Measurement of Telomere Length in Colorectal Cancers for Improved Molecular Diagnosis

 ,
,
Abstract

1. Introduction
2. Results
2.1. Description of the Tumors Analyzed
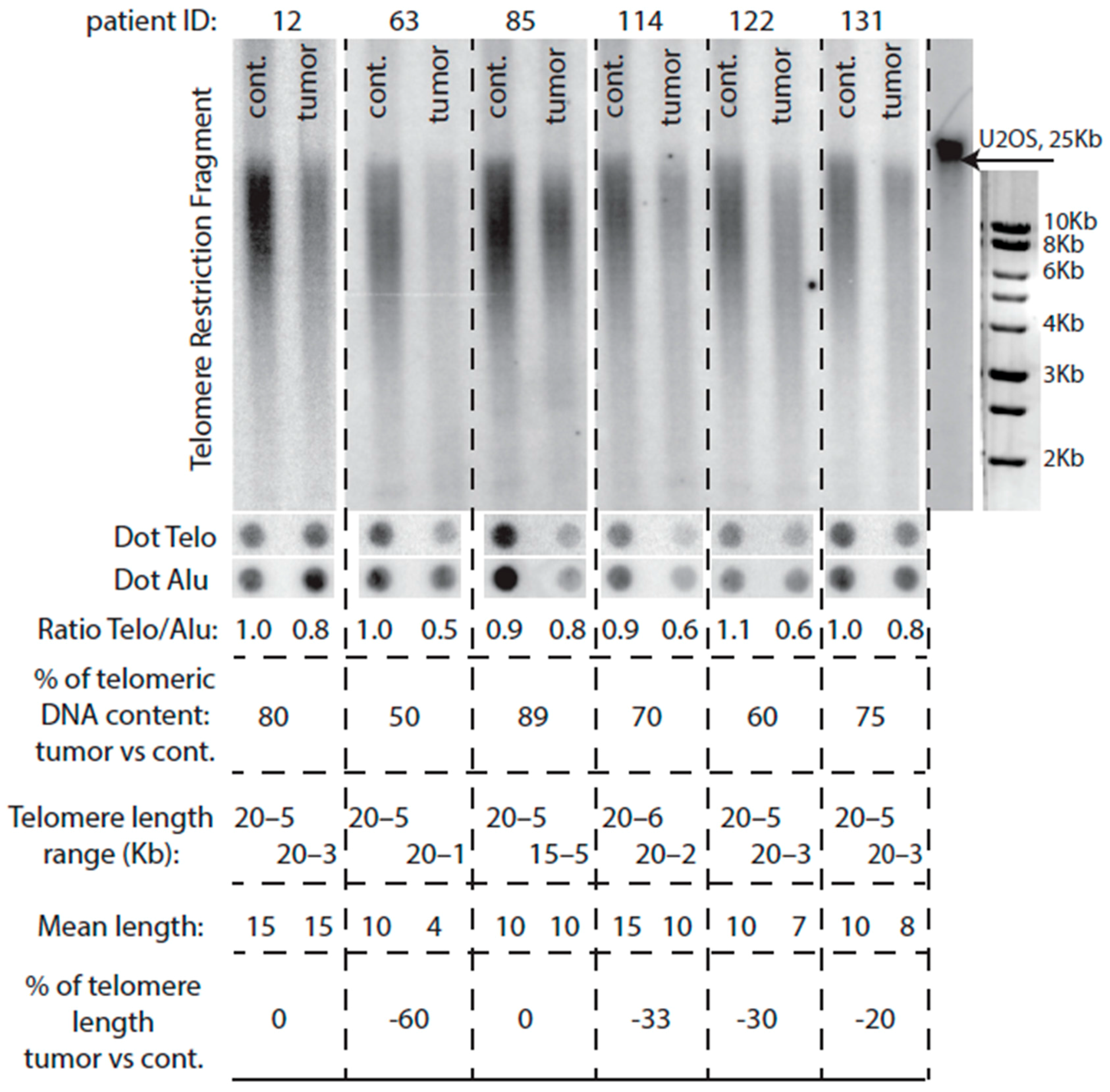
2.2. Telomeres Are Globally Much Shorter in Tumors Than in Healthy Tissue
2.3. A Class of Colorectal Cancers Does Not Exhibit Telomere Erosion
2.4. Comparison of Telomere Restriction Fragment and Telo/Alu Methods to Measure Telomere Length
2.5. Mutated KRAS Preferentially Associates with Short Telomeres
2.6. Telomere Lengthening in the Tumor Preferentially Occurs in Patients with Short Telomeres
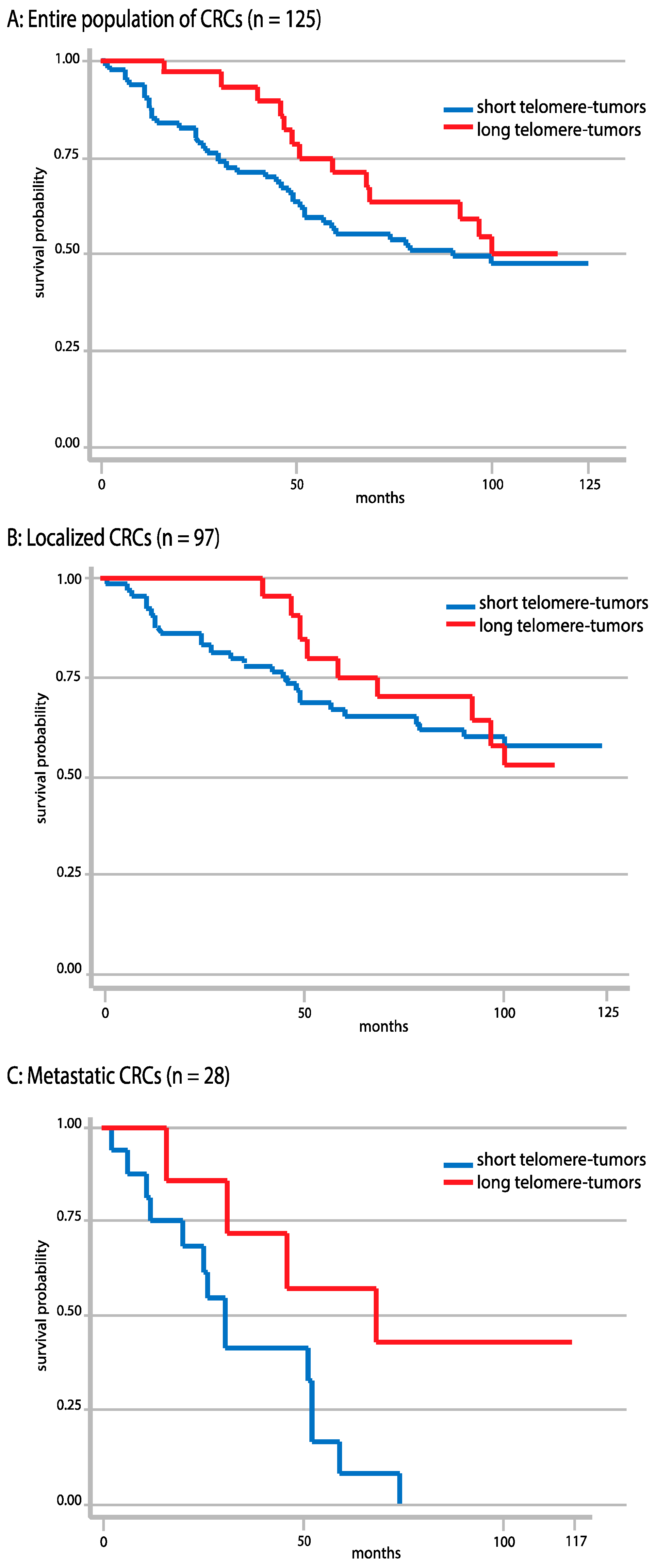
2.7. Correlations between Telomere Length and Overall Survival
2.8. Correlations between Telomere Length and Tumor Localization
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Collection
4.2. Genomic DNA Extraction, Mutational Analysis and MLH1 Promoter Methylation
4.3. Telomeric DNA Quantification by Dot Blot for Telomere Length Measurement
4.4. Telomere Length Measurement by Telomere Restriction Fragment (TRF) Southern Blot Analysis
4.5. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sideris, M.; Papagrigoriadis, S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer Res. 2014, 34, 2061–2068. [Google Scholar] [PubMed]
- Fearon, E.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Gyparaki, M.T.; Basdra, E.K.; Papavassiliou, A.G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. J. Mol. Med. 2013, 91, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Church, D.; Midgley, R.; Kerr, D. Biomarkers in early-stage colorectal cancer: Ready for prime time? Dig. Dis. 2012, 30, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.; Blackburn, E.H. The telomere syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Feuerhahn, S.; Chen, L.; Luke, B.; Porro, A. No DDRama at chromosome ends: TRF2 takes centre stage. Trends Biochem. Sci. 2015, 40, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.A.; Tomita, K. Fundamental mechanisms of telomerase action in yeasts and mammals: Understanding telomeres and telomerase in cancer cells. Open Biol. 2017, 7, 160338. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Renault, V.M.; Jamet, K.; Gilson, E. Transcriptional outcome of telomere signalling. Nat. Rev. Genet. 2014, 15, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; DePinho, R.A. Telomeres and telomerase in cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, J.; Cech, T.R. Finding the end: Recruitment of telomerase to telomeres. Nat. Rev. Mol. Cell. Biol. 2013, 14, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.T. Telomerase-independent paths to immortality in predictable cancer sub-types. J. Cancer 2012, 3, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Hastie, N.D.; Dempster, M.; Dunlop, M.G.; Thompson, A.M.; Green, D.K.; Allshire, R.C. Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, M.; Drullinsky, P.; Guillem, J.; Moore, M.A. Telomerase and telomere length in the development and progression of premalignant lesions to colorectal cancer. Clin. Cancer Res. 1997, 3, 1931–1941. [Google Scholar] [PubMed]
- Gertler, R.; Rosenberg, R.; Stricker, D.; Friederichs, J.; Hoos, A.; Werner, M.; Ulm, K.; Holzmann, B.; Nekarda, H.; Siewert, J.-R. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J. Clin. Oncol. 2004, 22, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Broccoli, D.; Godley, L.A.; Donehower, L.A.; Varmus, H.E.; de Lange, T. Telomerase activation in mouse mammary tumours: Lack of telomere shortening and evidence for regulation of telomerase with RNA cell proliferation. Mol. Cell. Biol. 1996, 16, 3765–3772. [Google Scholar] [CrossRef] [PubMed]
- Aubert, G.; Hills, M.; Lansdorp, P.M. Telomere length measurement—Caveats and a critical assessment of the available technologies and tools. Mut. Res. 2012, 730, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.C.Y.; Moshir, S.; Garini, Y.; Chuang, A.Y.; Young, I.T.; Vermolen, B.; van den Doel, R.; Mougey, V.; Perrin, M.; Braun, M.; et al. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol. 2004, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.Y.; Chuang, A.Y.C.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytom. Part A 2005, 67, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Vallente, R.; Klewes, L.; Righolt, C.; Wark, L.; Kongruttanachok, N.; Knecht, H.; Mai, S. Nuclear remodeling as a mechanism for genomic instability in cancer. Adv. Cancer Res. 2011, 112, 77–126. [Google Scholar] [PubMed]
- Katayama, S.; Shiota, G.; Oshimura, M.; Kawasaki, H. Clinical usefulness of telomerase activity and telomere length in the preoperative diagnosis of gastric and colorectal cancer. J. Cancer Res. Clin. Oncol. 1999, 125, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Kinouchi, Y.; Hiwatashi, N.; Chida, M.; Nagashima, F.; Takahashi, S.; Negoro, K.; Shimosegawa, T.; Toyota, T. Telomere shortening and the clinicopathologic characteristics of human colorectal carcinomas. Cancer 1999, 86, 1431–1436. [Google Scholar] [CrossRef]
- Nakamura, K.; Furugori, E.; Esaki, Y.; Arai, T.; Sawabe, M.; Okayasu, I.; Fujiwara, M.; Kammori, M.; Mafune, K.; Kato, M.; et al. Correlation of telomere lengths in normal and cancers tissue in the large bowel. Cancer Lett. 2000, 158, 179–184. [Google Scholar] [CrossRef]
- Plentz, R.R.; Wiemann, S.U.; Flemming, P.; Meier, P.N.; Kubicka, S.; Kreipe, H.; Manns, M.P.; Rudolph, K.L. Telomere shortening of epithelial cells characterises the adenoma-carcinoma transition of human colorectal cancer. Gut 2003, 52, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aranda, C.; de Juan, C.; Diaz-Lopez, A.; Sanchez-Pernaute, A.; Torres, A.; Diaz-Rubio, E.; Balibrea, J.L.; Benito, M.; Iniesta, P. Correlations of telomere length, telomerase activity and telomeric-repeat binding factor 1 expression in colorectal carcinoma. Cancer 2006, 106, 541–551. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Risques, R.A.; Mandelson, M.T.; Chen, L.; Brentnall, T.A.; Bronner, M.P.; Macmillan, M.P.; Feng, Z.; Siebert, J.R.; Potter, J.D.; et al. Telomere length in the colon declines with age: A relation to colorectal cancer? Cancer Epidemiol. Biomark. Prev. 2006, 15, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, C.M.; Jang, S.J.; Nuciforo, P.; Lantuejoul, S.; Brambilla, E.; Mounier, N.; Olaussen, K.A.; André, F.; Morat, L.; Sabatier, L.; et al. Telomere shortening is correlated with the DNA damage response and telomeric protein down-regulation in colorectal preneoplastic lesions. Ann. Oncol. 2008, 19, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Bertorelle, R.; Serra, L.; Terrin, L.; Candiotto, C.; Pucciarelli, S.; Del Bianco, P.; Nitti, D.; De Rossi, A. Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br. J. Cancer 2010, 102, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Valls, C.; Piñol, C.; Reñé, J.M.; Buenestado, J.; Viñas, J. Telomere length is a prognostic factor for overall survival in colorectal cancer. Colorectal Dis. 2011, 13, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Jones, R.E.; Heppel, N.H.; Williams, G.T.; Sampson, J.R.; Baird, D.M. Extensive telomere erosion in the initiation of colorectal adenomas and its association with chromosomal instability. J. Natl. Cancer Inst. 2013, 105, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, R.; Rampazzo, E.; Pucciarelli, S.; Nitti, D.; De Rossi, A. Telomeres, telomerase and colorectal cancer. World J. Gastroenterol. 2014, 20, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Kinouchi, Y.; Hiwatashi, N.; Nagashima, F.; Chida, M.; Takahashi, S.; Negoro, K.; Shimosegawa, T.; Toyota, T. Relationship between microsatellite instability and telomere shortening in colorectal cancer. Dis. Colon Rectum 2000, 43, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, Y.J.; Kim, H.J.; Kim, S.K.; Lee, J.H. Telomere length changes in colorectal cancers and polyps. J. Korean Med. Sci. 2002, 17, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Piñol-Felis, C.; Fernández-Marcelo, T.; Viñas-Salas, J.; Valls-Bautista, C. Telomeres and telomerase in the clinical management of colorectal cancer. Clin. Transl. Oncol. 2017, 19, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Nera, B.; Huang, H.; Lai, T.; Xu, L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat. Commun. 2015, 6, 10132. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Johnson, N.A.; Haliotis, T.; Lichtensztejn, D.; Mai, S. Disruption of direct 3D telomere-TRF2 interaction through two molecularly disparate mechanisms is a hallmark of primary Hodgkin and Reed-Sternberg cells. Lab. Investig. 2017, 97, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Kyo, S.; Kanaya, T.; Wang, Z.; Yatabe, N.; Tanaka, M.; Nakamura, M.; Ohmichi, M.; Gotoh, N.; Murakami, S.; et al. Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene 2002, 21, 4071–4079. [Google Scholar] [CrossRef] [PubMed]
- Heeg, S.; Hirt, N.; Queisser, A.; Schmieg, H.; Thaler, M.; Kunert, H.; Quante, M.; Goessel, G.; von Werder, A.; Harder, J.; et al. EGFR overexpression induces activation of telomerase via PI3K/AKT-mediated phosphorylation and transcriptional regulation through Hif1-α in a cellular model of oral-esophageal carcinogenesis. Cancer Sci. 2011, 102, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Augustine, T.A.; Baig, M.; Sood, A.; Budagov, T.; Atzmon, G.; Mariadason, J.M.; Aparo, S.; Maitra, R.; Goel, S. Telomere length is a novel predictive biomarker of sensitivity to anti-EGFR therapy in metastatic colorectal cancer. Br. J. Cancer 2015, 112, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Blackburn, E.H. Human cancer cells harbor t-stumps, a distinct class of extremely short telomeres. Mol. Cell 2007, 28, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Fortin, D.; Tsanaclis, A.M.; Garini, Y.; Katzir, N.; Wienburg, Y.; Yan, J.; Klewes, L.; Klonisch, T.; Drouin, R.; et al. Three-dimensional nuclear telomere architecture is associated with differential time to progression and overall survival in glioblastoma patients. Neoplasia 2010, 12, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Kongruttanachok, N.; Sawan, B.; Brossard, J.; Prévost, S.; Turcotte, E.; Lichtensztejn, Z.; Lichtensztejn, D.; Mai, S. Three-dimensional telomere signatures of Hodgkin- and Reed-Sternberg cells at diagnosis identify Patients with poor response to conventional chemotherapy. Transl. Oncol. 2012, 5, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D.; Srivatsan, A.; Nene, R.; Martinez, S.L.; Clotfelter, S.P.; Bell, S.N.; Somach, S.B.; de Souza, J.E.; Fonseca, A.F.; de Souza, S.J.; et al. A genetic network that suppresses genome rearrangements in Saccharomyces cerevisiae and contains defects in cancers. Nat. Commun. 2016, 7, 11256. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Truninger, K.; Menigatti, M.; Luz, J.; Russell, A.; Haider, R.; Gebbers, J.O.; Bannwart, F.; Yurtsever, H.; Neuweiler, J.; Riehle, H.M.; et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 2005, 128, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Fogli, A.; Demattei, M.V.; Corset, L.; Vaurs-Barrière, C.; Chautard, E.; Biau, J.; Kémény, J.L.; Godfraind, C.; Pereira, B.; Khalil, T.; et al. Detection of the alternative lengthening of telomeres pathway in malignant gliomas for improved molecular diagnosis. J. Neurooncol. 2017. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Right and Transverse Colon | Left and Sigmoid Colon | Rectum | Total |
|---|---|---|---|---|
| Stage I | 2 (1.6%) | 6 (4.8%) | 3 (2.4%) | 11 (8.8%) |
| Stage II | 15 (12.0%) | 28 (22.4%) | 1 (0.8%) | 44 (35.2%) |
| Stage III | 18 (14.4%) | 23 (18.4%) | 1 (0.8%) | 42 (33.6%) |
| Stage IV | 15 (12.0%) | 13 (10.4%) | 0 (0%) | 28 (22.4%) |
| Total | 50 (40.0%) | 70 (56.0%) | 5 (4.0%) | 125 (100%) |
| Gene Mutations | Stage I | Stage II | Stage III | Stage IV | Total |
|---|---|---|---|---|---|
| KRAS mutated | 2 | 12 | 15 | 13 | 42 (33.6%) |
| BRAF mutated | 0 | 8 | 8 | 1 | 17 (13.6%) |
| PIK3CA mutated | 0 | 4 | 3 | 6 | 13 (10.4%) |
| MSI a | 0 | 9 | 7 | 3 | 19 (15.2%) |
| Parameters | Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|---|
| Number of cases | 11 | 44 | 42 | 28 |
| Telomere length in tumor tissue | 6.8 Kb | 7.6 Kb | 7.2 Kb | 6.8 Kb |
| Telomere length in adjacent healthy tissue | 10.0 Kb | 9.7 Kb | 10.3 Kb | 8.6 Kb |
| Significance | p = 0.01 | p < 0.0001 | p < 0.0001 | p < 0.001 |
| Tumor and Control Classes | 91 Control (Average, n = 91) 10.0 ± 2.8 | 91 Tumor Group b (Average, n = 91) 6.5 ± 1.7 | 34 Control (Average, n = 34) 8.8 ± 2.4 | 34 Tumor Group c (Average, n = 34) 9.2 ± 2.2 | 125 Control (Average, n = 125) 9.7 ± 2.7 | Tumor (Average, n = 125) 7.2 ± 2.2 |
|---|---|---|---|---|---|---|
| Control (n = 91) | <0.0001 | 0.021 | 0.391 | |||
| Tumor (n = 91) | <0.0001 | <0.0001 | 0.008 | |||
| Control (n = 34) | 0.021 | 0.50 | 0.075 | |||
| Tumor (n = 34) | <0.0001 | 0.50 | 3.947 | |||
| Control (n = 125) | <0.0001 | |||||
| Tumor (n = 125) | <0.0001 |
| Parameters | Right and Transverse Colon | Left and Sigmoid Colon | Rectum |
|---|---|---|---|
| Number of cases | 50 | 70 | 5 |
| Telomere length in tumor tissue | 7.0 Kb | 7.3 Kb | 7.1 Kb |
| Telomere length in adjacent healthy tissue | 9.9 Kb | 9.5 Kb | 10.5 Kb |
| Significance | p < 0.0001 | p < 0.0001 | p = 0.07 b |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balc’h, E.L.; Grandin, N.; Demattei, M.-V.; Guyétant, S.; Tallet, A.; Pagès, J.-C.; Ouaissi, M.; Lecomte, T.; Charbonneau, M. Measurement of Telomere Length in Colorectal Cancers for Improved Molecular Diagnosis. Int. J. Mol. Sci. 2017, 18, 1871. https://doi.org/10.3390/ijms18091871
Balc’h EL, Grandin N, Demattei M-V, Guyétant S, Tallet A, Pagès J-C, Ouaissi M, Lecomte T, Charbonneau M. Measurement of Telomere Length in Colorectal Cancers for Improved Molecular Diagnosis. International Journal of Molecular Sciences. 2017; 18(9):1871. https://doi.org/10.3390/ijms18091871
Chicago/Turabian StyleBalc’h, Eric Le, Nathalie Grandin, Marie-Véronique Demattei, Serge Guyétant, Anne Tallet, Jean-Christophe Pagès, Mehdi Ouaissi, Thierry Lecomte, and Michel Charbonneau. 2017. "Measurement of Telomere Length in Colorectal Cancers for Improved Molecular Diagnosis" International Journal of Molecular Sciences 18, no. 9: 1871. https://doi.org/10.3390/ijms18091871
APA StyleBalc’h, E. L., Grandin, N., Demattei, M.-V., Guyétant, S., Tallet, A., Pagès, J.-C., Ouaissi, M., Lecomte, T., & Charbonneau, M. (2017). Measurement of Telomere Length in Colorectal Cancers for Improved Molecular Diagnosis. International Journal of Molecular Sciences, 18(9), 1871. https://doi.org/10.3390/ijms18091871