IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types



Abstract
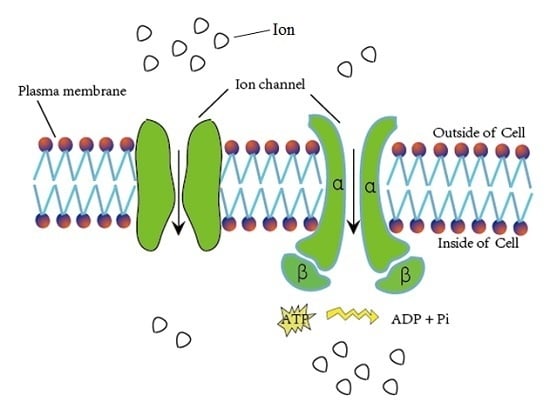
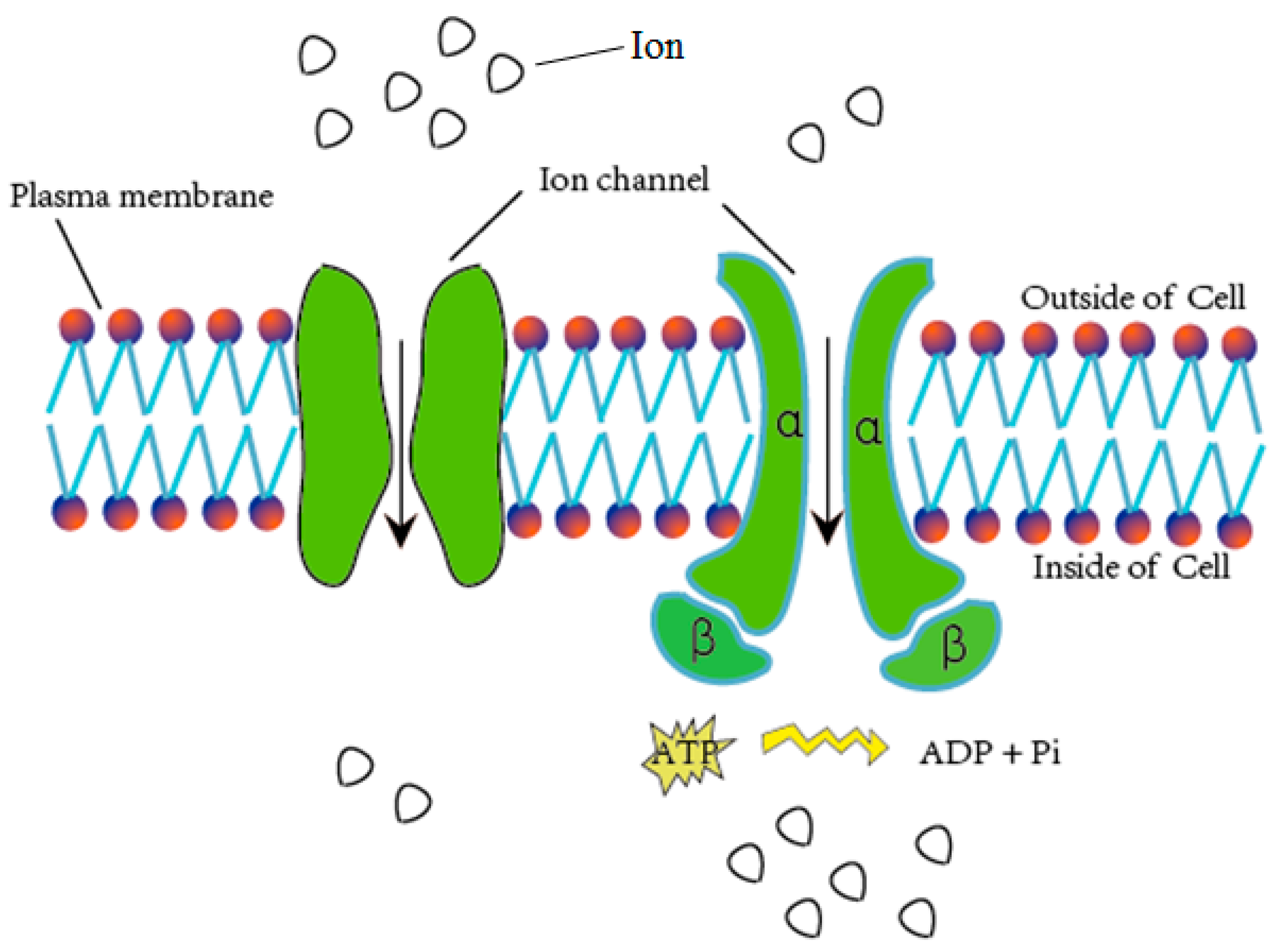
:
1. Introduction
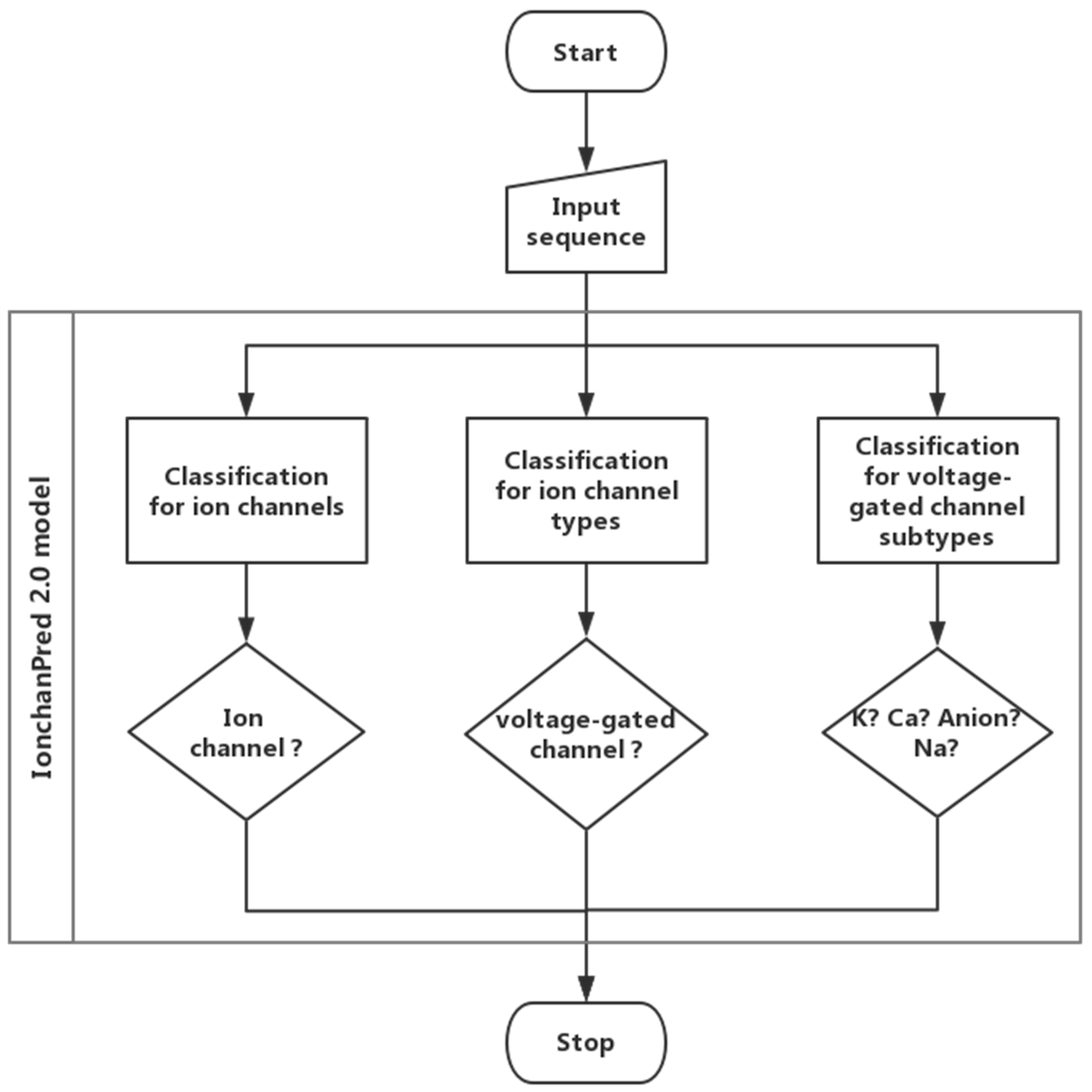
2. Results and Discussion
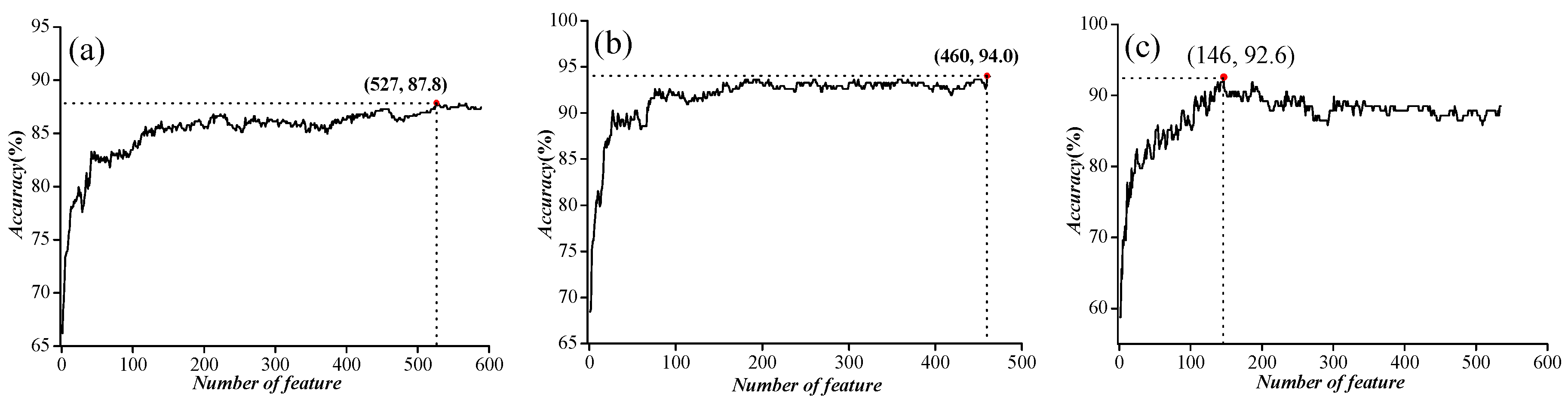
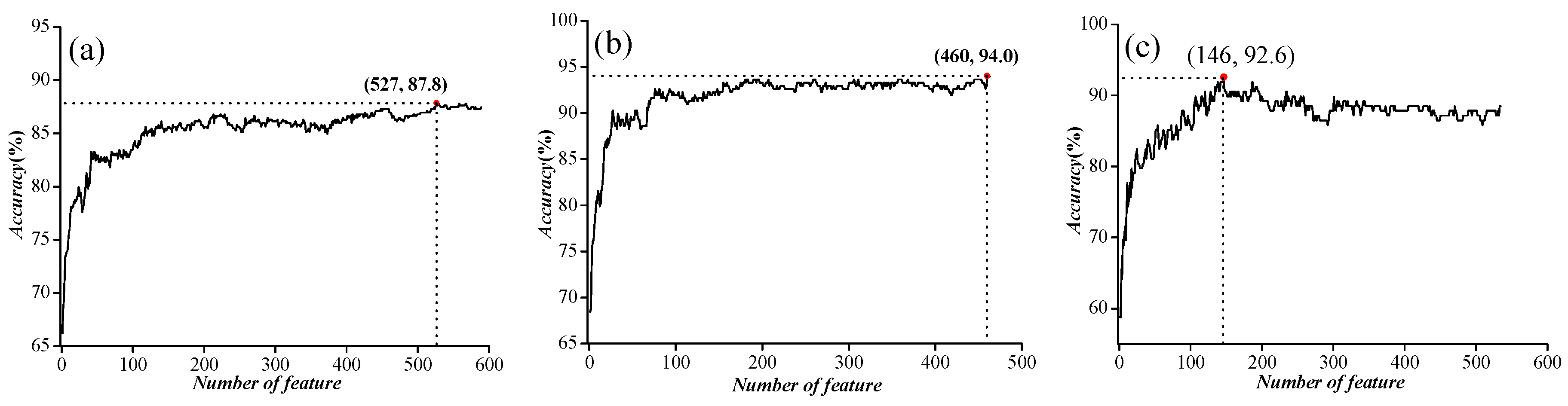
2.1. Parameter Optimization
2.2. Model Establishment
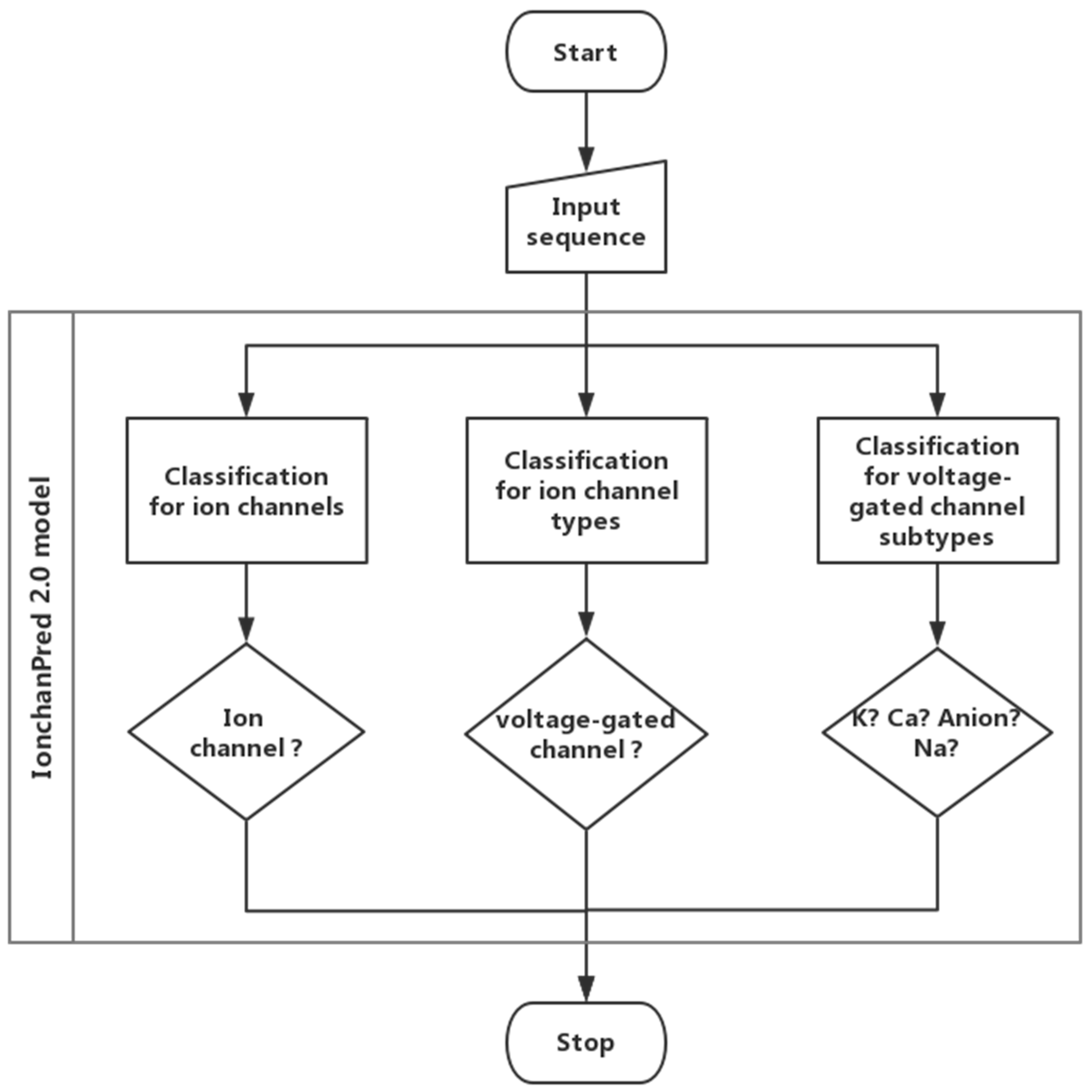
3. Materials and Methods
3.1. Benchmark Databases
3.2. Feature Extraction of Samples
3.3. Feature Selection
3.4. Support Vector Machine
3.5. Performance Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wulff, H.; Christophersen, P. Recent developments in ion channel pharmacology. Channels 2015, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Gabashvili, I.S.; Sokolowski, B.H.; Morton, C.C.; Giersch, A.B. Ion channel gene expression in the inner ear. J. Assoc. Res. Otolaryngol. 2007, 8, 305–328. [Google Scholar] [CrossRef] [PubMed]
- Ger, M.F.; Rendon, G.; Tilson, J.L.; Jakobsson, E. Domain-based identification and analysis of glutamate receptor ion channels and their relatives in prokaryotes. PLoS ONE 2010, 5, e12827. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, L.M.; Su, T.; He, N.; Lin, Z.J.; Wang, J.; Shi, Y.W.; Yi, Y.H.; Liao, W.P. Ion Channel Genes and Epilepsy: Functional Alteration, Pathogenic Potential, and Mechanism of Epilepsy. Neurosci. Bull. 2017, 33, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Knutson, K.; Alcaino, C.; Linden, D.R.; Gibbons, S.J.; Kashyap, P.; Grover, M.; Oeckler, R.; Gottlieb, P.A.; Li, H.J.; et al. Mechanosensitive ion channel Piezo2 is important for enterochromaffin cell response to mechanical forces. J. Phys. 2017, 595, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Huang, S.; Meynard, D.; Chaine, C.; Michel, R.; Roelfsema, M.R.G.; Guiderdoni, E.; Sentenac, H.; Very, A.A. A Dual Role for the OsK5.2 Ion Channel in Stomatal Movements and K+ Loading into Xylem Sap. Plant Phys. 2017, 174, 2409–2418. [Google Scholar] [CrossRef] [PubMed]
- Zubcevic, L.; Herzik, M.A., Jr.; Chung, B.C.; Liu, Z.; Lander, G.C.; Lee, S.Y. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 2016, 23, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Linsdell, P. Metal bridges to probe membrane ion channel structure and function. Biomol. Concepts 2015, 6, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Prindle, A.; Liu, J.; Asally, M.; Ly, S.; Garcia-Ojalvo, J.; Suel, G.M. Ion channels enable electrical communication in bacterial communities. Nature 2015, 527, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Li, M.L.; Tan, F.Y.; Lu, M.C.; Wang, K.L.; Guo, Y.Z.; Wen, Z.N.; Jiang, L. Local sequence information-based support vector machine to classify voltage-gated potassium channels. Acta Biochim. Biophys. Sin. 2006, 38, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Zack, J.; Singh, B.; Raghava, G.P. VGIchan: Prediction and classification of voltage-gated ion channels. Genom. Proteom. Bioinform. 2006, 4, 253–258. [Google Scholar] [CrossRef]
- Lin, H.; Ding, H. Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J. Theor. Biol. 2011, 269, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, H. Identification of voltage-gated potassium channel subfamilies from sequence information using support vector machine. Comput. Biol. Med. 2012, 42, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Deng, E.Z.; Chen, W.; Lin, H. Identifying the subfamilies of voltage-gated potassium channels using feature selection technique. Int. J. Mol. Sci. 2014, 15, 12940–12951. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Srivastava, R. An efficient approach for the prediction of ion channels and their subfamilies. Comput. Biol. Chem. 2015, 58, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cui, W.; Sheng, Y.; Ruan, J.; Kurgan, L. PSIONplus: Accurate Sequence-Based Predictor of Ion Channels and Their Types. PLoS ONE 2016, 11, e0152964. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, W.X.; He, J.; Liu, X.H.; Ding, H.; Chen, W. Predicting cancerlectins by the optimal g-gap dipeptides. Sci. Rep. 2015, 5, 16964. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, C. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Donizelli, M.; Djite, M.A.; le Novere, N. LGICdb: A manually curated sequence database after the genomes. Nucleic Acids Res. 2006, 34, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics 2016, 107, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-AI: Identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 2017, 8, 4208–4217. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Mao, Y.; Hu, L.; Wu, Y.; Ji, Z. miRClassify: An advanced web server for miRNA family classification and annotation. Comput. Biol. Med. 2014, 45, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, H. Prediction of midbody, centrosome and kinetochore proteins based on gene ontology information. Biochem. Biophys. Res. Commun. 2010, 401, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Lin, H. Prediction of ketoacyl synthase family using reduced amino acid alphabets. J. Ind. Microbiol. Biotechnol. 2012, 39, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Chou, K.C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using Chou′s pseudo amino acid composition with feature selection technique. Mol. BioSyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.W.; Lai, H.Y.; Tang, H.; Chen, W.; Lin, H. Prediction of phosphothreonine sites in human proteins by fusing different features. Sci. Rep. 2016, 6, 34817. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, J.; Wang, X. Protein remote homology detection by combining Chou′s distance-pair pseudo amino acid composition and principal component analysis. Mol. Genet. Genom. 2015, 290, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Feng, P.M.; Chen, W.; Lin, H. Identification of bacteriophage virion proteins by the ANOVA feature selection and analysis. Mol. BioSyst. 2014, 10, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Ju, Y.; Zou, Q. Prediction of G-protein-coupled receptors with SVM-Prot features and random forest. Scientifica 2016, 2016, 8309253. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ju, Y.; Zou, Q. Protein Folds Prediction with Hierarchical Structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.-C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Chen, X.X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169–28175. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines. Acm Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Chou, K.C.; Zhang, C.T. Prediction Of Protein Structural Classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, H.; Wang, X.; Chou, K.-C. Pse-Analysis a python package for DNA, RNA and protein peptide sequence analysis based on pseudo components and kernel methods. Oncotarget 2017, 8, 13338–13343. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liang, Z.Y.; Tang, H.; Chen, W. Identifying σ70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Tang, H.; Li, W.C.; Lin, H.; Chen, W.; Chou, K.C. iOri-Human: Identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 2016, 7, 69783–69793. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | λ | ω | OA (%) |
|---|---|---|---|
| IC vs. NIC | 21 | 0.20 | 87.5 |
| VGIC vs. LGIC | 7 | 0.30 | 93.9 |
| four types of VGIC | 9 | 0.15 | 89.1 |
| Datasets | Our Model | Previous Model [13] | |||||
|---|---|---|---|---|---|---|---|
| Sn | OA | AA | Sn | OA | AA | ||
| IC vs. NIC | IC | 80.2 | 87.8 | 87.8 | 85.9 | 86.6 | 86.6 |
| NIC | 95.3 | 87.3 | |||||
| VGIC vs. LGIC | VGIC | 94.7 | 94.0 | 94.0 | 94.6 | 92.6 | 92.7 |
| LGIC | 93.2 | 90.7 | |||||
| Types of VGIC | K+ | 97.5 | 92.6 | 87.7 | 92.6 | 87.8 | 83.7 |
| Ca2+ | 89.7 | 82.8 | |||||
| Na+ | 75.0 | 75.0 | |||||
| An− | 88.5 | 84.6 | |||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.-W.; Su, Z.-D.; Yang, W.; Lin, H.; Chen, W.; Tang, H. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. Int. J. Mol. Sci. 2017, 18, 1838. https://doi.org/10.3390/ijms18091838
Zhao Y-W, Su Z-D, Yang W, Lin H, Chen W, Tang H. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. International Journal of Molecular Sciences. 2017; 18(9):1838. https://doi.org/10.3390/ijms18091838
Chicago/Turabian StyleZhao, Ya-Wei, Zhen-Dong Su, Wuritu Yang, Hao Lin, Wei Chen, and Hua Tang. 2017. "IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types" International Journal of Molecular Sciences 18, no. 9: 1838. https://doi.org/10.3390/ijms18091838
APA StyleZhao, Y.-W., Su, Z.-D., Yang, W., Lin, H., Chen, W., & Tang, H. (2017). IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. International Journal of Molecular Sciences, 18(9), 1838. https://doi.org/10.3390/ijms18091838