Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health


Abstract
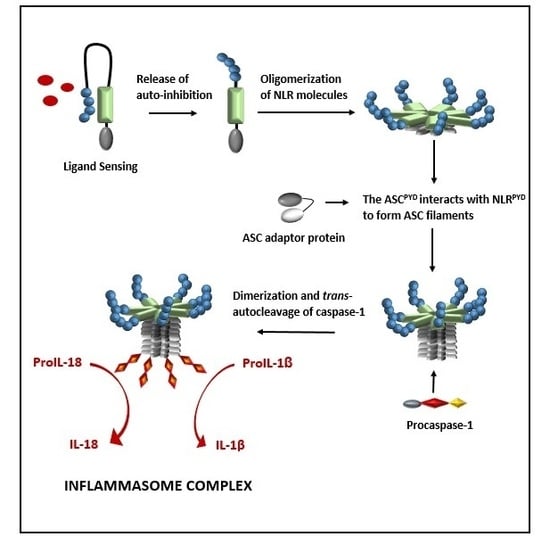
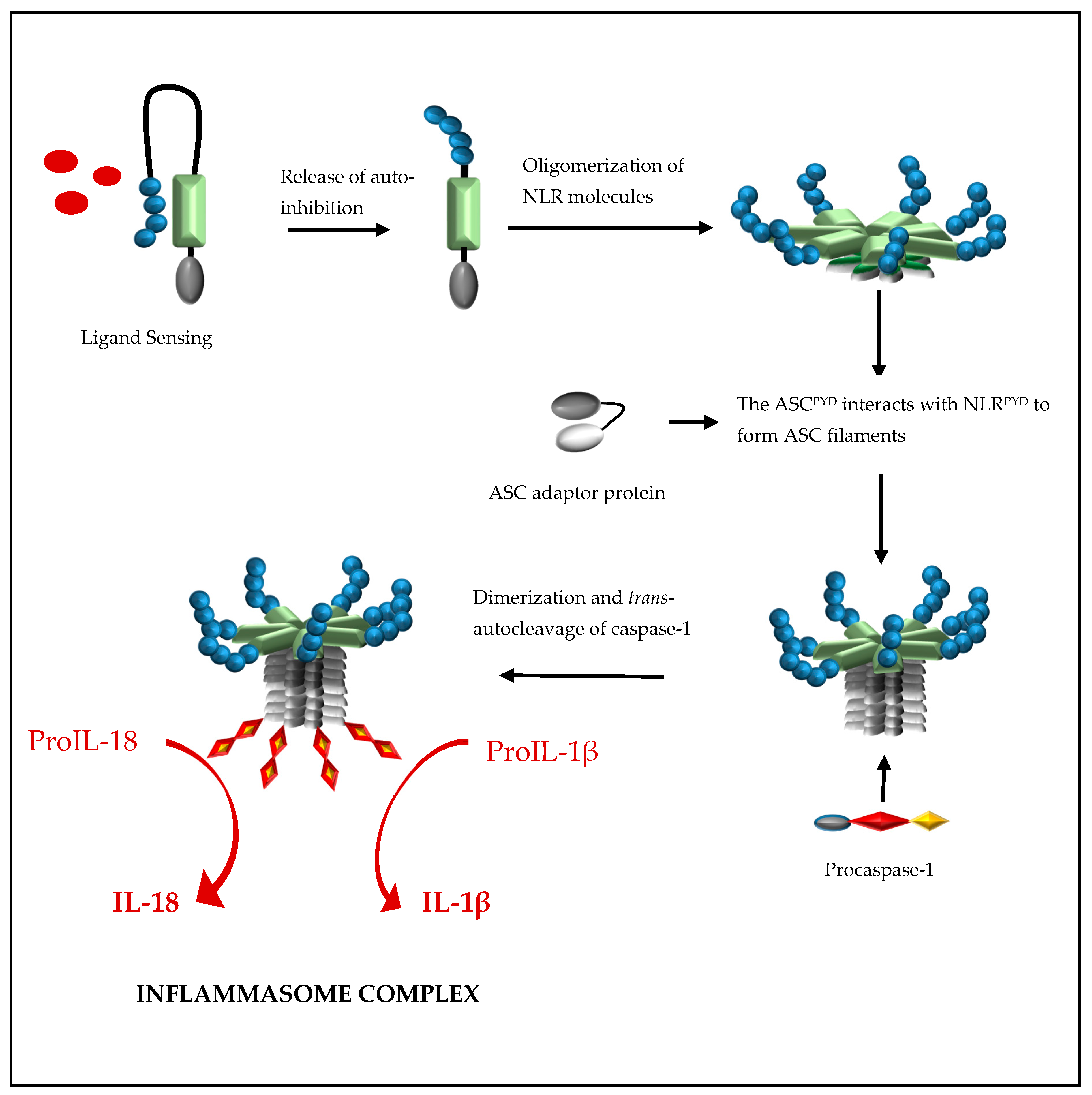
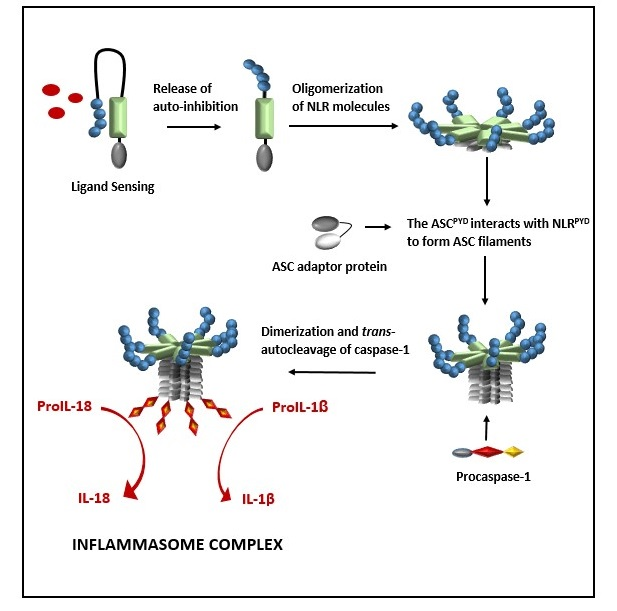{kind=link}
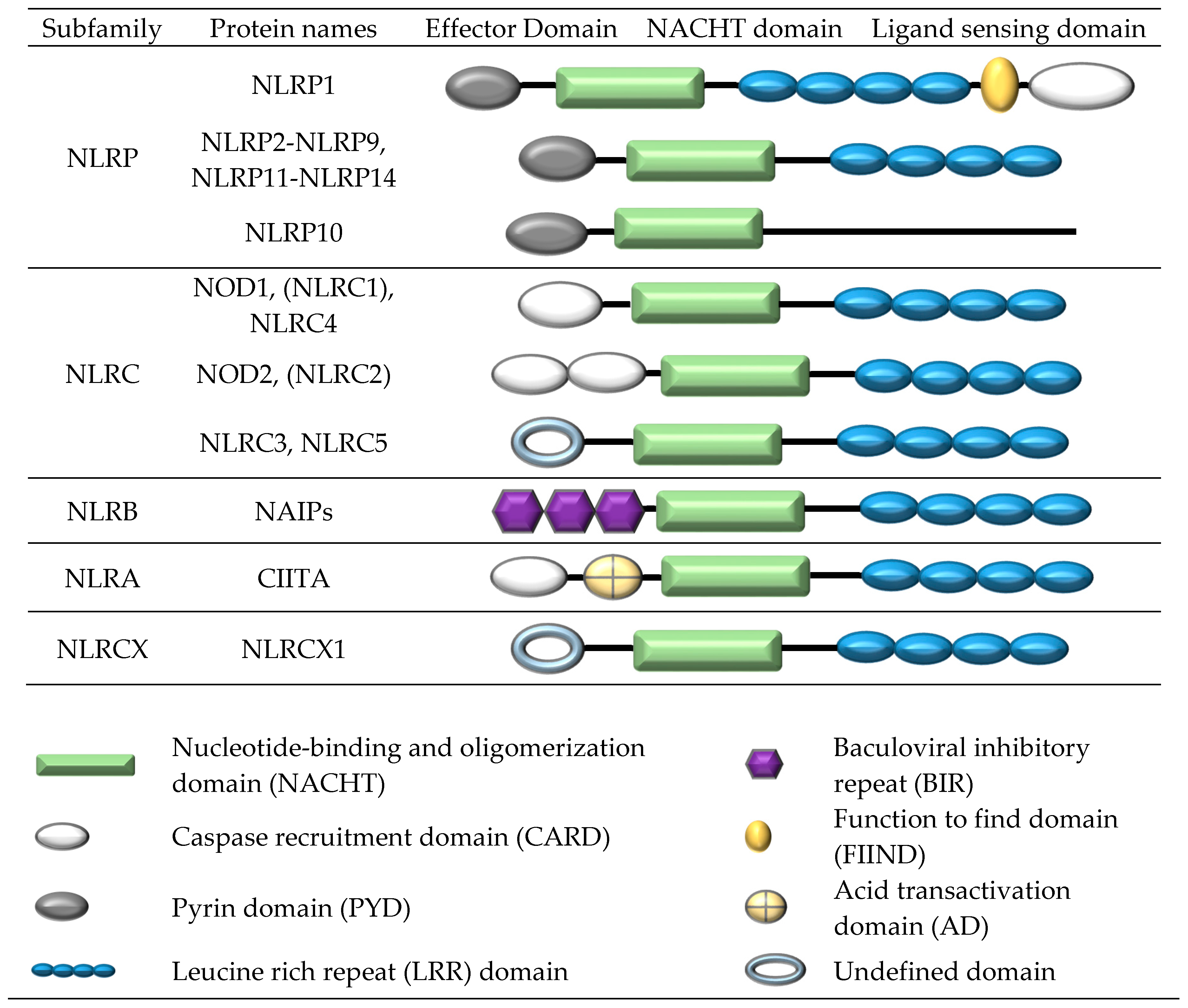{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Formation of a NOD-Like Receptor Protein (NLRP) Inflammasome Complex
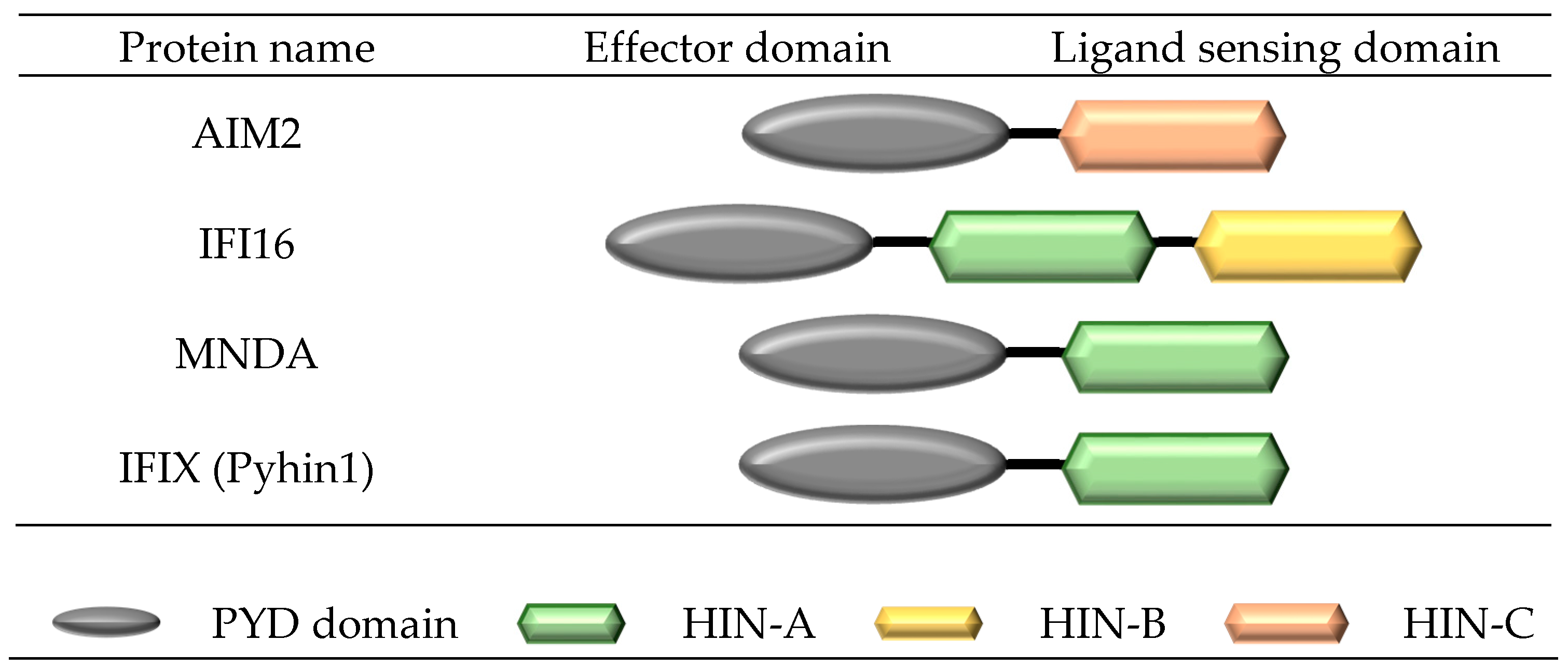
3. Structure and Formation of a Pyrin and HIN Domain (PYHIN) Inflammasome Complex
4. Ligand Sensing of Inflammasome Complexes
5. The NLRP1 Inflammasome
6. The NLRP3 Inflammasome
7. The NLRC4 Inflammasome
8. The AIM2 and IFI16 Inflammasome
9. The NLRP6, NLRP7, and NLRP12 Inflammasomes
10. Regulation of the Inflammasome Complex
11. Regulation by Autoinhibition of the Ligand-Sensing Domain
12. Priming Events That Regulate Activation
13. Regulation by POPs and COPs
14. Regulation by Type I Interferons
15. Regulation from Inflammasome Components
16. Inflammasome Complexes and the Intestinal Environment
17. Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bickston, S.J.; Bloomfeld, R.S. Handbook of Inflammatory Bowel Disease, 1st ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2010; 175p. [Google Scholar]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; So, A. Regulation of inflammasome activity. Immunology 2010, 130, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Abreu, M.T. What are toll-like receptors and what role may they have in IBD? Inflamm. Bowel Dis. 2008, 14, S90–S92. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, M.; Ceballos-Olvera, I.; del Barrio, L.; Re, F. Role of the Inflammasome, IL-1β, and IL-18 in Bacterial Infections. Sci. World J. 2011, 11, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- McGeough, M.D.; Pena, C.A.; Mueller, J.L.; Pociask, D.A.; Broderick, L.; Hoffman, H.M.; Brydges, S.D. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J. Immunol. 2012, 189, 2707–2711. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Leaf, I.A.; Hagar, J.A.; Fontana, M.F.; Campos, C.G.; Zak, D.E.; Tan, M.H.; Cotter, P.A.; Vance, R.E.; Aderem, A.; et al. Caspase-11 protects against bacteria that escape the vacuole. Science 2013, 339, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.S.; Guarda, G.; D’Ombrain, M.C.; Drexler, S.K. Inflammatory caspases in innate immunity and inflammation. J. Innate Immun. 2010, 2, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- De Alba, E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J. Biol. Chem. 2009, 284, 32932–32941. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.B.; Dorfleutner, A.; Kramer, S.J.; Yun, C.; Rojanasakul, Y.; Stehlik, C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J. Inflamm. 2010, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.E.; O’Neill, L.A. Inflammasomes in inflammatory disorders: The role of TLRs and their interactions with NLRs. Semin. Immunopathol. 2007, 29, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.D.; Mcdonald, T.T.; Blumberg, R.S. (Eds.) Principles of Mucosal Immunology; Garland Science, Taylor and Francis Group, LLC: New York, NY, USA, 2013. [Google Scholar]
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- D’Osualdo, A.; Weichenberger, C.X.; Wagner, R.N.; Godzik, A.; Wooley, J.; Reed, J.C. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS ONE 2011, 6, e27396. [Google Scholar] [CrossRef] [PubMed]
- Pathan, N.; Marusawa, H.; Krajewska, M.; Matsuzawa, S.; Kim, H.; Okada, K.; Torii, S.; Kitada, S.; Krajewski, S.; Welsh, K.; et al. TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J. Biol. Chem. 2001, 276, 32220–32229. [Google Scholar] [CrossRef] [PubMed]
- Stilo, R.; Leonardi, A.; Formisano, L.; di Jeso, B.; Vito, P.; Liguoro, D. TUCAN/CARDINAL and DRAL participate in a common pathway for modulation of NF-κB activation. FEBS Lett. 2002, 521, 165–169. [Google Scholar] [CrossRef]
- Tschopp, J.; Martinon, F.; Burns, K. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 2003, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef] [PubMed]
- Schattgen, S.A.; Fitzgerald, K.A. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 2011, 24, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Cridland, J.A.; Curley, E.Z.; Wykes, M.N.; Schroder, K.; Sweet, M.J.; Roberts, T.L.; Ragan, M.A.; Kassahn, K.S.; Stacey, K.J. The mammalian PYHIN gene family: Phylogeny, evolution and expression. BMC Evol. Biol. 2012, 12, 140. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Newman, Z.L.; Printz, M.P.; Liu, S.; Crown, D.; Breen, L.; Miller-Randolph, S.; Flodman, P.; Leppla, S.H.; Moayeri, M. Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog. 2010, 6, e1000906. [Google Scholar] [CrossRef] [PubMed]
- Terra, J.K.; Cote, C.K.; France, B.; Jenkins, A.L.; Bozue, J.A.; Welkos, S.L.; LeVine, S.M.; Bradley, K.A. Cutting edge: Resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J. Immunol. 2010, 184, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-C.; Ali, S.R.; McGillivray, S.; Tseng, P.-H.; Mariathasan, S.; Humke, E.W.; Eckmann, L.; Powell, J.J.; Nizet, V.; Dixit, V.M.; et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1 secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA 2008, 105, 7803–7808. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hsu, L.C.; Liu, H.; Bankston, L.A.; Iimura, M.; Kagnoff, M.F.; Eckmann, L.; Karin, M. Nod2 mutation in Crohn’s disease potentiates NF-κB activity and IL-1β processing. Science 2005, 307, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Malireddi, R.K.; Kanneganti, T.D. Role of the nlrp3 inflammasome in microbial infection. Front. Microbiol. 2011, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Nunez, G. Cutting edge: TNF-α mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Gombault, A.; Baron, L.; Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 2012, 3, 414. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Gao, X.; Huang, M.T.; O’Connor, B.P.; Thomas, C.E.; Willingham, S.B.; Bergstralh, D.T.; Jarvis, G.A.; Sparling, P.F.; Ting, J.P. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 2009, 182, 6460–6469. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.L.; Stehle, J.C.; Kopf, M.; Stamenkovic, I.; et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE 2009, 4, e6510. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Chiu, Y.H.; Ismail, A.S.; Behrendt, C.L.; Wight-Carter, M.; Hooper, L.V.; Chen, Z.J. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc. Natl. Acad. Sci. USA 2011, 108, 17390–17395. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Kanneganti, T.D.; Dubyak, G.R.; Nunez, G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 2007, 282, 18810–18818. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Franchi, L.; Munoz-Planillo, R.; Park, J.H.; Reimer, T.; Nunez, G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-κB activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Shi, J.; Shao, F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. USA 2013, 110, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010, 30, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Chen, W.; Li, N.; Hu, X.Z.; Du, C.T.; Yu, S.X.; Zhou, M.; Zhang, X.J.; Jiang, G.M.; Han, W.Y.; et al. Cytosolic double-stranded DNA induces nonnecroptotic programmed cell death in trophoblasts via IFI16. J. Infect. Dis. 2014, 210, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010, 11, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M.; et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y. Role of Nlrp6 and Nlrp12 in the maintenance of intestinal homeostasis. Eur. J. Immunol. 2014, 44, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Malireddi, R.K.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447S–455S. [Google Scholar] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Komuves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-xl regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Faustin, B.; Chen, Y.; Zhai, D.; le Negrate, G.; Lartigue, L.; Satterthwait, A.; Reed, J.C. Mechanism of Bcl-2 and Bcl-xl inhibition of NLRP1 inflammasome: Loop domain-dependent suppression of ATP binding and oligomerization. Proc. Natl. Acad. Sci. USA 2009, 106, 3935–3940. [Google Scholar] [CrossRef] [PubMed]
- Arlehamn, C.S.; Petrilli, V.; Gross, O.; Tschopp, J.; Evans, T.J. The role of potassium in inflammasome activation by bacteria. J.Biol. Chem. 2010, 285, 10508–10518. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, J.P.; Galante, P.A.; de Souza, S.J. Different evolutionary strategies for the origin of caspase-1 inhibitors. J. Mol. Evol. 2008, 66, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Stehlik, C.; Krajewska, M.; Welsh, K.; Krajewski, S.; Godzik, A.; Reed, J.C. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-κB and pro-caspase-1 regulation. Biochem. J. 2003, 373, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Bedoya, F.; Sandler, L.L.; Harton, J.A. Pyrin-only protein 2 modulates NF-κB and disrupts ASC:CLR interactions. J. Immunol. 2007, 178, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Stehlik, C.; Dorfleutner, A. COPs and POPs: Modulators of inflammasome activity. J. Immunol. 2007, 179, 7993–7998. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Denecker, G.; Kalai, M.; D’Hondt, K.; Meeus, A.; Declercq, W.; Saelens, X.; Vandenabeele, P. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1β generation. J. Biol. Chem. 2004, 279, 51729–51738. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Forster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Dostert, C.; Staehli, F.; Cabalzar, K.; Castillo, R.; Tardivel, A.; Schneider, P.; Tschopp, J. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 2009, 460, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Imamura, R.; Wang, Y.; Kinoshita, T.; Suzuki, M.; Noda, T.; Sagara, J.; Taniguchi, S.; Okamoto, H.; Suda, T. Anti-inflammatory activity of PYNOD and its mechanism in humans and mice. J. Immunol. 2010, 184, 5874–5884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hasegawa, M.; Imamura, R.; Kinoshita, T.; Kondo, C.; Konaka, K.; Suda, T. PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int. Immunol. 2004, 16, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Wang, Y.; Hasegawa, M.; Imamura, R.; Suda, T. PYPAF3, a PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1β secretion. J. Biol. Chem. 2005, 280, 21720–21725. [Google Scholar] [CrossRef] [PubMed]
- Messaed, C.; Akoury, E.; Djuric, U.; Zeng, J.; Saleh, M.; Gilbert, L.; Seoud, M.; Qureshi, S.; Slim, R. NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J. Biol. Chem. 2011, 286, 43313–43323. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science 1995, 267, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 20, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009, 30, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; Flavell, R.A. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Mamantopoulos, M.; Ronchi, F.; van Hauwermeiren, F.; Vieira-Silva, S.; Yilmaz, B.; Martens, L.; Saeys, Y.; Drexler, S.K.; Yazdi, A.S.; Raes, J.; et al. Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 2017, 47, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Loening-Baucke, V.; Vaneechoutte, M.; Doerffel, Y. Active Crohn’s disease and ulcerative colitis can be specifically diagnosed and monitored based on the biostructure of the fecal flora. Inflamm. Bowel Dis. 2008, 14, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Jarnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 2010, 139, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Neut, C.; Bulois, P.; Desreumaux, P.; Membre, J.M.; Lederman, E.; Gambiez, L.; Cortot, A.; Quandalle, P.; van Kruiningen, H.; Colombel, J.F. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Christophi, G.P.; Rong, R.; Holtzapple, P.G.; Massa, P.T.; Landas, S.K. Immune markers and differential signaling networks in ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2012, 18, 2342–2356. [Google Scholar] [CrossRef] [PubMed]
- McAlindon, M.E.; Hawkey, C.J.; Mahida, Y.R. Expression of interleukin 1β and interleukin 1β converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 1998, 42, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, W.; Peeters, P.M.; Staelens, D.; Schraenen, A.; van der Goten, J.; Cleynen, I.; de Schepper, S.; van Lommel, L.; Reynaert, N.L.; Schuit, F.; et al. Strong Upregulation of AIM2 and IFI16 Inflammasomes in the Mucosa of Patients with Active Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranson, N.; Kunde, D.; Eri, R. Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health. Int. J. Mol. Sci. 2017, 18, 2379. https://doi.org/10.3390/ijms18112379
Ranson N, Kunde D, Eri R. Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health. International Journal of Molecular Sciences. 2017; 18(11):2379. https://doi.org/10.3390/ijms18112379
Chicago/Turabian StyleRanson, Nicole, Dale Kunde, and Rajaraman Eri. 2017. "Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health" International Journal of Molecular Sciences 18, no. 11: 2379. https://doi.org/10.3390/ijms18112379
APA StyleRanson, N., Kunde, D., & Eri, R. (2017). Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health. International Journal of Molecular Sciences, 18(11), 2379. https://doi.org/10.3390/ijms18112379