gli1 at multiple stages of thymocyte development. Cell Cycle 2010, 9, 4144–4152. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Copland, M. Targeting hedgehog in hematologic malignancy. Blood 2012, 119, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Dorsch, M.; Warmuth, M. Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle 2010, 9, 3449–3456. [Google Scholar] [CrossRef] [PubMed]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; El Andaloussi, A.; Nimer, S.D.; Kee, B.L.; Taichman, R.; et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell 2009, 4, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, A.; Demeyer, S.; De Bie, J.; Radaelli, E.; Pauwels, D.; Degryse, S.; Gielen, O.; Vicente, C.; Vandepoel, R.; Geerdens, E.; et al. Hedgehog pathway activation in T-cell acute lymphoblastic leukemia predicts response to SMO and GLI1 inhibitors. Blood 2016, 128, 2642–2654. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, e102–e105. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Smoothening the controversial role of hedgehog in hematopoiesis. Cell Stem Cell 2009, 4, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, X.; Zhang, P.; Fan, Y.; Ma, A.; Pang, T.; Song, Z.; Jin, Y.; Hao, W.; Liu, F.; et al. Inhibition of hedgehog signaling by GANT58 induces apoptosis and shows synergistic antitumor activity with AKT inhibitor in acute T cell leukemia cells. Biochimie 2014, 101, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Integrative genomic analyses on GLI1: Positive regulation of GLI1 by Hedgehog-GLI, TGFβ-Smads, and RTK-PI3K-AKT signals, and negative regulation of GLI1 by NOTCH-CSL-HES/HEY, and GPCR-Gs-PKA signals. Int. J. Oncol. 2009, 35, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Xie, J. Non-canonical HH signaling in cancer-current understanding and future directions. Cancers 2015, 7, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Bueno, O.F.; Brandt, E.B.; Rothenberg, M.E.; Molkentin, J.D. Defective T cell development and function in calcineurin A β-deficient mice. Proc. Natl. Acad. Sci. USA 2002, 99, 9398–9403. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.R.; Winslow, M.M.; Hur, E.M.; Crabtree, G.R. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity 2004, 20, 255–266. [Google Scholar] [CrossRef]
- Avni, O.; Lee, D.; Macian, F.; Szabo, S.J.; Glimcher, L.H.; Rao, A. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat. Immunol. 2002, 3, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Macián, F.; García-Cózar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef]
- Gachet, S.; Ghysdael, J. Calcineurin/NFAT signaling in lymphoid malignancies. Gen. Physiol. Biophys. 2009, 28, F47–F54. [Google Scholar] [PubMed]
- Marafioti, T.; Pozzobon, M.; Hansmann, M.L.; Ventura, R.; Pileri, S.A.; Roberton, H.; Gesk, S.; Gaulard, P.; Barth, T.F.; Du, M.Q.; et al. The NFATc1 transcription factor is widely expressed in white cells and translocates from the cytoplasm to the nucleus in a subset of human lymphomas. Br. J. Haematol. 2005, 128, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Robbs, B.K.; Cruz, A.L.; Werneck, M.B.; Mognol, G.P.; Viola, J.P. Dual roles for NFAT transcription factor genes as oncogenes and tumor suppressors. Mol. Cell. Biol. 2008, 28, 7168–7181. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Alcalde, H.; Berthier, C.; Guillemin, M.C.; dos Santos, N.R.; Janin, A.; Decaudin, D.; de Thé, H.; Ghysdael, J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 2007, 13, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Gachet, S.; Genescà, E.; Passaro, D.; Irigoyen, M.; Alcalde, H.; Clémenson, C.; Poglio, S.; Pflumio, F.; Janin, A.; Lasgi, C.; et al. Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia 2013, 27, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Musson, R.E.; Cobbaert, C.M.; Smit, N.P. Molecular diagnostics of calcineurin-related pathologies. Clin. Chem. 2012, 58, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; De Jesus, C.D.C.; Lasgi, C.; Ghysdael, J. CXCR4 Is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Christopher, M.J.; Liu, F.; Hilton, M.J.; Long, F.; Link, D.C. Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood 2009, 114, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Sahin, A.O.; Buitenhuis, M. Molecular mechanisms underlying adhesion and migration of hematopoietic stem cells. Cell Adhes. Migr. 2012, 6, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.C.C.; Longhini, A.L.; Bigarella, C.L.; Baratti, M.O.; Traina, F.; Favaro, P.; de Melo Campos, P.; Saad, S.T. CXCR7 is highly expressed in acute lymphoblastic leukemia and potentiates CXCR4 response to CXCL12. PLoS ONE 2014, 9, e85926. [Google Scholar] [CrossRef] [PubMed]
- Tosello, V.; Saccomani, V.; Yu, J.Y.; Bordin, F.; Amadori, A.; Piovan, E. Calcineurin complex isolated from T-cell acute lymphoblastic leukemia (T-ALL) cells identifies new signaling pathways including mTOR/AKT/S6K whose inhibition synergize with calcineurin inhibition to promote T-ALL cell death. Oncotarget 2016, 7, 45715–45729. [Google Scholar] [CrossRef] [PubMed]
- Tosello, V.; Bordin, F.; Yu, J.; Agnusdei, V.; Indraccolo, S.; Basso, G.; Amadori, A.; Piovan, E. Calcineurin and GSK-3 inhibition sensitizes T-cell acute lymphoblastic leukemia cells to apoptosis through X-linked inhibitor of apoptosis protein degradation. Leukemia 2016, 30, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, F.; Price, E.R.; Milan, D.; McKeon, F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature 1996, 382, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Beals, C.R.; Sheridan, C.M.; Turck, C.W.; Gardner, P.; Crabtree, G.R. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 1997, 275, 1930–1934. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Kincade, P.W. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell 2009, 4, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signaling in development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Famili, F.; Garcia Perez, L.; Pike-Overzet, K. Aberrant Wnt Signaling in Leukemia. Cancers 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Dose, M.; Kovalovsky, D.; Chang, R.; O’Neil, J.; Look, A.T.; von Boehmer, H.; Khazaie, K.; Gounari, F. β-catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood 2007, 109, 5463–5472. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.H.; Erbilgin, Y.; Firtina, S.; Celkan, T.; Karakas, Z.; Aydogan, G.; Turkkan, E.; Yildirmak, Y.; Timur, C.; Zengin, E.; et al. Deregulated WNT signaling in childhood T-cell acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e192. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Baert, M.R.; Schonewille, T.; Brugman, M.H.; Famili, F.; Salvatori, D.C.; Meijerink, J.P.; Ozbek, U.; Clevers, H.; van Dongen, J.J.; et al. The nuclear effector of Wnt-signaling, Tcf1, functions as a T-cell-specific tumor suppressor for development of lymphomas. PLoS Biol. 2012, 10, e1001430. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhou, X.; Steinke, F.C.; Liu, C.; Chen, S.C.; Zagorodna, O.; Jing, X.; Yokota, Y.; Meyerholz, D.K.; Mullighan, C.G.; et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 2012, 37, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, C.; Reschly, E.J.; Zagort, D.E.; Yashiro-Ohtani, Y.; Beverly, L.J.; Capobianco, A.; Pear, W.S.; Kee, B.L. NOTCH1 co-opts lymphoid enhancer factor 1 for survival of murine T-cell lymphomas. Blood 2007, 110, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Sanda, T.; Ma, W.; Zhang, J.; Grebliunaite, R.; Dahlberg, S.; Neuberg, D.; Protopopov, A.; Winter, S.S.; Larson, R.S.; et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood 2010, 115, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.E.; Lam, S.H.; Hoofd, C.; Belmonte, M.; Wang, X.; Gusscott, S.; Gracias, D.; Weng, A.P. Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood 2015, 125, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.P.; Braun, R.D.; Gwinn, W.M.; Greer, P.K.; Dewhirst, M.W. Hypoxia in the thymus: Role of oxygen tension in thymocyte survival. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1467–H1477. [Google Scholar] [CrossRef] [PubMed]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Hoffmann, K.; Cummings, A.J.; de Klerk, N.H.; Kees, U.R. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia 2006, 20, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell. Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.; Chiarini, F.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Battistelli, M.; Falcieri, E.; Bortul, R.; Melchionda, F.; Iacobucci, I.; et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: Therapeutic implications. Leukemia 2012, 26, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, T.K.; Leclerc, G.M.; Hsieh-Kinser, T.T.; Leclerc, G.J.; Singh, I.; Barredo, J.C. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: Implication for targeted therapy. Mol. Cancer 2007, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ulbrich, J.; Muller, J.; Wustefeld, T.; Aeberhard, L.; Kress, T.R.; Muthalagu, N.; Rycak, L.; Rudalska, R.; Moll, R.; et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 2012, 483, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschenes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Accordi, B.; Galla, L.; Milani, G.; Curtarello, M.; Serafin, V.; Lissandron, V.; Viola, G.; te Kronnie, G.; De Maria, R.; Petricoin, E.F., 3rd; et al. AMPK inhibition enhances apoptosis in MLL-rearranged pediatric B-acute lymphoblastic leukemia cells. Leukemia 2013, 27, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bongiovanni, D.; Saccomani, V.; Piovan, E. Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2017, 18, 1904. https://doi.org/10.3390/ijms18091904
Bongiovanni D, Saccomani V, Piovan E. Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2017; 18(9):1904. https://doi.org/10.3390/ijms18091904
Chicago/Turabian StyleBongiovanni, Deborah, Valentina Saccomani, and Erich Piovan. 2017. "Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 18, no. 9: 1904. https://doi.org/10.3390/ijms18091904
APA StyleBongiovanni, D., Saccomani, V., & Piovan, E. (2017). Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 18(9), 1904. https://doi.org/10.3390/ijms18091904