CFTR-NHERF2-LPA2 Complex in the Airway and Gut Epithelia


Abstract
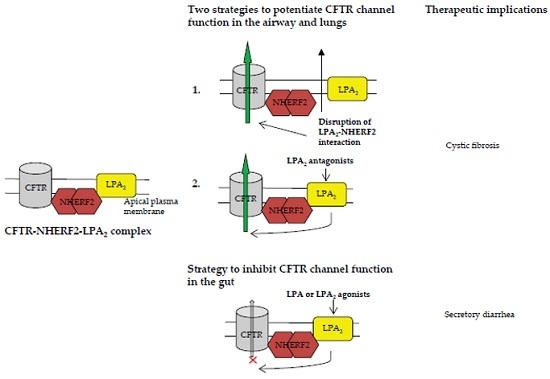
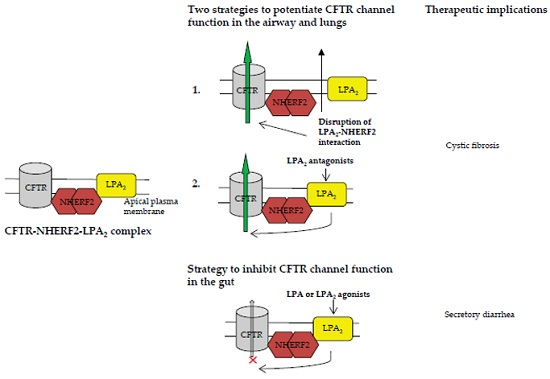
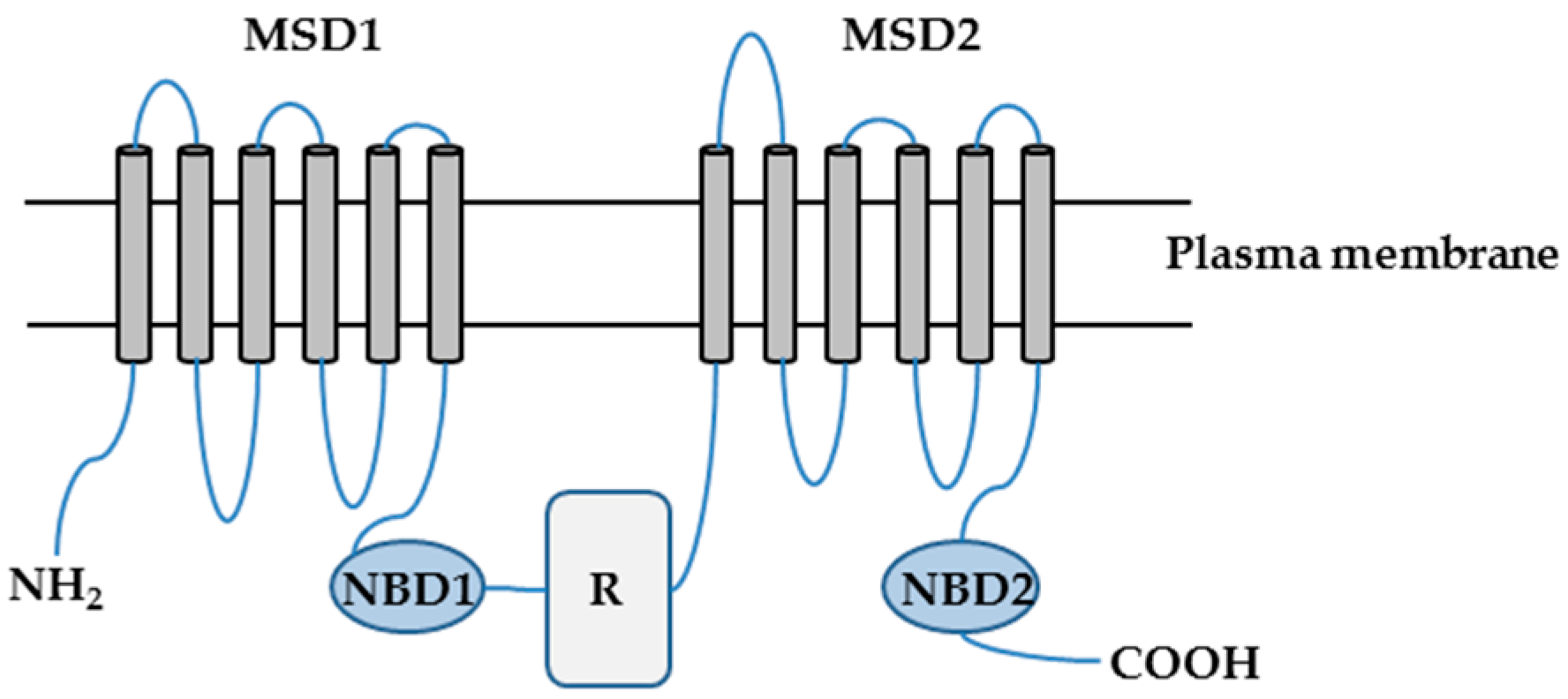
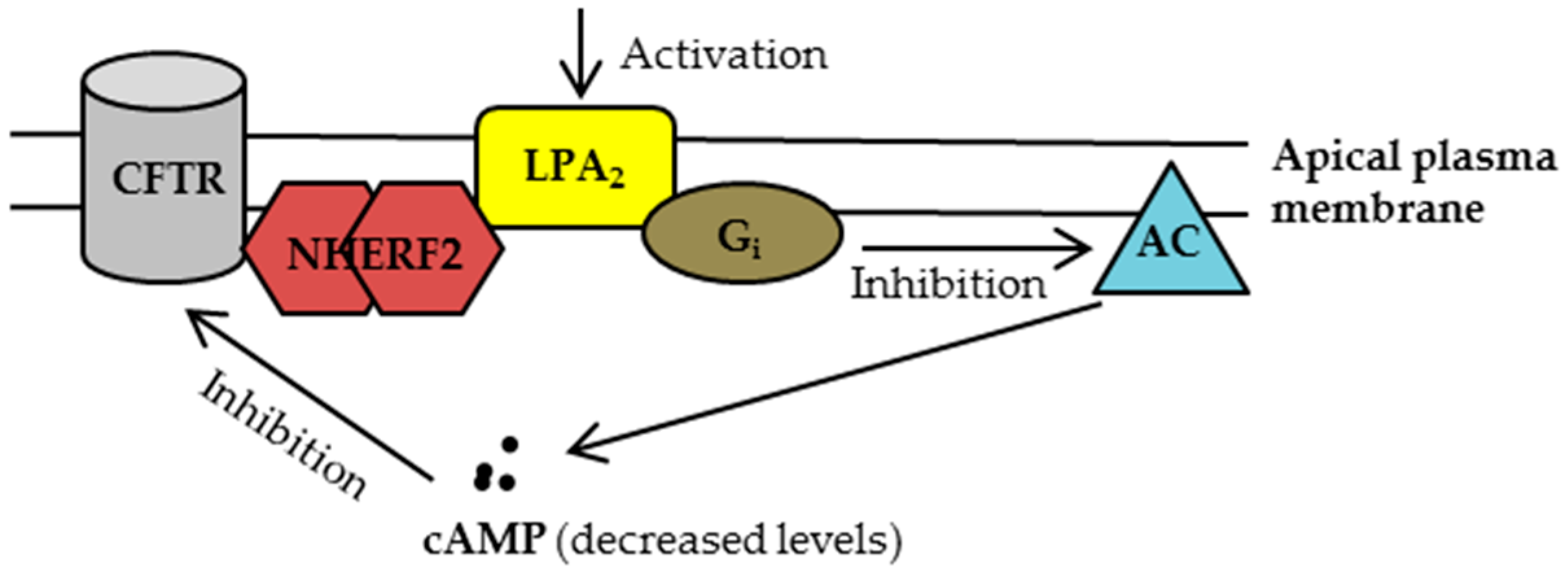
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
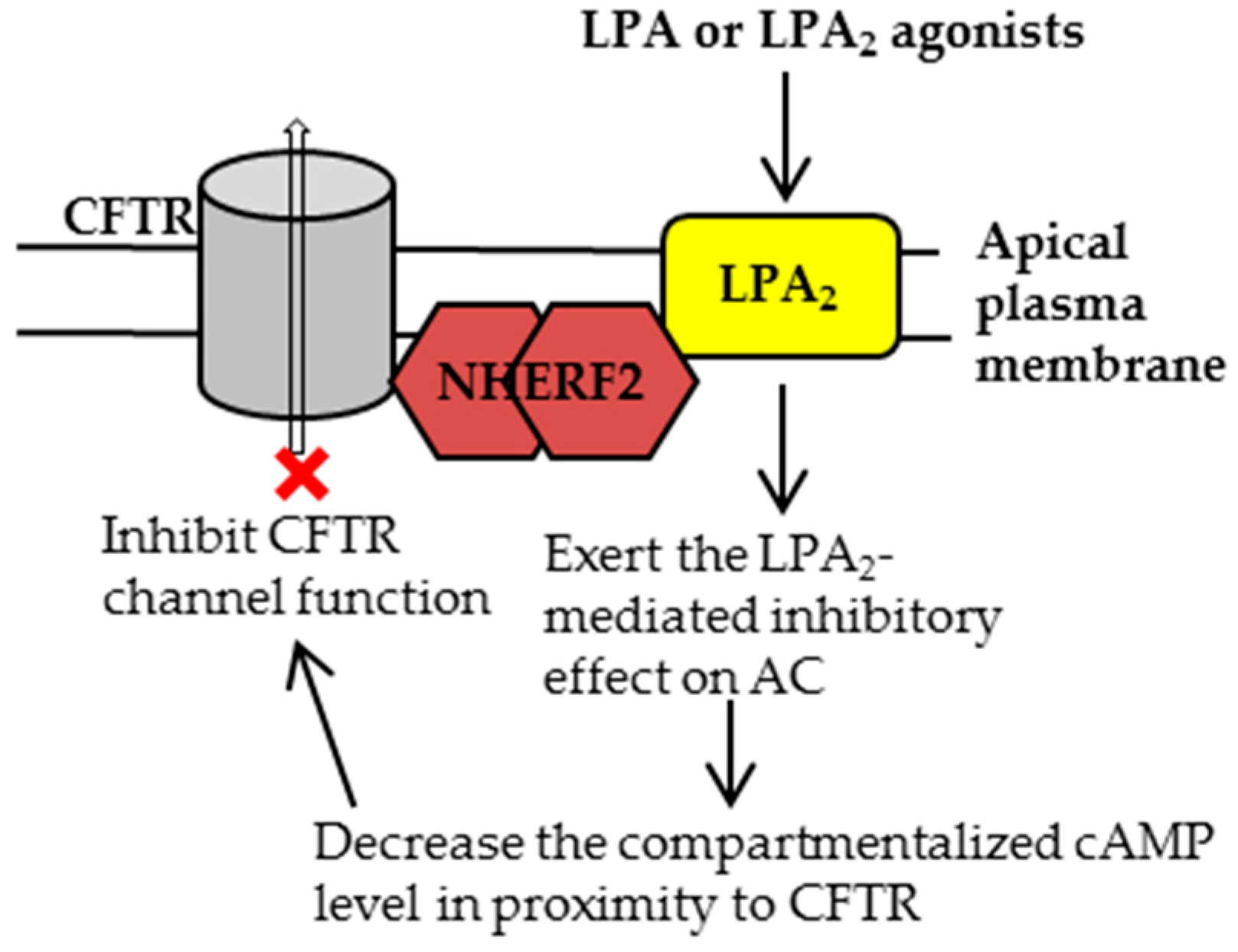
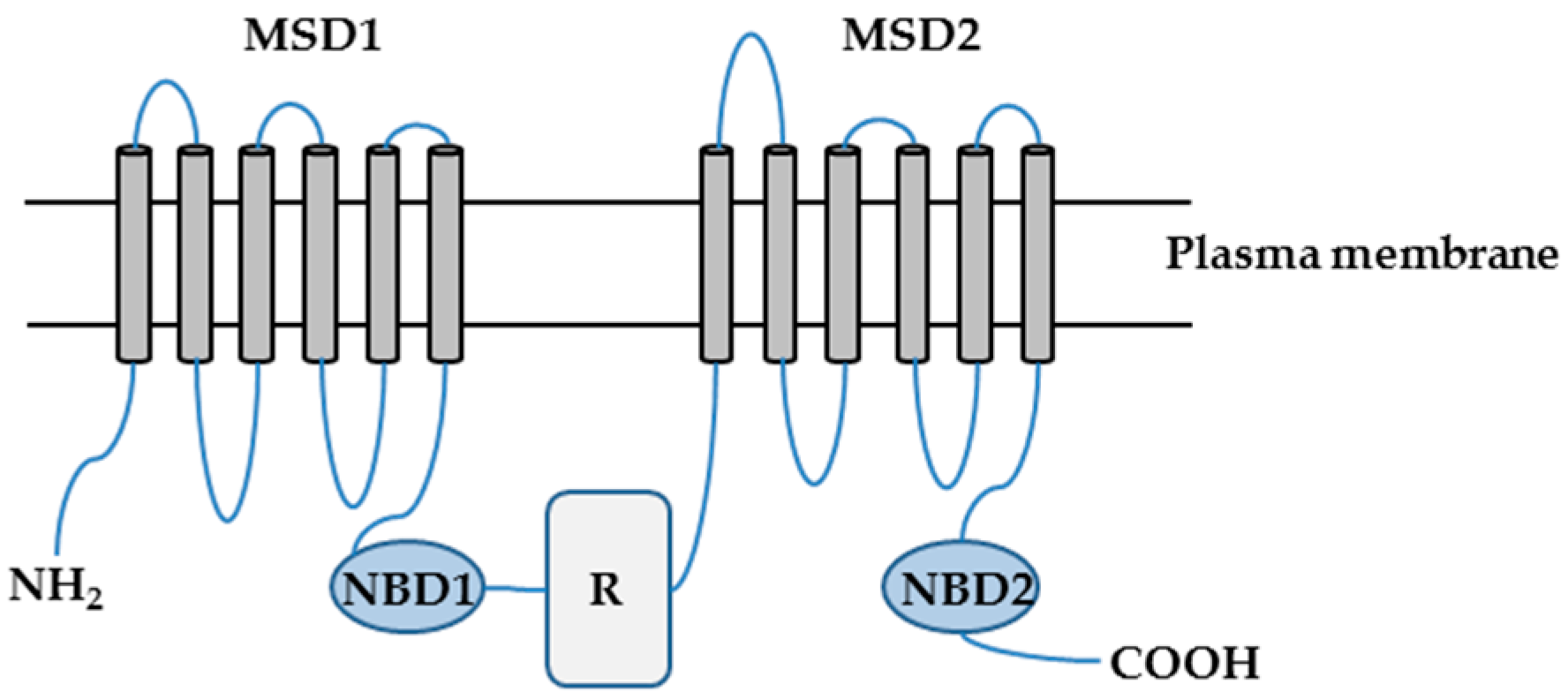
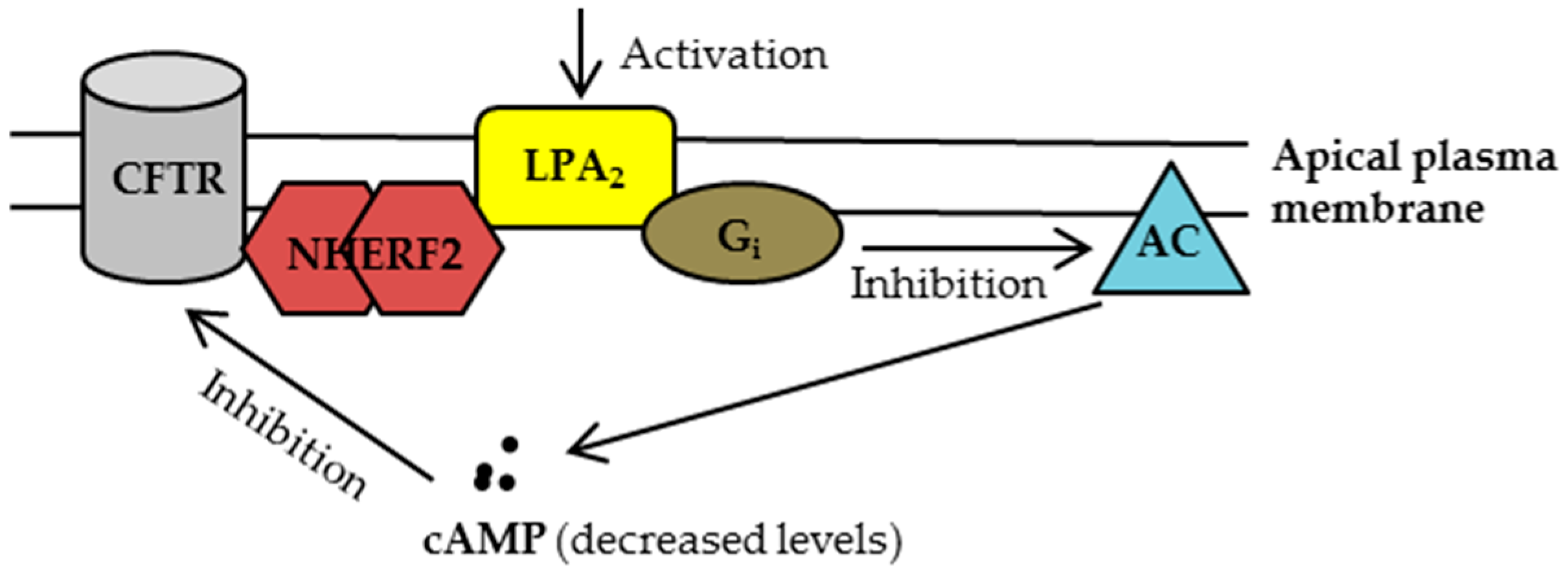
2. CFTR-NHERF2-LPA2 Complex in Airway and Gut Epithelial Cells
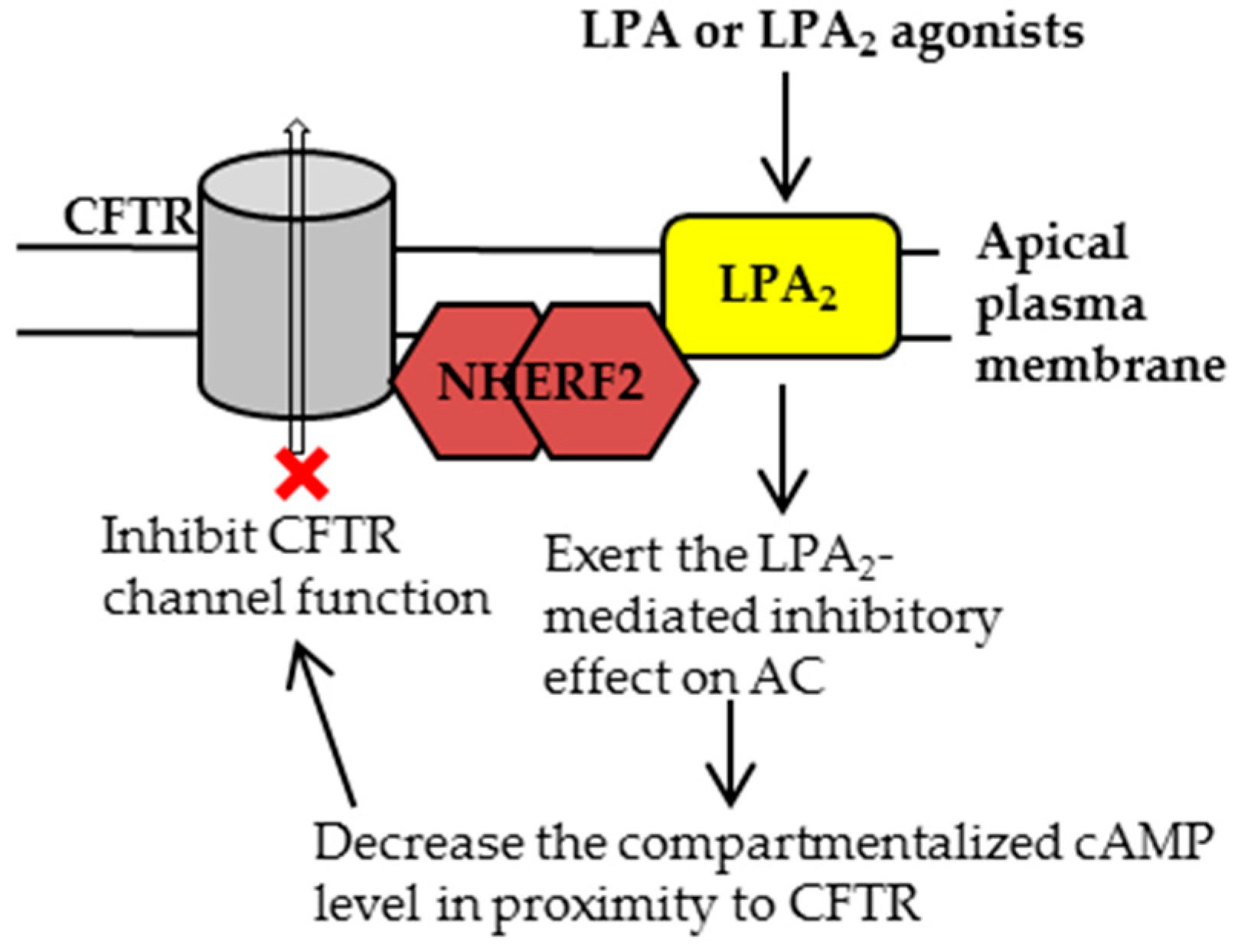
2.1. Characterization of CFTR-NHERF2-LPA2 Complex
2.2. The Involvment of CFTR in Two Major Human Diseases: Cystic Fibrosis (CF) and Secretory Diarrhea
3. Strategies to Target CFTR-NHERF2-LPA2 Complex for Possible Therapeutic Interventions of CF and Secretory Diarrhea
3.1. Strategies to Target CFTR-NHERF2-LPA2 Complex for Possible Therapeutic Interventions of CF
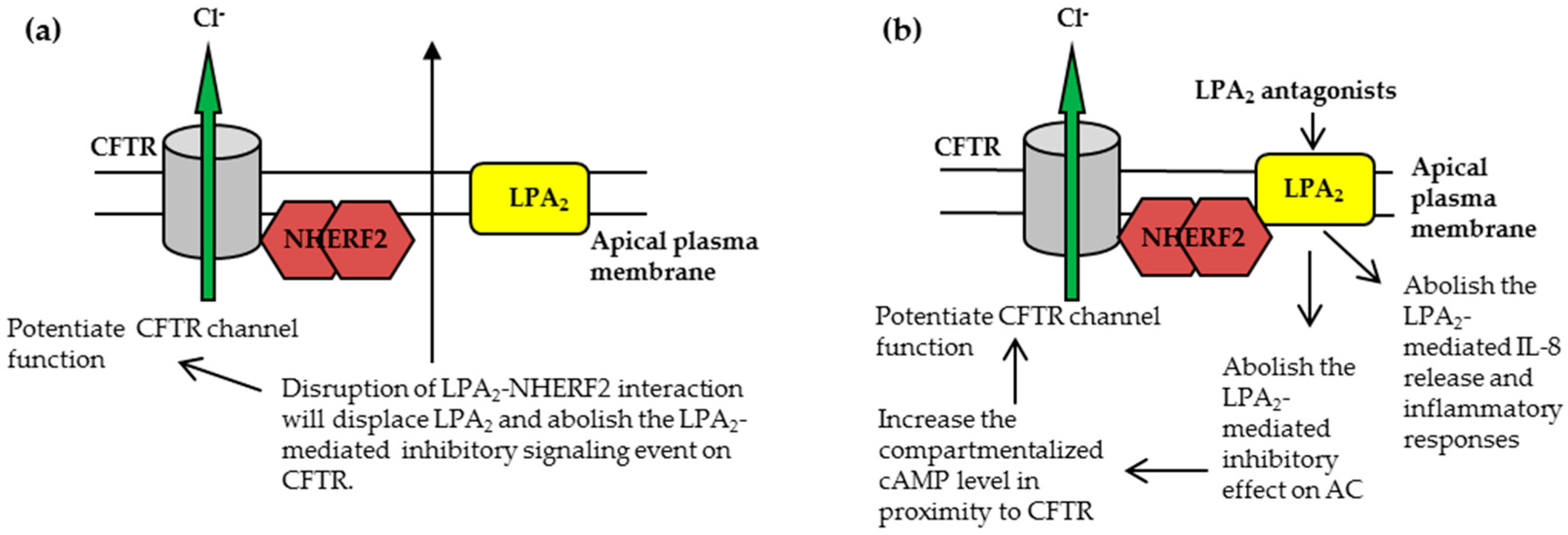
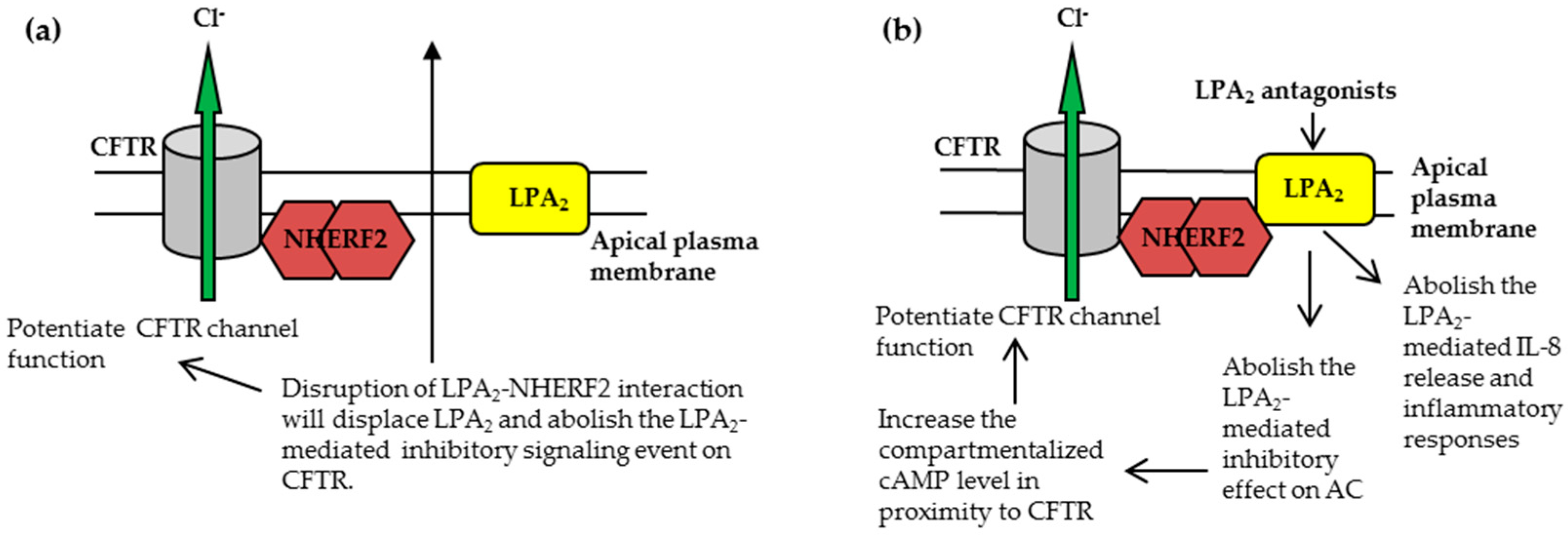
3.1.1. Disruption of NHERF2-LPA2 Interaction to Potentiate CFTR Channel Function
3.1.2. Targeting CFTR-NHERF2-LPA2 Complex to Suppress the Release of Interleukin 8 (IL-8)
3.2. Targeting CFTR–NHERF2–LPA2 Complexes in the Gut Epithelia for Possible Therapeutic Interventions of Secretory Diarrhea
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AC | Adenylyl cyclase |
| β2-AR | β2-Adrenergic receptor |
| ASL | Airway surface liquid |
| BAL | Bronchoalveolar lavage |
| CAL | CFTR-associated ligand |
| CF | Cystic fibrosis |
| CFBEo−- | CF bronchial epithelial cells |
| CFTR | CF transmembrane conductance regulator |
| CTX | Cholera toxin |
| ERM | Ezrin/radixin/moesin |
| FSK | Forskolin |
| IL-8 | Interleukin 8 |
| Isc | Short-circuit currents |
| LPA | Lysophosphatidic acids |
| LPA2 | LPA receptor 2 |
| MRP4 | Multidrug resistance protein 4 |
| MSD | Membrane-spanning domain |
| NBD | Nucleotide binding domain |
| NHE | Na+/H+ exchanger |
| NHERF2 | Na+/H+ exchanger regulatory factor 2 |
| PDZ | Postsynaptic density-95, discs large, zona occludens-1 |
| PLC-β3 | Phospholipase C-β3 |
| R domain | Regulatory domain |
| WT | Wild type |
References
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Phosphodiesterases and compartmentalized cAMP signaling in the heart. Eur. J. Cell Biol. 2006, 85, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Krishnamurthy, P.C.; Penmatsa, H.; Marrs, K.L.; Wang, X.Q.; Zaccolo, M.; Jalink, K.; Li, M.; Nelson, D.J.; Schuetz, J.D.; et al. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 2007, 131, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Gregory, R.J.; Thompson, S.; Souza, D.W.; Paul, S.; Mulligan, R.C.; Smith, A.E.; Welsh, M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991, 253, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Bear, C.E.; Li, C.H.; Kartner, N.; Bridges, R.J.; Jensen, T.J.; Ramjeesingh, M.; Riordan, J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992, 68, 809–818. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.C.; Kirk, K.L. The CFTR ion channel: Gating, regulation, and anion permeation. Cold Spring Harb. Perspect. Med. 2013, 3, a009498. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Naren, A.P. CFTR chloride channel in the apical compartments: Spatiotemporal coupling to its interacting partners. Integr. Biol. 2010, 2, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Mutation Databases. Available online: http://www.genet.sickkids.on.ca/StatisticsPage.html (accessed on 20 July 2017).
- Clancy, J.P.; Jain, M. Personalized medicine in cystic fibrosis: Dawning of a new era. Am. J. Respir. Crit. Care Med. 2012, 186, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Guggino, W.B.; Stanton, B.A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006, 7, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Naren, A.P.; Cobb, B.; Li, C.; Roy, K.; Nelson, D.; Heda, G.D.; Liao, J.; Kirk, K.L.; Sorscher, E.J.; Hanrahan, J.; et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. USA 2003, 100, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dandridge, K.S.; Di, A.; Marrs, K.L.; Harris, E.L.; Roy, K.; Jackson, J.S.; Makarova, N.V.; Fujiwara, Y.; Farrar, P.L.; et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhoea through CFTR-dependent protein interactions. J. Exp. Med. 2005, 202, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Shenolikar, S.; Weinman, E.J. NHERF: Targeting and trafficking membrane proteins. Am. J. Physiol. Ren. Physiol. 2001, 280, F389–F395. [Google Scholar]
- Li, C.; Naren, A.P. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 2005, 108, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Seidler, U.; Singh, A.K.; Cinar, A.; Chen, M.; Hillesheim, J.; Hogema, B.; Riederer, B. The role of the NHERF family of PDZ scaffolding proteins in the regulation of salt and water transport. Ann. N. Y. Acad. Sci. 2009, 1165, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.P.; Alexander, S.P.; Sharman, J.L.; Pawson, A.J.; Benson, H.E.; Monaghan, A.E.; Liew, W.C.; Mpamhanga, C.P.; Bonner, T.I.; Neubig, R.R.; et al. International union of basic and clinical pharmacology. LXXXVIII. G protein-coupled receptor list: Recommendations for new pairings with cognate ligands. Pharmacol. Rev. 2013, 65, 967–986. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Bleu, T.; Hallmark, O.G.; Goetzl, E.J. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J. Biol. Chem. 1998, 273, 7906–7910. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.T.; Lai, Y.J. Regulation of the LPA2 receptor signaling through the carboxyl-terminal tail-mediated protein-protein interactions. Biochim. Biophys. Acta 2008, 1781, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Jo, N.W.; Choi, J.W.; Kim, H.S.; Seo, S.W.; Kang, K.O.; Hwang, J.I.; Heo, K.; Kim, S.H.; Kim, Y.H.; et al. NHERF2 specifically interacts with LPA2 receptor and defines the specificity and efficiency of receptor-mediated phospholipase C-β3 activation. Mol. Cell Biol. 2004, 24, 5069–5079. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.C.; Sun, H.; Wang, D.; Rusovici, R.; Castleberry, A.; Hall, R.A.; Shim, H. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am. J. Physiol. Cell Physiol. 2005, 289, C2–C11. [Google Scholar] [CrossRef] [PubMed]
- E, S.; Lai, Y.J.; Tsukahara, R.; Chen, C.S.; Fujiwara, Y.; Yue, J.; Yu, J.H.; Guo, H.; Kihara, A.; Tigyi, G.; et al. Lysophosphatidic acid 2 receptor-mediated supramolecular complex formation regulates its antiapoptotic effect. J. Biol. Chem. 2009, 284, 14558–14571. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Riederer, B.; Krabbenhöft, A.; Rausch, B.; Bonhagen, J.; Lehmann, U.; de Jonge, H.R.; Donowitz, M.; Yun, C.; Weinman, E.; et al. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J. Clin. Investig. 2009, 119, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Zhang, Y.H.; Strokes, D.C.; Naren, A.P. Lumacaftor/ivacaftor combination for cystic fibrosis patients homozygous for Phe508del-CFTR. Drugs Today 2016, 52, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Moon, C.; Zhang, X.; Naren, A.P.; Zhang, W. The rescued F508del-CFTR forms an inhibitory complex with NHERF2 and LPA2 at the cell surface. Pediatr. Pulmonol. 2015, 50, 200. [Google Scholar]
- Zhang, W.; Fujii, N.; Naren, A.P. Recent advances and new perspectives in targeting CFTR for therapy of cystic fibrosis and enterotoxin-induced secretory diarrheas. Future Med. Chem. 2012, 4, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- About Cystic Fibrosis. Cystic Fibrosis Foundation [US]. Available online: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/ (accessed on 20 July 2017).
- Davies, J.C.; Alton, E.W.; Bush, A. Cystic fibrosis. BMJ 2007, 335, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Banks-Schlegel, S.; Accurso, F.J.; Boucher, R.C.; Cutting, G.R.; Engelhardt, J.F.; Guggino, W.B.; Karp, C.L.; Knowles, M.R.; Kolls, J.K.; et al. Future directions in early cystic fibrosis lung disease research: An NHLBI workshop report. Am. J. Respir. Crit. Care Med. 2012, 185, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Scholte, B.J.; Veltman, M.; Horati, H.; Koval, S.; Vreeken, R.; Harms, A.; Tiddens, H.; Janssens, H.; Triouvanziam, R.; Stick, S.M. Bioactive lipids in bronchoalveolar lavage of CF infants correlate with lung CF score, revealing therapeutic targets. Pediatr. Pulmonol. 2016, 51, 268. [Google Scholar]
- Gabriel, S.E.; Brigman, K.N.; Koller, B.H.; Boucher, R.C.; Stutts, M.J. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1994, 266, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Donowitz, M.; Verkman, A.S. Secretory diarrhoea: Mechanisms and emerging therapies. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Lukacs, G.L.; Galietta, L.J. CFTR chloride channel drug discovery—Inhibitors as antidiarrheals and activators for therapy of cystic fibrosis. Curr. Pharm. Des. 2006, 12, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Ko, E.A.; Tradtrantip, L.; Donowitz, M.; Verkman, A.S. Discovery and development of antisecretory drugs for treating diarrheal diseases. Clin. Gastroenterol. Hepatol. 2014, 12, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Drug Development Pipeline. Cystic Fibrosis Foundation [US]. Available online: https://www.cff.org/trials/pipeline (accessed on 20 July 2017).
- Ivacaftor (Kalydeco®). Cystic Fibrosis Foundation [US]. Available online: https://www.cff.org/Trials/Pipeline/details/49/Ivacaftor-Kalydeco (accessed on 20 July 2017).
- Lumacaftor + ivacaftor (Orkambi®). Cystic Fibrosis Foundation [US]. Available online: https://www.cff.org/Trials/Pipeline/details/86/Lumacaftor-ivacaftor-Orkambi- (accessed on 20 July 2017).
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Penmatsa, H.; Ren, A.; Punchihewa, C.; Lemoff, A.; Yan, B.; Fujii, N.; Naren, A.P. Functional regulation of cystic fibrosis transmembrane conductance regulator-containing macromolecular complexes: A small-molecule inhibitor approach. Biochem. J. 2011, 435, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Wolde, M.; Fellows, A.; Cheng, J.; Kivenson, A.; Coutermarsh, B.; Talebian, L.; Karlson, K.; Piserchio, A.; Mierke, D.F.; Stanton, B.A.; et al. Targeting CAL as a negative regulator of DeltaF508-CFTR cell-surface expression: An RNA interference and structure-based mutagenetic approach. J. Biol. Chem. 2007, 282, 8099–8109. [Google Scholar] [CrossRef] [PubMed]
- Cushing, P.R.; Vouilleme, L.; Pellegrini, M.; Boisguerin, P.; Madden, D.R. A stabilizing influence: CAL PDZ inhibition extends the half-life of ΔF508-CFTR. Angew. Chem. Int. Ed. Engl. 2010, 49, 9907–9911. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; Cushing, P.R.; Boisguerin, P.; Madden, D.R.; Donald, B.R. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput. Biol. 2012, 8, e1002477. [Google Scholar] [CrossRef] [PubMed]
- Amacher, J.F.; Zhao, R.; Spaller, M.R.; Madden, D.R. Chemically modified peptide scaffolds target the CFTR-associated ligand PDZ domain. PLoS ONE 2014, 9, e103650. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Xu, X.; Amacher, J.F.; Madden, D.R.; Cormet-Boyaka, E.; Pei, D. Intracellular delivery of peptidyl ligands by reversible cyclization: Discovery of a PDZ Domain inhibitor that rescues CFTR activity. Angew. Chem. Int. Ed. Engl. 2015, 54, 5874–5878. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hou, Y.; Sun, F.; Yang, Z.; Li, C. Dysregulated chemokine signaling in cystic fibrosis lung disease: A potential therapeutic target. Curr. Drug Targets 2016, 17, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kong, S.; Tigyi, G.J.; Zhang, W. A protein complex of Phe508del-CFTR, NHERF2, and LPA2 regulates IL-8 secretion from airway epithelial cells. Am. J. Respir. Crit. Care Med. 2017, 195, A1283. [Google Scholar]
- Lin, S.; Yeruva, S.; He, P.; Singh, A.K.; Zhang, H.; Chen, M.; Lamprecht, G.; de Jonge, H.R.; Tse, M.; Donowitz, M.; et al. Lysophosphatidic acid stimulates the intestinal brush border Na(+)/H(+) exchanger 3 and fluid absorption via LPA(5) and NHERF2. Gastroenterology 2010, 138, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.N.; Fells, J.I.; Gupte, R.; Lee, S.C.; Liu, J.; Nusser, N.; Lim, K.G.; Ray, R.M.; Lin, F.T.; Parrill, A.L.; et al. Virtual screening for LPA2-specific agonists identifies a nonlipid compound with antiapoptotic actions. Mol. Pharmacol. 2012, 82, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ren, A.; Naren, A.P. Identification of novel antidiarrheal agents by targeting LPA2 receptor. Gastroenterology 2012, 142, S-693. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Zhang, Z.; Zhang, Y.; Naren, A.P. CFTR-NHERF2-LPA2 Complex in the Airway and Gut Epithelia. Int. J. Mol. Sci. 2017, 18, 1896. https://doi.org/10.3390/ijms18091896
Zhang W, Zhang Z, Zhang Y, Naren AP. CFTR-NHERF2-LPA2 Complex in the Airway and Gut Epithelia. International Journal of Molecular Sciences. 2017; 18(9):1896. https://doi.org/10.3390/ijms18091896
Chicago/Turabian StyleZhang, Weiqiang, Zhihong Zhang, Yanhui Zhang, and Anjaparavanda P. Naren. 2017. "CFTR-NHERF2-LPA2 Complex in the Airway and Gut Epithelia" International Journal of Molecular Sciences 18, no. 9: 1896. https://doi.org/10.3390/ijms18091896
APA StyleZhang, W., Zhang, Z., Zhang, Y., & Naren, A. P. (2017). CFTR-NHERF2-LPA2 Complex in the Airway and Gut Epithelia. International Journal of Molecular Sciences, 18(9), 1896. https://doi.org/10.3390/ijms18091896