Abstract
Although its prevalence is declining, gastric cancer remains a significant public health issue. The bacterium Helicobacter pylori is known to colonize the human stomach and induce chronic atrophic gastritis, intestinal metaplasia, and gastric cancer. Results using a Mongolian gerbil model revealed that H. pylori infection increased the incidence of carcinogen-induced adenocarcinoma, whereas curative treatment of H. pylori significantly lowered cancer incidence. Furthermore, some epidemiological studies have shown that eradication of H. pylori reduces the development of metachronous cancer in humans. However, other reports have warned that human cases of atrophic metaplastic gastritis are already at risk for gastric cancer development, even after eradication of these bacteria. In this article, we discuss the effectiveness of H. pylori eradication and the morphological changes that occur in gastric dysplasia/cancer lesions. We further assess the control of gastric cancer using various chemopreventive agents.
1. Introduction
Although its prevalence is declining because of improved sanitation and antibiotic use, gastric cancer remains one of the leading causes of cancer-related deaths worldwide [1]. Thus, the prevention of gastric cancer is a substantial issue for cancer control programs. Various epidemiological, biological, and pathological characteristics of Helicobacter pylori-associated lesions have been evaluated in humans and animal models, especially in mice and Mongolian gerbils [2]. Recent health insurance program-supported efforts to eradicate H. pylori have been used for the prevention of gastric carcinogenesis, not only for patients with metachronous gastric cancer but also for those with chronic active gastric inflammation [3]. However, difficulties in demarcating cancerous lesions both endoscopically and histopathologically reveal that gastric cancer is still a major and challenging health issue [4]. In this article, we describe the challenges that exist in gastric cancer prevention strategies and compare human and animal lesions, with special attention to current pathological and biological findings.
2. Role of H. pylori Infection and Modifying Factors in Chronic Active Gastritis, Intestinal Metaplasia, and Gastric Carcinogenesis
2.1. Epidemiological Aspects
H. pylori was discovered in patients with chronic gastritis as Gram-negative, flagellated, microaerophilic bacilli, and was initially considered a species within the genus Campylobacter [5,6]. Strong clinical and epidemiological evidence has suggested that H. pylori is significantly correlated with active chronic gastritis, peptic ulcers, atrophic gastritis, intestinal metaplasia, and malignant lymphoma or cancer [7,8,9,10,11,12,13,14,15,16,17]. In a prospective study, Uemura et al. [18] confirmed that gastric cancer developed in only 2.9% of an H. pylori-infected symptomatic group compared to 0% in an uninfected group. The World Health Organization/International Agency for Research on Cancer evaluated H. pylori as a “definite biological carcinogen” based on epidemiological findings in 1994, requiring evidence of induction of gastric cancer in experimental animals [19].
2.2. Geographical Difference of H. pylori
H. pylori itself has several virulence factors. Among them, CagA has been reported to play an important role in gastric carcinogenesis. CagA is injected into gastric surface epithelial cells through the bacterial type IV secretion system, then is tyrosine-phosphorylated with Src and Abl [20] at variable EPIYA (Glu-Pro-Ile-Tyr-Ala) motif repeats region. These characteristic amino acids show structural diversity between East-Asian and Western countries [21]. H. pylori, found in the former, possess EPIYA-A, B, and D motifs, and the latter EPIYA-A, B, and C counterparts. Tyrosine phosphorylated EPIYA-C or D segments acquire the potential to interact with an oncoprotein, SHP2 phosphatase. East-Asian Cag A binds more strongly to SHP2 and induces morphological change, called the hummingbird phenotype, than does Western CagA [22]. This genetic variety may contribute to geographical difference for gastric carcinogenesis.
2.3. Animal Models
2.3.1. Mouse Models
Several animal models have been used to mimic human gastric cancer caused by H. pylori infection, but most have yielded unsatisfactory results [23,24]. Human clinical samples infected with H. pylori were inoculated into nude and euthymic mice to determine the causative factor of chronic active gastritis [25,26,27]. In addition to H. pylori, another Helicobacter species, H. felis, is present in the cat stomach. This organism can be inoculated into germ-free mice to induce acute and chronic inflammation [28]. Lee et al. [29] established an H. pylori strain, the Sydney strain (SS1), which has been used frequently in mice. Recently, Draper et al. [30] compared the inter- and intra-genomic variability of two reference strains of H. pylori, PMSS1 (pre-mouse SS1) [31], a parental strain isolated from a human gastric ulcer patient, and SS1, a PMSS1 descendant being passed through mice for better mice colonization. The CagA copy number was noted as 1 and 4 in SS1 and PMSS1, correlating with the protein expression level. The most substantial alteration in the PMSS1 strain was an insertion in cagY, a virB10 orthologue in the cag pathogenicity island (cagPAI) gene, which encodes a protein required for a type IV secretion system [32]. PMSS1 is now a valuable tool to study CagPAI, which requires the type IV secretion system [33].
Mice are resistant to a chemical carcinogen, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), which has been used to successfully induce gastric cancer in rats [34]. To study carcinogenesis in mice, investigators found that N-methyl-N-nitrosourea (MNU), another alkylating agent, could cause adenocarcinomas in the glandular stomachs of BALB/c [35] and C3H [36] mice. A gastric carcinogenesis model using this carcinogen was utilized in combination with H. pylori infection in later experiments to show that β-catenin activation may play an important role in distal carcinogenesis, especially in H. pylori-infected K19-C2mE transgenic mice compared to the non-treated K19-C2mE mice harboring predominantly proximal tumors. [37].
Genetic manipulation is used more successfully in mouse models than in other animal models [38]. To mimic H. pylori-induced inflammation, a transgenic mouse whose gastric epithelial cells simultaneously express both cyclooxygenase-2 (COX-2) and microsomal prostaglandin E synthase-1 under the control of the keratin 19 promoter, the K19-C2mE transgenic mouse, was established [39]. The combination treatment of K19-C2mE mice with MNU and H. pylori (Sydney strain, SS1) induced adenocarcinomas not only in the pyloric mucosa but also in the fundic glands, thus serving as a good model of proximal gastric cancer [37]. In addition to these inflammatory factors, the expression of Wnt1 was found to cause gastric lesions to become more dysplastic [40]. An interleukin-1β (IL-1β) polymorphism was reported to be involved in gastric carcinogenesis [41]. The overexpression of IL-1β, under the control of a parietal cell-specific H/K-ATPase promoter, caused transgenic mice to spontaneously develop chronic gastritis, intestinal metaplasia, and high-grade dysplasia/carcinoma with an accompanying H. felis infection compared to the control mice [42].
Ins-Gas mice harbor a chimeric insulin-gastrin (INS-GAS) transgene, in which the expression of the human gastrin gene is driven from the rat insulin I promoter [43]. Ins-Gas mice exhibited gastric metaplasia, dysplasia, carcinoma in situ, and gastric cancer with vascular invasion. H. felis infection accelerated cancer development with occasional submucosal invasion [44].
2.3.2. Mongolian Gerbil Model
To better mimic severe human H. pylori infection and inflammation, a Mongolian gerbil (Meriones unguiculatus) model was successfully established. Infected animals develop chronic active gastritis, peptic ulcers, and intestinal metaplasia, resembling human lesions [45]. Twenty-five weeks after inoculation with H. pylori, the gastric glands become hyperplastic (heterotopic proliferative glands), characterized by severe chronic active gastritis with occasional penetrance through the muscularis mucosae. Fifty weeks after infection, intestinal metaplastic cells, including Alcian blue-stained goblet cells and/or absorptive cells that possess a striated brush border, appear among the gastric epithelial cells. After 75 weeks, the gastric cell phenotype gradually decreases, whereas the intestinal cell phenotype increases, accompanied by the formation of a more complete intestinal metaplasia, sometimes containing Paneth cells, by 100 weeks [46]. Heterotopic proliferative glands often appear resembling differentiated or mucinous adenocarcinomas because of their unusual structural abnormalities [47,48].
As in mouse models, chemical gastric carcinogenesis induced by MNU and MNNG can be modeled using Mongolian gerbils [49]. H. pylori infection accelerates both MNU- and MNNG-induced gastric carcinogenesis in a wide variety of cell types, including differentiated or signet-ring cell carcinomas [50,51,52].
2.4. Pathological Changes Caused by H. pylori Infection
In humans, chronic atrophic gastritis and intestinal metaplasia progress simultaneously. For the classification of intestinal metaplasia, we have proposed two categories [53]. The first is gastric-and-intestinal-mixed, which consists of atrophying gastric cells, including mucin core protein (MUC) 5AC-positive foveolar cells and/or MUC6-expressing pyloric cells, and intestinal cells, including MUC2-expressing/Alcian blue-stained goblet cells and CD10/villin-positive absorptive cells. These cells are putative markers of the progression of both chronic atrophic gastritis and intestinal metaplasia. The second category of intestinal metaplasia is the solely intestinal type, which is the extreme stage of intestinal metaplasia progression in this classification with disappearance of gastric compartments, accompanied by the full expression of intestinal markers, including caudal type homeobox 2 (CDX2), MUC2, and CD10. Thus, these subtypes of intestinal metaplasia reflect the gradual changes in gene expression during the progression from gastric to intestinal characteristics. This serial mucin change would cause spontaneous eradication of H. pylori, since the bacteria could colonize only in MUC5AC positive surface foveolar mucin but not in MUC2 positive intestinal mucins [2].
In the Mongolian gerbil model, gastric-and-intestinal-mixed type intestinal metaplasia was found to appear first, followed by the solely-intestinal type with the appearance of Paneth cells during the overall course of H. pylori infection [46]. Summarizing these human and animal data, intestinal metaplasia might be caused by the gradual intestinalization of gastric gland cells from the gastric-and-intestinal-mixed type to the solely-intestinal type.
Regarding stomach adenocarcinomas, gastric cancers at early stages mainly consist of gastric type cancer cells, and a phenotypic shift from gastric to intestinal phenotypic expression is observed with progression [53]. In the Mongolian gerbil gastric carcinogenesis model, 56 advanced glandular stomach cancers were analyzed for the gastrointestinal phenotypes. In H. pylori-infected gerbils, 56% (28 out of 50 cases) harbored the intestinal phenotype, but all the lesions (6/6) were classified as gastric type in non-infected gerbils. These findings suggested that adenocarcinomas also intestinalized with H. pylori infection and inflammation like intestinal metaplasia [54].
2.5. Host and Environmental Factors
Smoking has been shown to be associated with many kinds of human cancers [55]. For gastric cancers, a Japanese 10-year study has revealed that past and current smokers showed an increased risk of differentiated type gastric cancer in the distal region compared to non-smokers at a relative risk of 2.0 and 2.1, respectively [56]. A systematic review confirmed that relative risk for current smokers was estimated to be 1.56 (95% CI 1.36–1.80) for the Japanese population and concluded that tobacco smoking moderately increases the risk of gastric cancer, with the sex difference being 1.79 (1.51–2.12) and 1.22 (1.07–1.38) in men and women, respectively [57]. Tamer et al. [58] analyzed glutathione S-transferases (GSTs) genotypes in association with smoking and revealed that the GSTM1 null genotype was associated with an increased gastric cancer risk for smokers (odds ratio (OR) = 2.15; 95% CI, 1.02–4.52), whereas no significant differences in the distributions of any of the other GST genes, GSTT1 and GSTP1, existed in the Turkish population.
Males are at a higher risk of developing gastric cancer than females [59]. Androgen receptor in stromal cells was significantly higher in the advanced stage of gastric cancer in males, which might explain the gender difference [60].
2.6. Dietary Factors
2.6.1. Salt
Among various food ingredients, salt and salted foods are probable risk factors for gastric cancer, based on evidence from a large number of case-control and ecological studies [61,62,63,64]. Tajima et al. [61] revealed that fondness for salted foods including pickled vegetable and dried and salted fishes, typical traditional Japanese foods, and showed a significantly positive association with stomach cancer at relative risk = 2.60. Several biologic markers in blood and urine were analyzed in ecological studies and revealed a significant and strong correlation between the amount of salt excreted in urine and stomach cancer mortality in both men and women in Japan [62], as well as worldwide [63].
Researchers have attempted to reveal how salted diet enhanced gastric carcinogenesis using an experimental model. In the pre-Helicobacter era, sodium chloride (NaCl) was found to enhance the carcinogenic effects of chemical carcinogens such as MNNG and 4-nitroquinoline 1-oxide (4-NQO) in the rat glandular stomach [65], possibly due to the reduction of the mucus viscosity and the impairment of the protective mucous barrier. Later, after the discovery of the bacteria in the human stomach, Nozaki et al. showed [66] how a high-salt diet enhanced the effects of H. pylori infection on gastric carcinogenesis. Although high salt intake alone had a minor influence on MNU induced gastric carcinogenesis, H. pylori infection and consequent inflammation acted synergistically with a high salt intake to promote the development of stomach cancers in the Mongolian gerbil model [67]. In H. pylori-infected gerbils, a high salt diet was associated with elevation of anti-H. pylori antibody titers, serum gastrin levels, and inflammatory cell infiltration in a dose-dependent fashion. The high salt diet upregulated the amount of surface mucous cell mucin, suitable for H. pylori colonization, but decreased the amount of gland mucous cell mucin, acting against H. pylori infection by inhibiting the bacterial cell wall component [68]. The incidences of glandular stomach cancers were 15% in the normal diet group and 33%, 36%, and 63% in the 2.5%, 5%, and 10% NaCl diet groups, showing a dose-dependent increase. The reduction of salt intake could thus be one of the most important strategies for the reduction of human gastric cancer.
2.6.2. Green Tea
A comparative case-referent study revealed that the OR of stomach cancer decreased to 0.69 (95% confidence interval (CI) = 0.48–1.00) with a high intake of green tea (seven cups or more per day) [69]. A cross-sectional study was conducted on 636 subjects in Japan to examine the relationship among green tea consumption and H. pylori-induced chronic atrophic gastritis, and revealed that high green tea consumption (more than 10 cups per day) was negatively associated with the risk of chronic atrophic gastritis [70]. Many polyphenolic compounds have demonstrated anticarcinogenic activities, which included flavanone, flavonols, isoflavone, and catechins [71]. Epigallocatechin-3-gallate (EGCG), the major polyphenol in green tea, could affect carcinogenesis and the development of many cancers. Besides the anti-oxidative activity, EGCG inhibits the canonical Wnt/β-catenin signaling [72]. Ohno et al. [73] evaluated the protective effect of green tea catechins using Ins-Gas mice. Although catechin supplementation did not affect inflammation, dysplasia was significantly diminished histopathologically.
2.6.3. Mastic Gum
Mastic gum is a resinous exudate obtained from Pistacia lentiscus which showed bactericidal activity against H. pylori in vitro [74]. An in vivo trial revealed no significant alleviation of H. pylori infection [75]. Another human trial illustrated the dose-dependent trend of mastic gum on H. pylori eradication, although this was not statistically significant [76].
In the mouse model infected with H. pylori SS1, the animals were administered with 2 g of mastic for seven days but failed to eradicate the infection [77]. In the other trial, administration of the total mastic extract without polymer at 0.75 mg/day to H. pylori SS1-infected mice for three months led to an approximate 30-fold reduction in the H. pylori colonization. However, no attenuation was observed in the H. pylori-associated inflammatory infiltration and the activity of chronic gastritis [78].
2.6.4. Ginseng
Korean red ginseng extract, a herbal medicine, is widely used in Asian countries for various biological activities including its anti-inflammatory effect. Ginseng inhibits H. pylori-induced gastric inflammation in Mongolian gerbils by suppressing induction of inflammatory cytokines such as IL-1β, inducible nitric oxide synthase (iNOS), myeloperoxidase, and lipid peroxidase levels in H. pylori-infected gastric mucosa, although ginseng did not affect viable bacterial colonization in the stomach [79]. In vitro analysis revealed that Ginseng extract had strong anti-proliferative and pro-apoptotic effects on KATO3 human gastric cancer cells via the upregulation of Bax (B-cell lymphoma 2-associated X protein), IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α) proteolysis, and the blocking of mTOR (mammalian target of rapamycin) and protein kinase B signaling [80]. In a case-control study, Yun et al. showed the preventive effect of ginseng intake against various human cancers including stomach cancer [81]. However, others did not illustrate clear results; further evaluation in Asian cohort studies may help clarify the role of ginseng in gastric carcinogenesis [82].
2.6.5. Spices
H. pylori is known to play a causative role in gastric carcinogenesis, but wide variations in incidence have been noticed in Asian countries. H. pylori infection is more frequent in developing countries such as India, Pakistan, and Bangladesh than in other countries including Japan, China, and South Korea. Nonetheless, the frequency of gastric cancer is typically higher in the latter countries. This discrepancy is designated “the Asian enigma”, which may result from the genetic diversity of the infective H. pylori strains and differences in the genetic backgrounds of the various ethnic groups studied, as well as from their dietary habits [83]. To assess this problem, dietary spices were evaluated for the relief of H. pylori-induced inflammation. Capsaicin and piperine, but not curcumin, were found to have anti-inflammatory effects on H. pylori-induced gastritis in Mongolian gerbils, independent of direct antibacterial effects, and may thus function as chemopreventive agents for H. pylori-associated gastric carcinogenesis [84].
3. Effects of Eradication of H. pylori on Gastric Inflammation, Intestinal Metaplasia, and Carcinogenesis
3.1. Humans
Many researchers have attempted to clarify whether and how far the serial process of atrophic gastritis and intestinal metaplasia can be reversed after the eradication of H. pylori. As observed by endoscopic analysis, the enlarged or elongated pit patterns in H. pylori-positive specimens were improved to small, oval, pinhole-sized, or round pits after bacterial eradication, with decreased densities of fine, irregular vessels; such changes were not observed in specimens from subjects with severe gastric atrophy and intestinal metaplasia [85]. However, other reports have not always shown histological improvements in gastric atrophy and intestinal metaplasia after the eradication of H. pylori [86,87,88,89]. In contrast, some studies have reported that eradication effectively improves gastric lesions in the antrum or corpus [87,90,91,92].
After the eradication of H. pylori, the number of neutrophils drastically decreased, in contrast to the number of mononuclear cells, which gradually decreased (Figure 1). Eradication also alleviated the hyperplastic and hypertrophic enlargement of the surface foveolar epithelium in the gastric type, but not in the intestinal type, of metaplastic glands, as suggested by the endoscopic results mentioned above. It is currently unclear how bacterial eradication affects the amounts of the mucin core proteins, MUC5AC and MUC6, in these cells. In terms of morphological changes, the length of the proliferative zone and the number of Ki-67-positive cells were both significantly decreased in gastric-type glands [86,93] but not in intestinal metaplastic glands [86,94]. However, both gastric-and-intestinal-mixed and solely intestinal types of intestinal metaplasia always harbored larger numbers of mitotic cells, being positive for phosphorylated histone H3 protein at serine 28, than did gastric-type cells, regardless of the presence of H. pylori infection. Thus, the initial development of intestinal metaplasia could represent an irreversible change with atrophic gastritis [86].
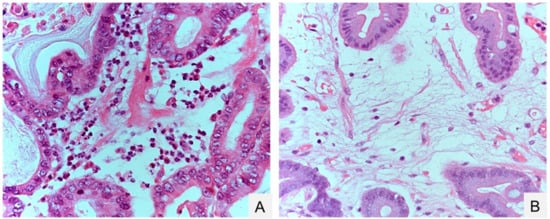
Figure 1.
Gastric inflammation before and after eradication of Helicobacter pylori. (A) Neutrophil inflammation before H. pylori eradication; (B) edematous stroma after H. pylori eradication. Hematoxylin-Eosin (HE) staining. Original magnification, 400× (A,B).
The eradication of H. pylori has been approved for both the prevention of metachronous cancer and cases of chronic atrophic gastritis [3]. Long term follow up after treatment of H. pylori infection revealed the regression of preneoplastic gastric lesions, including intestinal metaplasia [95,96,97]. Both prospective [98,99] and retrospective [100] studies have documented that the successful eradication of H. pylori might reduce the occurrence of metachronous gastric cancer after the endoscopic resection of early lesions over a 3-year period. However, a 7.5-year randomized controlled trial in China revealed that the eradication of this organism significantly decreased the incidence of gastric adenocarcinoma in a subgroup of patients without atrophy, intestinal metaplasia, or dysplasia, whereas the overall incidence did not improve significantly between the eradication and placebo groups [101]. Another meta-analysis [102] supported the idea that eradication of H. pylori is only effective in a subgroup of patients without intestinal metaplasia or dysplasia. A prospective study monitored serum pepsinogen levels and the pepsinogen I/II ratio to determine the degree of chronic gastritis; these authors observed a significant reduction in cancer incidence in pepsinogen test-negative subjects with mild gastritis after H. pylori eradication over a mean period of 9.3 ± 0.7 years [103].
Endoscopic findings have revealed that the gastric tumor area has a gastritis-like appearance rather than typical malignant characteristics [4]. An histopathological analysis of gastric dysplasia (as in the Western category [104], which is intramucosal adenocarcinomas according to the Japanese criteria [105]) revealed significant and rapid alterations in tumor morphology and proliferative characteristics after the eradication of bacteria (Nakagawa et al., manuscript submitted) (Figure 2 and Figure 3). Additionally, gastric tumors appeared to be covered with normal [106] or low-grade atypical epithelium [107] after treatment with antibiotics. These morphological changes make the diagnosis of gastric dysplasia difficult using either endoscopic or histopathologic methods.
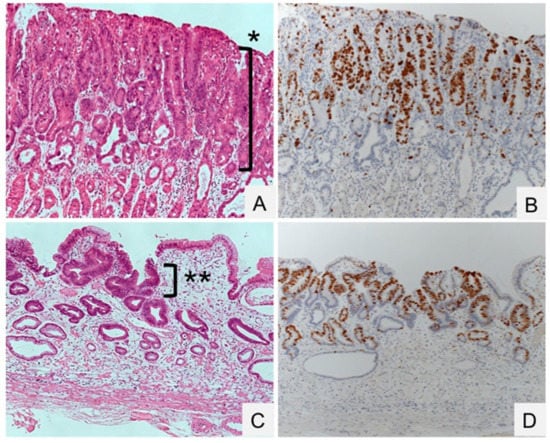
Figure 2.
Gastric dysplasia (intramucosal adenocarcinoma) before and after eradication of Helicobacter pylori. (A,B) Dysplasia proliferating to the surface of the mucosa in an H. pylori-positive specimen (*). (C,D) Regression of dysplasia, localized beneath the normal surface epithelium in an H. pylori-negative specimen (**). HE staining (A,B) and Ki-67 immunostaining (C,D). Original magnification, 100×.
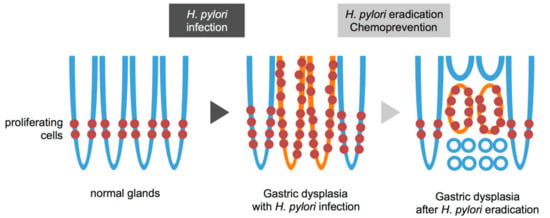
Figure 3.
Schematic view of gastric dysplasia (intramucosal adenocarcinoma) before and after eradication of Helicobacter pylori (H. pylori). Normal glands have proliferating cells in the lower narrow region (left). H. pylori infection widens proliferating zone in the normal glands (middle, blue line). Gastric dysplasia shows expanding proliferation with H. pylori infection and inflammation (middle, orange line). The tumor is shown around the proliferative zone (right, orange line) with subsequent regression at the top and bottom regions that are then occupied by adjacent normal epithelia (right, blue line) after eradication.
3.2. Animals
Several studies based on detailed histopathological assessments have reported a lack of carcinomas in animals subjected only to H. pylori infection [49,50,51,52,108]. With eradication therapy, the sizes of heterotopic proliferative glands were dramatically reduced, with only mucins remaining within them [46], indicating that H. pylori is a stronger promoter of gastric carcinogenesis than are carcinogens.
In the Mongolian gerbil model involving H. pylori infection and carcinogen treatment, H. pylori eradication provided direct evidence that gastric cancer can be prevented [108]. The incidence of adenocarcinoma was significantly lower after curative treatment of H. pylori infection than before treatment. Additional experiments using H. pylori-infected and carcinogen-treated Mongolian gerbils showed that earlier H. pylori eradication resulted in less carcinogenesis [109]. Animal models support the hypothesis that H. pylori eradication is useful for the prevention of gastric carcinogenesis, especially when performed during the early stages of cancer development.
4. Chemoprevention of Gastric Carcinogenesis
4.1. Oxygen Radical Scavengers
Natural products are believed to lower gastric cancer risk in humans [110]. Inflammation and subsequent oxidative stress play important roles in gastric carcinogenesis as mediators of DNA damage and carcinogen production [111]. The combination of bacterial eradication and the reduction of inflammation may be a more reasonable approach for the prevention of gastric cancer development, since the most important factor affecting gastric carcinogenesis is the severity of inflammation [112]. Using the Mongolian gerbil model, one of the most potent antioxidative compounds obtained from crude canola oil, 4-vinyl-2,6-dimethoxyphenol (canolol), was examined for its preventive effects against gastric inflammation and carcinogenesis in H. pylori-infected and carcinogen-treated animals. Canolol (0.1%) was mixed into food to suppress COX-2, iNOS, and 8-hydroxy-2′-deoxyguanosine, resulting in the marked reduction of the incidence of gastric adenocarcinoma, although the number of viable H. pylori was not changed [113]. Canolol also suppressed spontaneous gastric tumor development in K19-C2mE transgenic mice by reducing Cox-2, IL-1β, and IL-12β levels, possibly via the reactivation of tumor suppressor miR-7 microRNA [114]. Taking these results into account, the level of inflammation, rather than the existence of H. pylori, may be the most important factor in the process of carcinogenesis.
4.2. COX-2 Inhibitors
COX-2 and its downstream products play essential roles in the inflammatory microenvironment and tumorigenesis [115]. In mouse models, the overexpression of COX-2 has been shown to be associated with gastric and colorectal adenocarcinomas [37,39,40,116,117]. COX-2-selective inhibitors such as etodolac and celecoxib may have chemopreventive effects [118,119], not only suppressing inflammation but also causing tumor regression [120,121]. Considering the prevention of metachronous gastric cancer in patients that already have extensive metaplastic gastritis, COX-2 inhibitors could induce the regression of precancerous lesions and prevent gastric cancer occurrence after H. pylori eradication. In a nonrandomized trial, Yanaoka et al. [122] administered etodolac to serum pepsinogen test-positive and H. pylori antibody-negative patients, and found an effective reduction of metachronous cancer development. Another intervention trial with a COX-2 inhibitor, celecoxib, in combination with the eradication of H. pylori was conducted and showed the regression of gastric lesions, revealing the importance of the COX-2/prostaglandin E2 (PGE2) pathway [123].
5. Conclusions
Since the discovery of H. pylori in the human stomach, infection by these bacteria has been shown to be strongly associated with gastric lesions, including chronic atrophic gastritis, intestinal metaplasia, and gastric cancer. Epidemiological studies, in combination with results from animal models, confirm that eradication of H. pylori effectively prevents gastric carcinogenesis and mild gastritis without severe atrophy or intestinal metaplasia. However, bacterial eradication raises the issue of regression of gastric dysplasia (intramucosal adenocarcinoma), which might be underdiagnosed as a regenerating gland. Only by precise diagnoses, chemopreventive approaches, and H. pylori eradication can gastric cancer be conquered.
Acknowledgments
This study was supported, in part, by Grants-in-Aid from the Ministry of Education, Science, Sports, and Culture of Japan (15K08960).
Author contributions
Tetsuya Tsukamoto wrote the paper; Mitsuru Nakagawa and Yuka Kiriyama prepared the figures and contributed to valuable discussion; Takeshi Toyoda and Xueyuan Cao contributed to discussion and criticism. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare no conflict of interest.
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Toyoda, T.; Mizoshita, T.; Tatematsu, M. Helicobacter pylori infection and gastric carcinogenesis in rodent models. Semin. Immunopathol. 2013, 35, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Asaka, M.; Mabe, K. Strategies for eliminating death from gastric cancer in Japan. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Saka, A.; Yagi, K.; Nimura, S. Endoscopic and histological features of gastric cancers after successful Helicobacter pylori eradication therapy. Gastric Cancer 2016, 19, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.R.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, 1, 1273–1275. [Google Scholar] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Hu, P.J.; Li, Y.Y.; Zhou, M.H.; Chen, M.H.; Du, G.G.; Huang, B.J.; Mitchell, H.M.; Hazell, S.L. Helicobacter pylori associated with a high prevalence of duodenal ulcer disease and a low prevalence of gastric cancer in a developing nation. Gut 1995, 36, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Craanen, M.E.; Dekker, W.; Blok, P.; Ferwerda, J.; Tytgat, G.N. Intestinal metaplasia and Helicobacter pylori: An endoscopic bioptic study of the gastric antrum. Gut 1992, 33, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.H.; Kato, I.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991, 325, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; Newell, D.G.; Fullerton, F.; Yarnell, J.W.; Stacey, A.R.; Wald, N.; Sitas, F. Association between infection with Helicobacter pylori and risk of gastric cancer: Evidence from a prospective investigation. BMJ 1991, 302, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Lew, G.M.; Klein, P.D.; Evans, D.G.; Evans, D.J., Jr.; Saeed, Z.A.; Malaty, H.M. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann. Intern. Med. 1992, 116, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Uyterlinde, A.M.; Pena, A.S.; Roosendaal, R.; Pals, G.; Nelis, G.F.; Festen, H.P.; Meuwissen, S.G. Long-term sequelae of Helicobacter pylori gastritis. Lancet 1995, 345, 1525–1528. [Google Scholar] [CrossRef]
- Asaka, M.; Kato, M.; Kudo, M.; Katagiri, M.; Nishikawa, K.; Koshiyama, H.; Takeda, H.; Yoshida, J.; Graham, D.Y. Atrophic changes of gastric mucosa are caused by Helicobacter pylori infection rather than aging: Studies in asymptomatic Japanese adults. Helicobacter 1996, 1, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Sridhar, S.; Chen, Y.; Hunt, R.H. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998, 114, 1169–1179. [Google Scholar] [CrossRef]
- Parsonnet, J.; Hansen, S.; Rodriguez, L.; Gelb, A.B.; Warnke, R.A.; Jellum, E.; Orentreich, N.; Vogelman, J.H.; Friedman, G.D. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 1994, 330, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- The Eurogast Study Group. An international association between Helicobacter pylori infection and gastric cancer. Lancet 1993, 341, 1359–1363. [Google Scholar]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Infection with Helicobacter pylori. In Schistosomes, Liver Flukes and Helibacter Pylori; World Health Organization/International Agency for Research on Cancer: Lyon, France, 1994; pp. 177–241. [Google Scholar]
- Backert, S.; Selbach, M. Tyrosine-phosphorylated bacterial effector proteins: The enemies within. Trends Microbiol. 2005, 13, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Anthropological and clinical implications for the structural diversity of the Helicobacter pylori CagA oncoprotein. Cancer Sci. 2011, 102, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Yamazaki, T.; Tsutsumi, R.; Higashi, H.; Onoe, K.; Yamazaki, S.; Azuma, T.; Hatakeyama, M. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology 2006, 130, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Krakowka, S.; Morgan, D.R.; Kraft, W.G.; Leunk, R.D. Establishment of gastric Campylobacter pylori infection in the neonatal gnotobiotic piglet. Infect. Immun. 1987, 55, 2789–2796. [Google Scholar] [PubMed]
- Radin, M.J.; Eaton, K.A.; Krakowka, S.; Morgan, D.R.; Lee, A.; Otto, G.; Fox, J. Helicobacter pylori gastric infection in gnotobiotic beagle dogs. Infect. Immun. 1990, 58, 2606–2612. [Google Scholar] [PubMed]
- Karita, M.; Kouchiyama, T.; Okita, K.; Nakazawa, T. New small animal model for human gastric Helicobacter pylori infection: Success in both nude and euthymic mice. Am. J. Gastroenterol. 1991, 86, 1596–1603. [Google Scholar] [PubMed]
- Karita, M.; Li, Q.; Cantero, D.; Okita, K. Establishment of a small animal model for human Helicobacter pylori infection using germ-free mouse. Am. J. Gastroenterol. 1994, 89, 208–213. [Google Scholar] [PubMed]
- Marchetti, M.; Arico, B.; Burroni, D.; Figura, N.; Rappuoli, R.; Ghiara, P. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science 1995, 267, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Fox, J.G.; Otto, G.; Murphy, J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology 1990, 99, 1315–1323. [Google Scholar] [CrossRef]
- Lee, A.; O’Rourke, J.; de Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Draper, J.L.; Hansen, L.M.; Bernick, D.L.; Abedrabbo, S.; Underwood, J.G.; Kong, N.; Huang, B.C.; Weis, A.M.; Weimer, B.C.; van Vliet, A.H.; et al. Fallacy of the unique genome: Sequence diversity within single Helicobacter pylori strains. MBio 2017, 8, e02321–e02337. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.J.; Danon, S.J.; Wilson, J.E.; O’Rourke, J.L.; Salama, N.R.; Falkow, S.; Mitchell, H.; Lee, A. Chronic Helicobacter pylori infection with Sydney strain 1 and a newly identified mouse-adapted strain (Sydney strain 2000) in C57BL/6 and BALB/c mice. Infect. Immun. 2004, 72, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Lina, T.T.; Alzahrani, S.; House, J.; Yamaoka, Y.; Sharpe, A.H.; Rampy, B.A.; Pinchuk, I.V.; Reyes, V.E. Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS ONE 2015, 10, e0121841. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, T.; Fujimura, S. Tumour production in glandular stomach of rat by N-methyl-N′-nitro-N-nitrosoguanidine. Nature 1967, 216, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Ogawa, K.; Hoshiya, T.; Shichino, Y.; Kato, T.; Imaida, K.; Ito, N. Induction of adenocarcinomas in the glandular stomach of BALB/c mice treated with N-methyl-N-nitrosourea. Jpn. J. Cancer Res. 1992, 83, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Yamamoto, M.; Iwata, H.; Fukami, H.; Yuasa, H.; Tezuka, N.; Masui, T.; Nakanishi, H. Induction of glandular stomach cancers in C3H mice treated with N-methyl-N-nitrosourea in the drinking water. Jpn. J. Cancer Res. 1993, 84, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Takasu, S.; Tsukamoto, T.; Cao, X.Y.; Toyoda, T.; Hirata, A.; Ban, H.; Yamamoto, M.; Sakai, H.; Yanai, T.; Masegi, T.; et al. Roles of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 expression and β-catenin activation in gastric carcinogenesis in N-methyl-N-nitrosourea-treated K19-C2mE transgenic mice. Cancer Sci. 2008, 99, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yu, Y. Transgenic and gene knockout mice in gastric cancer research. Oncotarget 2017, 8, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Oshima, M.; Inaba, K.; Taketo, M.M. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004, 23, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature 2001, 412, 99. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.C.; Brand, S.J. Function and regulation of gastrin in transgenic mice: A review. Yale J. Biol. Med. 1992, 65, 705–713, discussion 737–740. [Google Scholar] [PubMed]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Hirayama, F.; Takagi, S.; Yokoyama, Y.; Iwao, E.; Ikeda, Y. Establishment of gastric Helicobacter pylori infection in Mongolian gerbils. J. Gastroenterol. 1996, 31 (Suppl. S9), 24–28. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Shimizu, N.; Tsukamoto, T.; Inada, K.; Cao, X.; Ikehara, Y.; Kaminishi, M.; Sugiyama, A.; Tatematsu, M. Reversibility of Heterotopic proliferative glands in glandular stomach of Helicobacter pylori-infected mongolian gerbils on eradication. Jpn. J. Cancer Res. 2002, 93, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Fujioka, T.; Tokieda, M.; Satoh, R.; Nishizono, A.; Nasu, M. Development of Helicobacter pylori-induced gastric carcinoma in Mongolian gerbils. Cancer Res. 1998, 58, 4255–4259. [Google Scholar] [PubMed]
- Watanabe, T.; Tada, M.; Nagai, H.; Sasaki, S.; Nakao, M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 1998, 115, 642–648. [Google Scholar] [CrossRef]
- Tatematsu, M.; Yamamoto, M.; Shimizu, N.; Yoshikawa, A.; Fukami, H.; Kaminishi, M.; Oohara, T.; Sugiyama, A.; Ikeno, T. Induction of glandular stomach cancers in Helicobacter pylori-sensitive Mongolian gerbils treated with N-methyl-N-nitrosourea and N-methyl-N′-nitro-N-nitrosoguanidine in drinking water. Jpn. J. Cancer Res. 1998, 89, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Maruta, F.; Ikeno, T.; Ishida, K.; Kawasaki, S.; Katsuyama, T.; Shimizu, N.; Tatematsu, M. Helicobacter pylori infection enhances N-methyl-N-nitrosourea-induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res. 1998, 58, 2067–2069. [Google Scholar] [PubMed]
- Shimizu, N.; Inada, K.; Nakanishi, H.; Tsukamoto, T.; Ikehara, Y.; Kaminishi, M.; Kuramoto, S.; Sugiyama, A.; Katsuyama, T.; Tatematsu, M. Helicobacter pylori infection enhances glandular stomach carcinogenesis in Mongolian gerbils treated with chemical carcinogens. Carcinogenesis 1999, 20, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Inada, K.I.; Tsukamoto, T.; Nakanishi, H.; Ikehara, Y.; Yoshikawa, A.; Kaminishi, M.; Kuramoto, S.; Tatematsu, M. New animal model of glandular stomach carcinogenesis in Mongolian gerbils infected with Helicobacter pylori and treated with a chemical carcinogen. J. Gastroenterol. 1999, 34, 61–66. [Google Scholar] [PubMed]
- Tatematsu, M.; Tsukamoto, T.; Inada, K. Stem cells and gastric cancer-role of gastric and intestinal mixed intestinal metaplasia. Cancer Sci. 2003, 94, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Mizoshita, T.; Tsukamoto, T.; Takenaka, Y.; Cao, X.; Kato, S.; Kaminishi, M.; Tatematsu, M. Gastric and intestinal phenotypes and histogenesis of advanced glandular stomach cancers in carcinogen-treated, Helicobacter pylori-infected Mongolian gerbils. Cancer Sci. 2006, 97, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tsuji, I.; Wakai, K.; Nagata, C.; Mizoue, T.; Tanaka, K.; Tsugane, S. Evaluation based on systematic review of epidemiological evidence among Japanese populations: Tobacco smoking and total cancer risk. Jpn. J. Clin. Oncol. 2005, 35, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Sasazuki, S.; Sasaki, S.; Tsugane, S. Cigarette smoking, alcohol consumption and subsequent gastric cancer risk by subsite and histologic type. Int. J. Cancer 2002, 101, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Inoue, M.; Tsuji, I.; Wakai, K.; Nagata, C.; Mizoue, T.; Tanaka, K.; Tsugane, S. Tobacco smoking and gastric cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn. J. Clin. Oncol. 2006, 36, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Tamer, L.; Ates, N.A.; Ates, C.; Ercan, B.; Elipek, T.; Yildirim, H.; Camdeviren, H.; Atik, U.; Aydin, S. Glutathione S-transferase M1, T1 and P1 genetic polymorphisms, cigarette smoking and gastric cancer risk. Cell Biochem. Funct. 2005, 23, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wan, H.; Lin, Y.; Xie, X.; Li, Z.; Tan, G. Androgen receptor may be responsible for gender disparity in gastric cancer. Med. Hypotheses 2013, 80, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Jukic, Z.; Radulovic, P.; Stojkovic, R.; Mijic, A.; Grah, J.; Kruslin, B.; Ferencic, Z.; Fucic, A. Gender difference in distribution of estrogen and androgen receptors in intestinal-type gastric cancer. Anticancer Res. 2017, 37, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Tominaga, S. Dietary habits and gastro-intestinal cancers: A comparative case-control study of stomach and large intestinal cancers in Nagoya, Japan. Jpn. J. Cancer Res. 1985, 76, 705–716. [Google Scholar] [PubMed]
- Tsugane, S.; Tsuda, M.; Gey, F.; Watanabe, S. Cross-sectional study with multiple measurements of biological markers for assessing stomach cancer risks at the population level. Environ. Health Perspect. 1992, 98, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Joossens, J.V.; Hill, M.J.; Elliott, P.; Stamler, R.; Lesaffre, E.; Dyer, A.; Nichols, R.; Kesteloot, H. Dietary salt, nitrate and stomach cancer mortality in 24 countries. Int. J. Epidemiol. 1996, 25, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Kono, S.; Hirohata, T. Nutrition and stomach cancer. Cancer Causes Control 1996, 7, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Takahashi, M.; Fukushima, S.; Hananouchi, M.; Shirai, T. Effects in rats of sodium chloride on experimental gastric cancers induced by N-methyl-N-nitro-N-nitrosoguanidine or 4-nitroquinoline-1-oxide. J. Natl. Cancer Inst. 1975, 55, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tsukamoto, T.; Mizoshita, T.; Tanaka, H.; Kumagai, T.; Ota, H.; Katsuyama, T.; Asaka, M.; Tatematsu, M. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int. J. Cancer 2006, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Shimizu, N.; Inada, K.; Tsukamoto, T.; Inoue, M.; Kumagai, T.; Sugiyama, A.; Mizoshita, T.; Kaminishi, M.; Tatematsu, M. Synergistic Promoting effects of Helicobacter pylori infection and high-salt diet on gastric carcinogenesis in mongolian gerbils. Jpn. J. Cancer Res. 2002, 93, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, M.; Ito, Y.; Okimura, Y.; Kobayashi, M.; Sakura, K.; Kasama, S.; Fukuda, M.N.; Fukuda, M.; Katsuyama, T.; Nakayama, J. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science 2004, 305, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tajima, K.; Hirose, K.; Hamajima, N.; Takezaki, T.; Kuroishi, T.; Tominaga, S. Tea and coffee consumption and the risk of digestive tract cancers: Data from a comparative case-referent study in Japan. Cancer Causes Control 1998, 9, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Moriyama, M.; Fukushima, T.; Kaetsu, A.; Miyazaki, M.; Une, H. Green tea consumption and chronic atrophic gastritis: A cross-sectional study in a green tea production village. J. Epidemiol. 2000, 10, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Lee, M.J.; Chen, L.; Yang, G.Y. Polyphenols as inhibitors of carcinogenesis. Environ. Health Perspect. 1997, 105, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Du, W.; Yang, D. Inhibition of green tea polyphenol EGCG((−)-epigallocatechin-3-gallate) on the proliferation of gastric cancer cells by suppressing canonical Wnt/β-catenin signalling pathway. Int. J. Food Sci. Nutr. 2016, 67, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Ohtani, M.; Suto, H.; Ohta, M.; Imamura, Y.; Matsuda, H.; Hiramatsu, K.; Nemoto, T.; Nakamoto, Y. Effect of green tea catechins on gastric mucosal dysplasia in insulin-gastrin mice. Oncol. Rep. 2016, 35, 3241–3247. [Google Scholar] [CrossRef] [PubMed]
- Marone, P.; Bono, L.; Leone, E.; Bona, S.; Carretto, E.; Perversi, L. Bactericidal activity of pistacia lentiscus mastic gum against Helicobacter pylori. J. Chemother. 2001, 13, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Bebb, J.R.; Bailey-Flitter, N.; Ala’Aldeen, D.; Atherton, J.C. Mastic gum has no effect on Helicobacter pylori load in vivo. J. Antimicrob. Chemother. 2003, 52, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Dabos, K.J.; Sfika, E.; Vlatta, L.J.; Giannikopoulos, G. The effect of mastic gum on Helicobacter pylori: A randomized pilot study. Phytomedicine 2010, 17, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, M.F.; Ala’Aldeen, D.A.; Jenks, P.J. Monotherapy with mastic does not eradicate Helicobacter pylori infection from mice. J. Antimicrob. Chemother. 2003, 51, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Paraschos, S.; Magiatis, P.; Mitakou, S.; Petraki, K.; Kalliaropoulos, A.; Maragkoudakis, P.; Mentis, A.; Sgouras, D.; Skaltsounis, A.L. In vitro and in vivo activities of Chios mastic gum extracts and constituents against Helicobacter pylori. Antimicrob. Agents Chemother. 2007, 51, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Jang, S.; Lim, J.W.; Kang, J.; Bak, E.J.; Cha, J.H.; Kim, H. Protective effect of korean red ginseng extract against Helicobacter pylori-induced gastric inflammation in Mongolian gerbils. J. Ginseng Res. 2014, 38, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Baek, Y.M.; Jang, I.S.; Yang, K.E.; Lee, D.G.; Yoon, S.J.; Rho, J.; Cho, C.K.; Lee, Y.W.; Kwon, K.R.; et al. An enzymatically fortified ginseng extract inhibits proliferation and induces apoptosis of KATO3 human gastric cancer cells via modulation of Bax, mTOR, PKB and IκBα. Mol. Med. Rep. 2015, 11, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.K.; Choi, S.Y. Preventive effect of ginseng intake against various human cancers: A case-control study on 1987 pairs. Cancer Epidemiol. Biomarkers Prev. 1995, 4, 401–408. [Google Scholar] [PubMed]
- Kamangar, F.; Gao, Y.T.; Shu, X.O.; Kahkeshani, K.; Ji, B.T.; Yang, G.; Li, H.L.; Rothman, N.; Chow, W.H.; Zheng, W. Ginseng intake and gastric cancer risk in the Shanghai Women’s Health Study cohort. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Miwa, H.; Go, M.F.; Sato, N. H. pylori and gastric cancer: The Asian enigma. Am. J. Gastroenterol. 2002, 97, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Shi, L.; Takasu, S.; Cho, Y.M.; Kiriyama, Y.; Nishikawa, A.; Ogawa, K.; Tatematsu, M.; Tsukamoto, T. Anti-inflammatory effects of capsaicin and piperine on Helicobacter pylori-induced chronic gastritis in mongolian gerbils. Helicobacter 2016, 21, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Tahara, T.; Shibata, T.; Nakamura, M.; Yoshioka, D.; Maeda, Y.; Yonemura, J.; Ishizuka, T.; Arisawa, T.; Hirata, I. Changes in gastric mucosal patterns seen by magnifying NBI during H. pylori eradication. J. Gastroenterol. 2011, 46, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Tahara, T.; Shibata, T.; Okubo, M.; Nakagawa, M.; Okabe, A.; Ohmiya, N.; Kuroda, M.; Sugioka, A.; Ichinose, M.; et al. Gastric-and-intestinal mixed intestinal metaplasia is irreversible point with eradication of Helicobacter pylori. Open J. Pathol. 2016, 6, 93–104. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, T.H.; Chiu, H.M.; Shun, C.T.; Chiang, H.; Liu, T.Y.; Wu, M.S.; Lin, J.T. The benefit of mass eradication of Helicobacter pylori infection: A community-based study of gastric cancer prevention. Gut 2013, 62, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Annibale, B.; Aprile, M.R.; D’Ambra, G.; Caruana, P.; Bordi, C.; delle Fave, G. Cure of Helicobacter pylori infection in atrophic body gastritis patients does not improve mucosal atrophy but reduces hypergastrinemia and its related effects on body ECL-cell hyperplasia. Aliment. Pharmacol. Ther. 2000, 14, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Forbes, G.M.; Warren, J.R.; Glaser, M.E.; Cullen, D.J.; Marshall, B.J.; Collins, B.J. Long-term follow-up of gastric histology after Helicobacter pylori eradication. J. Gastroenterol. Hepatol. 1996, 11, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Murakami, K.; Okimoto, T.; Sato, R.; Uchida, M.; Abe, T.; Shiota, S.; Nakagawa, Y.; Mizukami, K.; Fujioka, T. Ten-year prospective follow-up of histological changes at five points on the gastric mucosa as recommended by the updated Sydney system after Helicobacter pylori eradication. J. Gastroenterol. 2012, 47, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Haruma, K.; Kamada, T.; Mihara, M.; Kim, S.; Kitadai, Y.; Sumii, M.; Tanaka, S.; Yoshihara, M.; Chayama, K. Helicobacter pylori eradication therapy improves atrophic gastritis and intestinal metaplasia: A 5-year prospective study of patients with atrophic gastritis. Aliment. Pharmacol. Ther. 2002, 16, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, T.; Suwaki, K.; Miyake, Y.; Nakatsu, M.; Ando, M. Eradication of Helicobacter pylori infection improved gastric mucosal atrophy and prevented progression of intestinal metaplasia, especially in the elderly population: A long-term prospective cohort study. J. Gastroenterol. Hepatol. 2010, 25, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Fujioka, T.; Kodama, R.; Kubota, T.; Tokieda, M.; Nasu, M. Helicobacter pylori infection accelerates human gastric mucosal cell proliferation. J. Gastroenterol. 1997, 32, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Erkan, G.; Gonul, I.I.; Kandilci, U.; Dursun, A. Evaluation of apoptosis along with BCL-2 and Ki-67 expression in patients with intestinal metaplasia. Pathol. Res. Pract. 2012, 208, 89–93. [Google Scholar] [CrossRef] [PubMed]
- You, W.C.; Brown, L.M.; Zhang, L.; Li, J.Y.; Jin, M.L.; Chang, Y.S.; Ma, J.L.; Pan, K.F.; Liu, W.D.; Hu, Y.; et al. Randomized double-blind factorial trial of three treatments to reduce the prevalence of precancerous gastric lesions. J. Natl. Cancer Inst. 2006, 98, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Mera, R.; Fontham, E.T.; Bravo, L.E.; Bravo, J.C.; Piazuelo, M.B.; Camargo, M.C.; Correa, P. Long term follow up of patients treated for Helicobacter pylori infection. Gut 2005, 54, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Lin, S.R.; Ching, J.Y.; To, K.F.; Ng, E.K.; Chan, F.K.; Lau, J.Y.; Sung, J.J. Factors predicting progression of gastric intestinal metaplasia: Results of a randomised trial on Helicobacter pylori eradication. Gut 2004, 53, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Mukai, T.; Okamoto, S.; Yamaguchi, S.; Mashiba, H.; Taniyama, K.; Sasaki, N.; Haruma, K.; Sumii, K.; Kajiyama, G. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer. Cancer Epidemiol. Biomarkers Prev. 1997, 6, 639–642. [Google Scholar] [CrossRef]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef]
- Bae, S.E.; Jung, H.Y.; Kang, J.; Park, Y.S.; Baek, S.; Jung, J.H.; Choi, J.Y.; Kim, M.Y.; Ahn, J.Y.; Choi, K.S.; et al. Effect of Helicobacter pylori eradication on metachronous recurrence after endoscopic resection of gastric neoplasm. Am. J. Gastroenterol. 2014, 109, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.C.; Lam, S.K.; Wong, W.M.; Chen, J.S.; Zheng, T.T.; Feng, R.E.; Lai, K.C.; Hu, W.H.; Yuen, S.T.; Leung, S.Y.; et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: A randomized controlled trial. Jama 2004, 291, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.N.; Wang, Z.; Li, X.; Zhou, Z.G. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: Evidence from a meta-analysis. Gastric Cancer 2016, 19, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Yanaoka, K.; Oka, M.; Ohata, H.; Yoshimura, N.; Deguchi, H.; Mukoubayashi, C.; Enomoto, S.; Inoue, I.; Iguchi, M.; Maekita, T.; et al. Eradication of Helicobacter pylori prevents cancer development in subjects with mild gastric atrophy identified by serum pepsinogen levels. Int. J. Cancer 2009, 125, 2697–2703. [Google Scholar] [CrossRef] [PubMed]
- Schlemper, R.J.; Kato, Y.; Stolte, M. Diagnostic criteria for gastrointestinal carcinomas in Japan and Western countries: Proposal for a new classification system of gastrointestinal epithelial neoplasia. J. Gastroenterol. Hepatol. 2000, 15, G49–G57. [Google Scholar] [CrossRef] [PubMed]
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011, 14, 101–112. [Google Scholar]
- Ito, M.; Tanaka, S.; Takata, S.; Oka, S.; Imagawa, S.; Ueda, H.; Egi, Y.; Kitadai, Y.; Yasui, W.; Yoshihara, M.; et al. Morphological changes in human gastric tumours after eradication therapy of Helicobacter pylori in a short-term follow-up. Aliment. Pharmacol. Ther. 2005, 21, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Ito, M.; Matsuo, T.; Boda, T.; Oka, S.; Yoshihara, M.; Tanaka, S.; Chayama, K. Characteristic epithelium with low-grade atypia appears on the surface of gastric cancer after successful Helicobacter pylori eradication therapy. Helicobacter 2014, 19, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Ikehara, Y.; Inada, K.; Nakanishi, H.; Tsukamoto, T.; Nozaki, K.; Kaminishi, M.; Kuramoto, S.; Sugiyama, A.; Katsuyama, T.; et al. Eradication diminishes enhancing effects of Helicobacter pylori infection on glandular stomach carcinogenesis in Mongolian gerbils. Cancer Res. 2000, 60, 1512–1514. [Google Scholar] [PubMed]
- Nozaki, K.; Shimizu, N.; Ikehara, Y.; Inoue, M.; Tsukamoto, T.; Inada, K.; Tanaka, H.; Kumagai, T.; Kaminishi, M.; Tatematsu, M. Effect of early eradication on Helicobacter pylori-related gastric carcinogenesis in Mongolian gerbils. Cancer Sci. 2003, 94, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Sasazuki, S. Diet and the risk of gastric cancer: Review of epidemiological evidence. Gastric Cancer 2007, 10, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Yoshikawa, T. Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radic. Biol. Med. 2002, 33, 323–336. [Google Scholar] [CrossRef]
- Cao, X.; Tsukamoto, T.; Nozaki, K.; Tanaka, H.; Cao, L.; Toyoda, T.; Takasu, S.; Ban, H.; Kumagai, T.; Tatematsu, M. Severity of gastritis determines glandular stomach carcinogenesis in Helicobacter pylori-infected Mongolian gerbils. Cancer Sci. 2007, 98, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tsukamoto, T.; Seki, T.; Tanaka, H.; Morimura, S.; Cao, L.; Mizoshita, T.; Ban, H.; Toyoda, T.; Maeda, H.; et al. 4-Vinyl-2,6-dimethoxyphenol (canolol) suppresses oxidative stress and gastric carcinogenesis in Helicobacter pylori-infected carcinogen-treated Mongolian gerbils. Int. J. Cancer 2008, 122, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Jiang, J.; Tsukamoto, T.; Liu, R.; Ma, L.; Jia, Z.; Kong, F.; Oshima, M.; Cao, X. Canolol inhibits gastric tumors initiation and progression through COX-2/PGE2 pathway in K19-C2mE transgenic mice. PLoS ONE 2015, 10, e0120938. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Hirose, O.; Maeda, Y.; Oshima, M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016, 107, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Sung, J.J. Chemoprevention of gastric cancer. Eur. J. Gastroenterol. Hepatol. 2006, 18, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Prescott, S.M.; Fitzpatrick, F.A. Cyclooxygenase-2 and carcinogenesis. Biochim. Biophys. Acta 2000, 1470, M69–M78. [Google Scholar] [CrossRef]
- Futagami, S.; Suzuki, K.; Hiratsuka, T.; Shindo, T.; Hamamoto, T.; Tatsuguchi, A.; Ueki, N.; Shinji, Y.; Kusunoki, M.; Wada, K.; et al. Celecoxib inhibits Cdx2 expression and prevents gastric cancer in Helicobacter pylori-infected Mongolian gerbils. Digestion 2006, 74, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Magari, H.; Shimizu, Y.; Inada, K.; Enomoto, S.; Tomeki, T.; Yanaoka, K.; Tamai, H.; Arii, K.; Nakata, H.; Oka, M.; et al. Inhibitory effect of etodolac, a selective cyclooxygenase-2 inhibitor, on stomach carcinogenesis in Helicobacter pylori-infected Mongolian gerbils. Biochem. Biophys. Res. Commun. 2005, 334, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.H.; McEntee, M.F.; Whelan, J. Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Res. 1997, 57, 4267–4273. [Google Scholar] [PubMed]
- Reddy, B.S.; Maruyama, H.; Kelloff, G. Dose-related inhibition of colon carcinogenesis by dietary piroxicam, a nonsteroidal antiinflammatory drug, during different stages of rat colon tumor development. Cancer Res. 1987, 47, 5340–5346. [Google Scholar] [PubMed]
- Yanaoka, K.; Oka, M.; Yoshimura, N.; Deguchi, H.; Mukoubayashi, C.; Enomoto, S.; Maekita, T.; Inoue, I.; Ueda, K.; Utsunomiya, H.; et al. Preventive effects of etodolac, a selective cyclooxygenase-2 inhibitor, on cancer development in extensive metaplastic gastritis, a Helicobacter pylori-negative precancerous lesion. Int. J. Cancer 2010, 126, 1467–1473. [Google Scholar] [PubMed]
- Zhang, Y.; Pan, K.F.; Zhang, L.; Ma, J.L.; Zhou, T.; Li, J.Y.; Shen, L.; You, W.C. Helicobacter pylori, cyclooxygenase-2 and evolution of gastric lesions: Results from an intervention trial in China. Carcinogenesis 2015, 36, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).