Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders




Abstract
1. Introduction
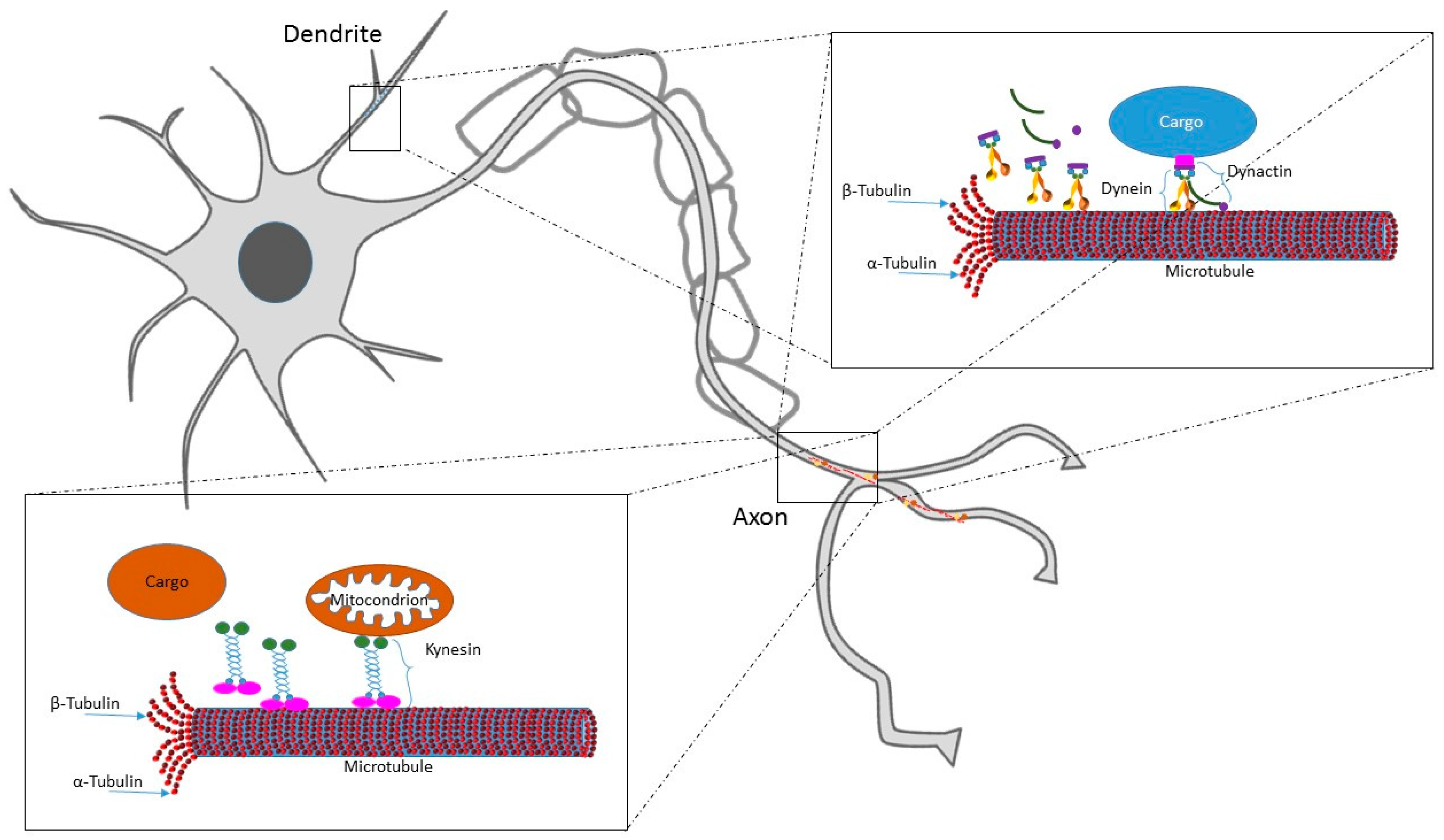
1.1. MTs Role in Neuron Lifecycle
1.2. Pathways Involved in MTs Dynamics Regulation
2. MTs-Linked NDDs
2.1. Autism
2.2. Schizophrenia
2.3. Down Syndrome
2.4. Epilepsy
2.5. Lissencephaly
2.6. Microcephaly
2.7. Intellectual Disability
3. MTs Stabilizing Agents in NDDs
3.1. Brain-Penetrant Microtubule-Stabilizing Compounds: Epothilone D
3.2. Non-Taxane MTs Stabilizers: NAP and D-SAL
3.3. Risperidone
3.4. New Experimental MTs-Acting Compounds
4. Conclusions
Acknowledgments
Authors Contribution
Conflicts of Interest
References
- Casanova, M.F.; Buxhoeveden, D.P.; Brown, C. Clinical and macroscopic correlates of minicolumnar pathology in autism. J. Child Neurol. 2002, 17, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.L.; Casanova, M.F. Autism and dyslexia: A spectrum of cognitive styles as defined by minicolumnar morphometry. Med. Hypotheses 2010, 74, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Beasley, C.L.; Chana, G.; Honavar, M.; Landau, S.; Everall, I.P.; Cotter, D. Evidence for altered neuronal organisation within the planum temporale in major psychiatric disorders. Schizophr. Res. 2005, 73, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Hardan, A.Y.; Jou, R.J.; Keshavan, M.S.; Varma, R.; Minshew, N.J. Increased frontal cortical folding in autism: A preliminary MRI study. Psychiatry Res. 2004, 131, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Palferman, S.; Heavey, L.; Le Couteur, A. Autism: The phenotype in relatives. J. Autism Dev. Disord. 1998, 28, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration; and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Tolić-Nørrelykke, I.M. Force and length regulation in the microtubule cytoskeleton: Lessons from fission yeast. Curr. Opin. Cell Biol. 2010, 22, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.; Gallo, G. Actin filament-microtubule interactions in axon initiation and branching. Brain Res. Bull. 2016, 126 Pt 3, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Ferrari-Toninelli, G.; Montinaro, M.; Memo, M. Notch signalling in adult neurons: A potential target for microtubule stabilization. Ther. Adv. Neurol. Disord. 2013, 6, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.; Hoogenraad, C.C. Centrosomes; microtubules and neuronal development. Mol. Cell Neurosci. 2011, 48, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Stiess, M.; Maghelli, N.; Kapitein, L.C.; Gomis-Rüth, S.; Wilsch-Bräuninger, M.; Hoogenraad, C.C.; Tolić-Nørrelykke, I.M.; Bradke, F. Axon extension occurs independently of centrosomal microtubule nucleation. Science 2010, 327, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, M. The 3Ms of central spindle assembly: Microtubules; motors and MAPs. Nat. Rev. Mol. Cell Biol. 2009, 10, 9–20. [Google Scholar] [CrossRef] [PubMed]
- De Anda, F.C.; Pollarolo, G.; Da Silva, J.S.; Camoletto, P.G.; Feiguin, F.; Dotti, C.G. Centrosome localization determines neuronal polarity. Nature 2005, 436, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J. Neuropathol. Exp. Neurol. 2007, 66, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Rivas, R.J.; Hatten, M.E. Motility and cytoskeletal organization of migrating cerebellar granule neurons. J. Neurosci. 1995, 15, 981–989. [Google Scholar] [PubMed]
- Asada, N.; Sanada, K. LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J. Neurosci. 2010, 30, 8852–8885. [Google Scholar] [CrossRef] [PubMed]
- Ka, M.; Jung, E.M.; Mueller, U.; Kim, W.Y. MACF1 regulates the migration of pyramidal neurons via microtubule dynamics and GSK-3 signaling. Dev. Biol. 2014, 395, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, J.; Fang, A.; Li, R.; Bai, Y.; Kriegstein, A.R.; Wang, X. The dynamics of neuronal migration. Adv. Exp. Med. Biol. 2014, 800, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.P.; Polleux, F. Establishment of axon-dendrite polarity in developing neurons. Annu. Rev. Neurosci. 2009, 32, 347–381. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [PubMed]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.W.; Cai, D.; Verhey, K.J. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xia, J.T.; Feng, Y. Microtubule stability and MAP1B upregulation control neuritogenesis in CAD cells. Acta Pharmacol. Sin. 2006, 27, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Trotta, N.; Orso, G.; Rossetto, M.G.; Daga, A.; Broadie, K. The hereditary spastic paraplegia gene; spastin; regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 2004, 14, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.I.; Moonen, G.; Nguyen, L. Neurotransmitters regulate cell migration in the telencephalon. Eur. J. Neurosci. 2007, 26, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Bhide, P.G. Dopamine, cocaine and the development of cerebral cortical cytoarchitecture: A review of current concepts. Semin. Cell Dev. Biol. 2009, 20, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.F.; Kriegstein, A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, L.; Timmerman, V.; Janssens, S. Microtubule dynamics in the peripheral nervous system: A matter of balance. Bioarchitecture 2011, 1, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.M.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. Modulation of microtubule dynamics by tau in living cells: Implications for development and neurodegeneration. Mol. Biol. Cell 2004, 15, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 2000, 275, 3577–3582. [Google Scholar] [CrossRef] [PubMed]
- Inada, H.; Goto, H.; Tanabe, K.; Nishi, Y.; Kaibuchi, K.; Inagaki, M. Rho-associated kinase phosphorylates desmin; the myogenic intermediate filament protein; at unique amino-terminal sites. Biochem. Biophys. Res. Commun. 1998, 253, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, K.; Kosako, H.; Azuma, I.; Inagaki, N.; Inagaki, M. Possible regulation of intermediate filament proteins by Rho-binding kinases. Subcell. Biochem. 1998, 31, 423–435. [Google Scholar] [PubMed]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef] [PubMed]
- Schofield, A.V.; Steel, R.; Bernard, O. Rho-associated coiled-coil kinase (ROCK) protein controls microtubule dynamics in a novel signaling pathway that regulates cell migration. J. Biol. Chem. 2012, 287, 43620–43629. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, L.; Wei, L. Rho-kinase in development and heart failure: Insights from genetic models. Pediatr. Cardiol. 2011, 32, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Mori, D.; Yamada, M.; Mimori-Kiyosue, Y.; Shirai, Y.; Suzuki, A.; Ohno, S.; Saya, H.; Wynshaw-Boris, A.; Hirotsune, S. An essential role of the aPKC-Aurora A-NDEL1 pathway in neurite elongation by modulation of microtubule dynamics. Nat. Cell Biol. 2009, 11, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, G.K.; Gleeson, J.G. Aurora A moonlights in neurite extension. Nat. Cell Biol. 2009, 11, 1053–1054. [Google Scholar] [CrossRef] [PubMed]
- Ciani, L.; Salinas, P.C. c-Jun N-terminal kinase (JNK) cooperates with Gsk3beta to regulate Dishevelled-mediated microtubule stability. BMC Cell Biol. 2007, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.S.; Hsiao, Y.L.; Lin, C.C.; Yu, C.T.; Hsu, C.M.; Chang, M.S.; Lee, C.I.; Huang, C.Y.; Howng, S.L.; Hong, Y.R. Glycogen synthase kinase 3beta interacts with and phosphorylates the spindle-associated protein astrin. J. Biol. Chem. 2008, 283, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J.G.; Stephens, D.J.; Tavaré, J.M. A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. J. Cell Sci. 2003, 116 Pt 4, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Munji, R.N.; Choe, Y.; Li, G.; Siegenthaler, J.A.; Pleasure, S.J. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J. Neurosci. 2011, 31, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fothergill, T.; Hutchins, B.I.; Dent, E.W.; Kalil, K. Wnt5a evokes cortical axon outgrowth and repulsive guidance by tau mediated reorganization of dynamic microtubules. Dev. Neurobiol. 2014, 74, 797–817. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.; Honavar, M.; Lovestone, S.; Raymond, L.; Kerwin, R.; Anderton, B.; Everall, I. Disturbance of Notch-1 and Wnt signalling proteins in neuroglial balloon cells and abnormal large neurons in focal cortical dysplasia in human cortex. Acta Neuropathol. 1999, 98, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.J.; Martinez-Garay, I.; Gil-Sanz, C.; Harkins-Perry, S.R.; Müller, U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 2011, 69, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Shahwan, S.A.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96, Erratum in 2001, 27, 225. [Google Scholar]
- Persico, A.M.; D’Agruma, L.; Maiorano, N.; Totaro, A.; Militerni, R.; Bravaccio, C.; Wassink, T.H.; Schneider, C.; Melmed, R.; Trillo, S.; et al. Collaborative Linkage Study of Autism. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 2001, 6, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Torii, K.; Torii, M.; Sarkisian, M.R.; Bartley, C.M.; Shen, J.; Radtke, F.; Gridley, T.; Sestan, N.; Rakic, P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron 2008, 60, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Gaiano, N. Strange bedfellows: Reelin and Notch signaling interact to regulate cell migration in the developing neocortex. Neuron 2008, 60, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Toninelli, G.; Bonini, S.A.; Uberti, D.; Napolitano, F.; Stante, M.; Santoro, F.; Minopoli, G.; Zambrano, N.; Russo, T.; Memo, M. Notch activation induces neurite remodeling and functional modifications in SH-SY5Y neuronal cells. Dev. Neurobiol. 2009, 69, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Toninelli, G.; Bonini, S.A.; Bettinsoli, P.; Uberti, D.; Memo, M. Microtubule stabilizing effect of notch activation in primary cortical neurons. Neuroscience 2008, 154, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Memo, M. Nuclear factor-kappaB/Rel proteins: A point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochem. Pharmacol. 1999, 57, 1–7. [Google Scholar] [CrossRef]
- Gutierrez, H.; Davies, A.M. Regulation of neural process growth; elaboration and structural plasticity by NF-κB. Trends Neurosci. 2011, 34, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Gavaldà, N.; Gutierrez, H.; Davies, A.M. Developmental switch in NF-kappaB signalling required for neurite growth. Development 2009, 136, 3405–3412. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Ferrari-Toninelli, G.; Uberti, D.; Montinaro, M.; Buizza, L.; Lanni, C.; Grilli, M.; Memo, M. Nuclear factor κB-dependent neurite remodeling is mediated by Notch pathway. J. Neurosci. 2011, 31, 11697–11705. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Mastinu, A.; Maccarinelli, G.; Mitola, S.; Premoli, M.; la Rosa, L.R.; Ferrari-Toninelli, G.; Grilli, M.; Memo, M. Cortical Structure Alterations and Social Behavior Impairment in p50-Deficient Mice. Cereb. Cortex 2016, 26, 2832–2849. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, X.; Ma, Y.; Calicchio, M.L.; Kong, D.; Teng, Y.D.; Yu, L.; Crain, A.M.; Vartanian, T.K.; Pasqualini, R.; et al. Nna1 mediates Purkinje cell dendritic development via lysyl oxidase propeptide and NF-κB signaling. Neuron 2010, 68, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Tischfield, M.A.; Cederquist, G.Y.; Gupta, M.L., Jr.; Engle, E.C. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 2011, 21, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; van den Ende, J.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Kapitansky, O.; Ivashco-Pachima, Y.; Malishkevich, A.; Giladi, E.; Skalka, N.; Rosin-Arbesfeld, R.; Mittelman, L.; Segev, O.; Hirsch, J.A.; et al. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 2014, 19, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Ivashko-Pachima, Y.; Gozes, I. The ADNP derived peptide; NAP modulates the tubulin pool: Implication for neurotrophic and neuroprotective activities. PLoS ONE 2012, 7, e51458. [Google Scholar] [CrossRef] [PubMed]
- Jouroukhin, Y.; Ostritsky, R.; Assaf, Y.; Pelled, G.; Giladi, E.; Gozes, I. NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: Protection against impairments in axonal transport. Neurobiol. Dis. 2013, 56, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Bakos, J.; Bacova, Z.; Grant, S.G.; Castejon, A.M.; Ostatnikova, D. Are Molecules Involved in Neuritogenesis and Axon Guidance Related to Autism Pathogenesis? Neuromol. Med. 2015, 17, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Kozma, R.; Sarner, S.; Ahmed, S.; Lim, L. Rho family GTPases and neuronal growth cone remodelling: Relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell Biol. 1997, 17, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Lee, C.; Chen, L.; Galvan, L.; Cepeda, C.; Chen, J.Y.; Peñagarikano, O.; Stein, J.L.; Li, A.; Oguro-Ando, A.; et al. JAKMIP1; a Novel Regulator of Neuronal Translation; Modulates Synaptic Function and Autistic-like Behaviors in Mouse. Neuron 2015, 88, 1173–1191. [Google Scholar] [CrossRef] [PubMed]
- Steindler, C.; Li, Z.; Algarté, M.; Alcover, A.; Libri, V.; Ragimbeau, J.; Pellegrini, S. Jamip1 (marlin-1) defines a family of proteins interacting with janus kinases and microtubules. J. Biol. Chem. 2004, 279, 43168–43177. [Google Scholar] [CrossRef] [PubMed]
- Deloulme, J.C.; Gory-Fauré, S.; Mauconduit, F.; Chauvet, S.; Jonckheere, J.; Boulan, B.; Mire, E.; Xue, J.; Jany, M.; Maucler, C.; et al. Microtubule-associated protein 6 mediates neuronal connectivity through Semaphorin 3E-dependent signalling for axonal growth. Nat. Commun. 2015, 6, 7246. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ma, Y.; Liu, J.; Ding, C.; Hu, F.; Yu, L. Proteomic analysis of cortical brain tissue from the BTBR mouse model of autism: Evidence for changes in STOP and myelin-related proteins. Neuroscience 2016, 312, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Sun, S.; Li, Y.; Yu, S. Reduced plasma levels of microtubule-associated STOP/MAP6 protein in autistic patients. Psychiatry Res. 2016, 245, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, A.; Salin, P.A.; Vernet, M.; Kujala, P.; Baratier, J.; Gory-Fauré, S.; Bosc, C.; Pointu, H.; Proietto, D.; Schweitzer, A.; et al. The suppression of brain cold-stable microtubules in mice induces synaptic defects associated with neuroleptic-sensitive behavioral disorders. Genes Dev. 2002, 16, 2350–2364. [Google Scholar] [CrossRef] [PubMed]
- Fournet, V.; Schweitzer, A.; Chevarin, C.; Deloulme, J.C.; Hamon, M.; Giros, B.; Andrieux, A.; Martres, M.P. The deletion of STOP/MAP6 protein in mice triggers highly altered mood and impaired cognitive performances. J. Neurochem. 2012, 121, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Sowell, S.M.; Luo, Y.; Chaubey, A.; Cameron, R.S.; Kim, H.G.; Srivastava, A.K. Autism and Intellectual Disability-Associated KIRREL3 Interacts with Neuronal Proteins MAP1B and MYO16 with Potential Roles in Neurodevelopment. PLoS ONE 2015, 10, e0123106. [Google Scholar] [CrossRef] [PubMed]
- Guerin, A.; Stavropoulos, D.J.; Diab, Y.; Chénier, S.; Christensen, H.; Kahr, W.H.; Babul-Hirji, R.; Chitayat, D. Interstitial deletion of 11q-implicating the KIRREL3 gene in the neurocognitive delay associated with Jacobsen syndrome. Am. J. Med. Genet. A 2012, 158A, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Henríquez, D.R.; Bodaleo, F.J.; Montenegro-Venegas, C.; González-Billault, C. The light chain 1 subunit of the microtubule-associated protein 1B (MAP1B) is responsible for Tiam1 binding and Rac1 activation in neuronal cells. PLoS ONE 2012, 7, e53123. [Google Scholar] [CrossRef] [PubMed]
- Pangratz-Fuehrer, S.; Bubna-Littitz, H.; Propst, F.; Reitsamer, H. Mice deficient in microtubule-associated protein MAP1B show a distinct behavioral phenotype and altered retina function. Behav. Brain Res. 2005, 164, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Yu, C.E.; Yu, J.; Munson, J.; Chen, D.; Hua, W.; Estes, A.; Cortes, F.; de la Barra, F.; Yu, D.; et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 2002, 80, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Hoshino, M. Neuronal Migration and AUTS2 Syndrome. Brain Sci. 2017, 7, E54. [Google Scholar] [CrossRef]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, T.; Chihama, K.; Nabeshima, Y.; Hoshino, M. The in vivo roles of STEF/Tiam1; Rac1 and JNK in cortical neuronal migration. EMBO J. 2003, 22, 4190–4201. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.A.; Liedén, A.; Westerlund, J.; Bremer, A.; Wincent, J.; Sahlin, E.; Gillberg, C.; Fernell, E.; Anderlid, B.M. Rare copy number variants are common in young children with autism spectrum disorder. Acta Paediatr. 2015, 104, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Cornell, B.; Wachi, T.; Zhukarev, V.; Toyo-Oka, K. Regulation of neuronal morphogenesis by 14–3-3epsilon (Ywhae) via the microtubule binding protein; doublecortin. Hum. Mol. Genet. 2016, 25, 4610. [Google Scholar] [CrossRef] [PubMed]
- Matenia, D.; Mandelkow, E.M. The tau of MARK: A polarized view of the cytoskeleton. Trends Biochem. Sci. 2009, 34, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Bernard, L.P.; Zhang, H. MARK/Par1 Kinase Is Activated Downstream of NMDA Receptors through a PKA-Dependent Mechanism. PLoS ONE 2015, 10, e0124816. [Google Scholar] [CrossRef] [PubMed]
- Maussion, G.; Carayol, J.; Lepagnol-Bestel, A.M.; Tores, F.; Loe-Mie, Y.; Milbreta, U.; Rousseau, F.; Fontaine, K.; Renaud, J.; Moalic, J.M.; et al. Convergent evidence identifying MAP/microtubule affinity-regulating kinase 1 (MARK1) as a susceptibility gene for autism. Hum. Mol. Genet. 2008, 17, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Fricano-Kugler, C.J.; Getz, S.A.; Skelton, P.D.; Lee, J.; Rizzuto, C.P.; Geller, J.S.; Li, M.; Luikart, B.W. A Retroviral CRISPR-Cas9 System for Cellular Autism-Associated Phenotype Discovery in Developing Neurons. Sci. Rep. 2016, 6, 25611. [Google Scholar] [CrossRef] [PubMed]
- Karam, C.S.; Ballon, J.S.; Bivens, N.M. Signaling Pathways in Schizophrenia: Emerging targets and therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Landek-Salgado, M.A.; Faust, T.E.; Sawa, A. Molecular substrates of schizophrenia: Homeostatic signaling to connectivity. Mol. Psychiatry 2016, 21, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.K.; Bishop, J.R.; Palumbo, D.; Sweeney, J.A. Effect of second-generation antipsychotics on cognition: Current issues and future challenges. Expert Rev. Neurother. 2010, 10, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of Schizophrenia; revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; van Leeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in Schizophrenia. Neurosci. 2013, 251, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Moehle, M.S.; Luduena, R.F.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Regional differences in expression of β-tubulin isoforms in schizophrenia. Schizophr. Res. 2012, 135, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Bulinski, J.C. Post-translational regulation of the microtubule cytoskeleton: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton 2016, 73, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Pu, J.; Hunter, I.; Liu, M.; Martin-Granados, C.; Reilly, T.J.; Gao, G.D.; Guan, Z.L.; Li, W.D.; Shi, Y.Y.; et al. Recurrent deletions of ULK4 in schizophrenia: A gene crucial for neuritogenesis and neuronal motility. J. Cell Sci. 2014, 127 Pt 3, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Magiera, M.M.; Janke, C. Post-translational modifications of tubulin. Curr. Biol. 2014, 24, R351–R354. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Brady, S.T. Posttranslational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Fullston, T.; Gabb, B.; Callen, D.; Ullmann, R.; Woollatt, E.; Bain, S.; Ropers, H.H.; Cooper, M.; Chandler, D.; Carter, K.; et al. Inherited balanced translocation t(9; 17)(q33.2; q25.3) concomitant with a 16p13.1 duplication in a patient with schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Sweet, R.A. Auditory dysfunction in schizophrenia: Integrating clinical and basic features. Nat. Rev. Neurosci. 2015, 16, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.A.; Newman, J.T.; Gu, H.; Sampson, A.R.; Fish, K.N.; MacDonald, M.L.; Moyer, C.E.; DiBitetto, J.V.; Dorph-Petersen, K.A.; Penzes, P.; et al. Loss of Microtubule-Associated Protein 2 Immunoreactivity Linked to Dendritic Spine Loss in Schizophrenia. Biol. Psychiatry 2015, 78, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, J.; Savarin, P.; Gans, P.; Hamon, L.; Clément, M.J.; David, M.O.; Bosc, C.; Andrieux, A. Curmi, Structural Basis for the Association of MAP6 Protein with Microtubules and Its Regulation by Calmodulin. J. Biol. Chem. 2013, 288, 24910–24922. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, N.J.; Porteous, D.J. DISC1-binding proteins in neural development; signalling and schizophrenia. Neuropharmacology 2012, 62, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Jin, C.; Zhou, Z.; Zhang, F.; Yuan, J.; Liu, X.; Cheng, Z. Association study of DISC1 genetic variants with the risk of schizophrenia. Psychiatr. Genet. 2016, 26, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Dresner, E.; Agam, G.; Gozes, I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: Deregulation in schizophrenia. Eur. Neuropsychopharmacol. 2011, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Merenlender-Wagner, A.; Malishkevich, A.; Shemer, Z.; Udawela, M.; Gibbons, A.; Scarr, E.; Dean, B.; Levine, J.; Agam, G.; Gozes, I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry 2015, 20, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Itoh, T.J.; Kimura, T.; Ménager, C.; Nishimura, T.; Shiromizu, T.; Watanabe, H.; Inagaki, N.; Iwamatsu, A.; Hotani, H.; et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 2002, 4, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Fallin, M.D.; Lasseter, V.K.; Liu, Y.; Avramopoulos, D.; McGrath, J.; Wolyniec, P.S.; Nestadt, G.; Liang, K.Y.; Chen, P.L.; Valle, D.; et al. Linkage and association on 8p21.2-p21.1 in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Edgar, P.F.; Douglas, J.E.; Cooper, G.J.; Dean, B.; Kydd, R.; Faull, R.L. Comparative proteome analysis of the hippocampus implicates chromosome 6q in schizophrenia. Mol. Psychiatry 2000, 5, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E.; Lyle, R.; Dermitzakis, E.T.; Reymond, A.; Deutsch, S. Chromosome 21 and down syndrome: From genomics to pathophysiology. Nat. Rev. Genet. 2004, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Engidawork, E.; Lubec, G. Molecular changes in fetal Down syndrome brain. J. Neurochem. 2003, 84, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Nielsen, J.A.; Ferguson, M.A.; Burback, M.C.; Cox, E.T.; Dai, L.; Gerig, G.; Edgin, J.O.; Korenberg, J.R. Abnormal brain synchrony in Down Syndrome. NeuroImage Clin. 2013, 2, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Falsafi, S.K.; Dierssen, M.; Ghafari, M.; Pollak, A.; Lubec, G. Reduced cortical neurotransmitter receptor complex levels in fetal Down syndrome brain. Amino Acids 2016, 48, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Gulesserian, T.; Kim, S.H.; Fountoulakis, M.; Lubec, G. Aberrant expression of centractin and capping proteins; integral constituents of the dynactin complex; in fetal down syndrome brain. Biochem. Biophys. Res. Commun. 2002, 291, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Okabayashi, S.; Ono, F. Dynein dysfunction disrupts intracellular vesicle trafficking bidirectionally and perturbs synaptic vesicle docking via endocytic disturbances a potential mechanism underlying age-dependent impairment of cognitive function. Am. J. Pathol. 2012, 180, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Soppa, U.; Schumacher, J.; Florencio Ortiz, V.; Pasqualon, T.; Tejedor, F.J.; Becker, W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27Kip1 and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014, 13, 2084–2100. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chung, K.C. New Perspectives of Dyrk1A Role in Neurogenesis and Neuropathologic Features of Down Syndrome. Exp. Neurobiol. 2013, 22, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Dowjat, K.; Adayev, T.; Kaczmarski, W.; Wegiel, J.; Hwang, Y.-W. Gene-Dosage-Dependent Association of DYRK1A with the Cytoskeleton in the Brain and Lymphocytes of Down Syndrome Patients. J. Neuropathol. Exp. Neurol. 2012, 71, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Yang, E.J.; Yoon, J.H.; Chung, K.C. Dyrk1A overexpression in immortalized hippocampal cells produces the neuropathological features of Down syndrome. Mol. Cell Neurosci. 2007, 36, 270–279. [Google Scholar] [CrossRef] [PubMed]
- De Martinez Lagran, M.; Benavides-Piccione, R.; Ballesteros-Yañez, I.; Calvo, M.; Morales, M.; Fillat, C.; Defelipe, J.; Ramakers, G.J.; Dierssen, M. Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb. Cortex 2012, 22, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, F.J.; Hämmerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy: Definitions proposed by the International League against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Leventer, R.J.; Guerrini, R.; Dobyns, W.B. Malformations of cortical development and epilepsy. Dialogues Clin. Neurosci. 2008, 10, 47–62. [Google Scholar] [PubMed]
- Kim, Y.O.; Nam, T.S.; Park, C.; Kim, S.K.; Yoon, W.; Choi, S.Y.; Kim, M.K.; Woo, Y.J. Familial pachygyria in both genders related to a DCX mutation. Brain Dev. 2016, 38, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, A.; van Haren, J.; Ribeiro, S.A.; Wittmann, T. Doublecortin is excluded from growing microtubule ends and recognizes the GDP-microtubule lattice. Curr. Biol. 2016, 26, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Huang, M.; Xiao, F.; Xi, Z. Echinoderm microtubule-associated protein -like protein 5 in anterior temporal neocortex of patients with intractable epilepsy. Iran. J. Basic Med. Sci. 2015, 18, 1008–1013. [Google Scholar] [PubMed]
- Eichenmuller, B.; Everley, P.; Palange, J.; Lepley, D.; Suprenant, K.A. The human EMAP-like protein-70 (ELP70) is a microtubule destabilizer that localizes to the mitotic apparatus. J. Biol. Chem. 2002, 277, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Thom, M.; Sisodiya, S.M.; Beckett, A.; Martinian, L.; Lin, W.R.; Harkness, W.; Mitchell, T.N.; Craig, J.; Duncan, J.; Scaravilli, F. Cytoarchitectural abnormalities in hippocampal sclerosis. J. Neuropathol. Exp. Neurol. 2002, 61, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Longo, B.; Vezzani, A.; Mello, L.E. Growth-associated protein 43 expression in hippocampal molecular layer of chronic epileptic rats treated with cycloheximide. Epilepsia 2005, 46 (Suppl. 5), 125–128. [Google Scholar] [CrossRef] [PubMed]
- An, S.J.; See, M.O.; Kim, H.S.; Park, S.K.; Hwang, I.K.; Won, M.H.; Kang, T.C. Accumulation of microtubule-associated proteins in the hippocampal neurons of seizure-sensitive gerbils. Mol. Cells 2003, 15, 200–207. [Google Scholar] [PubMed]
- Yang, J.W.; Czech, T.; Felizardo, M.; Baumgartner, C.; Lubec, G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids 2006, 30, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, X.F.; Mo, X.A.; Li, J.M.; Yuan, J.; Zheng, J.O.; Feng, Y.; Tang, M. Expression of laminin β1 and integrin α2 in the anterior temporal neocortex tissue of patients with intractable epilepsy. Int. J. Neurosci. 2011, 121, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.L.; Xing, S.G.; Deng, C.Y.; Ju, X.C.; Jiang, X.Y.; Luo, Z.G. Laminin/β1 integrin signal triggers axon formation by promoting microtubule assembly and stabilization. Cell Res. 2012, 22, 954–972. [Google Scholar] [CrossRef] [PubMed]
- Bonar, N.A.; Petersen, C.P. Integrin suppresses neurogenesis and regulates brain tissue assembly in planarian regeneration. Development 2017, 144, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hu, Y.; Xiong, Y.; Li, Z.; Wang, W.; Du, C.; Yang, Y.; Zhang, Y.; Xiao, F.; Wang, X. Association of Microtubule Dynamics with Chronic Epilepsy. Mol. Neurobiol. 2016, 53, 5013–5024. [Google Scholar] [CrossRef] [PubMed]
- Sapir, T.; Elbaum, M.; Reiner, O. Reduction of microtubule catastrophe events by LIS1; platelet-activating factor acetylhydrolase subunit. EMBO J. 1997, 16, 6977–6984. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.S.; Niethammer, M.; Ayala, R.; Zhou, Y.; Gambello, M.J.; Wynshaw-Boris, A.; Tsai, L.H. Regulation of cytoplasmic dynein behaviour and microtubule organization bymammalian Lis1. Nat. Cell. Biol. 2000, 2, 767–775. [Google Scholar] [PubMed]
- Reiner, O.; Sapir, T. LIS1 functions in normal development and disease. Curr. Opin. Neurobiol. 2013, 23, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Pilz, D.T.; Matsumoto, N.; Minnerath, S.; Mills, P.; Gleeson, J.G.; Allen, K.M.; Walsh, C.A.; Barkovich, A.J.; Dobyns, W.B.; Ledbetter, D.H.; et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly; but different patterns of malformation. Hum. Mol. Genet. 1998, 7, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Des Portes, V.; Carrié, A.; Billuart, P.; Kieffer, V.; Bienvenu, T.; Vinet, M.C.; Beldjord, C.; Kahn, A.; Ponsot, G.; Chelly, J.; et al. Inherited microdeletion in Xp21.3–22.1 involved in non-specific mental retardation. Clin. Genet. 1998, 53, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Moores, C.A.; Perderiset, M.; Francis, F.; Chelly, J.; Houdusse, A.; Milligan, R.A. Mechanism of microtubule stabilization by doublecortin. Mol. Cell 2004, 14, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Prokscha, A.; Ungewickell, E.; Eichele, G. Doublecortin association with actin filaments is regulated by neurabin II. J. Biol. Chem. 2005, 280, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Segal, M.; Reiner, O. Doublecortin supports the development of dendritic arbors in primary hippocampal neurons. Dev. Neurosci. 2008, 30, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z.; et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007, 128, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Keays, D.A.; Francis, F.; Saillour, Y.; Bahi, N.; Manouvrier, S.; Fallet-Bianco, C.; Pasquier, L.; Toutain, A.; Tuy, F.P.; et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum. Mutat. 2007, 28, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Jaglin, X.H.; Poirier, K.; Saillour, Y.; Buhler, E.; Tian, G.; Bahi-Buisson, N.; Fallet-Bianco, C.; Phan-Dinh-Tuy, F.; Kong, X.P.; bo Mont, P.; et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009, 41, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Frotscher, M. How does Reelin signaling regulate the neuronal cytoskeleton during migration? Neurogenesis 2016, 3, e1242455. [Google Scholar] [CrossRef] [PubMed]
- Thornton, G.K.; Woods, C.G. Primary microcephaly: Do all roads lead to Rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Mochida, G.H. Genetics and biology of microcephaly and lissencephaly. Semin. Pediatr. Neurol. 2009, 16, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Zollo, M.; Ahmed, M.; Ferrucci, V.; Salpietro, V.; Asadzadeh, F.; Carotenuto, M.; Maroofian, R.; Al-Amri, A.; Singh, R.; Scognamiglio, I.; et al. PRUNE is crucial for normal brain development and mutated in microcephaly with neurodevelopmental impairment. Brain 2017, 140, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Lockrow, J.P.; Holden, K.R.; Dwivedi, A.; Matheus, M.G.; Lyons, M.J. LIS1 duplication: Expanding the phenotype. J. Child Neurol. 2012, 27, 791–795. [Google Scholar] [CrossRef] [PubMed]
- McNeely, K.C.; Cupp, T.D.; Little, J.N.; Janisch, K.M.; Shrestha, A.; Dwyer, N.D. Mutation of Kinesin-6 Kif20b causes defects in cortical neuron polarization and morphogenesis. Neural Dev. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Wen, X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Larti, F.; Kahrizi, K.; Musante, L.; Hu, H.; Papari, E.; Fattahi, Z.; Bazazzadegan, N.; Liu, Z.; Banan, M.; Garshasbi, M.; et al. A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur. J. Hum. Genet. 2015, 23, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Kevenaar, J.T.; Bianchi, S.; van Spronsen, M.; Olieric, N.; Lipka, J.; Frias, C.P.; Mikhaylova, M.; Harterink, M.; Keijzer, N.; Wulf, P.S.; et al. Kinesin-Binding Protein Controls Microtubule Dynamics and Cargo Trafficking by Regulating Kinesin Motor Activity. Curr. Biol. 2016, 26, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.H.; Ba, W.; Wissink-Lindhout, W.M.; de Brouwer, A.P.; Haas, S.A.; Bienek, M.; Hu, H.; Vissers, L.E.; van Bokhoven, H.; Kalscheuer, V.; et al. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 2014, 51, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Esmaeeli Nieh, S.; Madou, M.R.; Sirajuddin, M.; Fregeau, B.; McKnight, D.; Lexa, K.; Strober, J.; Spaeth, C.; Hallinan, B.E.; Smaoui, N.; et al. De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Ann. Clin. Transl. Neurol. 2015, 2, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Hempel, M.; Cremer, K.; Ockeloen, C.W.; Lichtenbelt, K.D.; Herkert, J.C.; Denecke, J.; Haack, T.B.; Zink, A.M.; Becker, J.; Wohlleber, E.; et al. De Novo mutations in CHAMP1 cause intellectual disability with severe speech impairment. Am. J. Hum. Genet. 2015, 97, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Isidor, B.; Küry, S.; Rosenfeld, J.A.; Besnard, T.; Schmitt, S.; Joss, S.; Davies, S.J.; Lebel, R.R.; Henderson, A.; Schaaf, C.P.; et al. De novo truncating mutations in the Kinetochore-microtubules attachment gene CHAMP1 cause syndromic intellectual disability. Hum. Mutat. 2016, 37, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.J.; Cho, M.T.; Retterer, K.; Jones, J.R.; Nowak, C.; Douglas, J.; Jiang, Y.H.; McConkie-Rosell, A.; Schaefer, G.B.; Kaylor, J.; et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay; intellectual disability; and dysmorphic facial features. Cold Spring Harb. Mol. Case Stud. 2016, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Bartholdi, D.; Stray-Pedersen, A.; Azzarello-Burri, S.; Kibaek, M.; Kirchhoff, M.; Oneda, B.; Rødningen, O.; Schmitt-Mechelke, T.; Rauch, A.; Kjaergaard, S. A newly recognized 13q12.3 microdeletion syndrome characterized by intellectual disability; microcephaly; and eczema/atopic dermatitis encompassing the HMGB1 and KATNAL1 genes. Am. J. Med. Genet. A 2014, 164A, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.; Lassi, G.; Hoerder-Suabedissen, A.; Tinarelli, F.; Simon, M.M.; Wilcox, A.; Lau, P.; Lawson, T.N.; Johnson, S.; Rutman, A.; et al. A missense mutation in Katnal1 underlies behavioural; neurological and ciliary anomalies. Mol. Psychiatry 2017, in press. [Google Scholar] [CrossRef]
- Gholkar, A.A.; Senese, S.; Lo, Y.C.; Vides, E.; Contreras, E.; Hodara, E.; Capri, J.; Whitelegge, J.P.; Torres, J.Z. The X-Linked-Intellectual-Disability-Associated Ubiquitin Ligase Mid2 Interacts with Astrin and Regulates Astrin Levels to Promote Cell Division. Cell Rep. 2016, 14, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kolman, A. Epothilone D (Kosan/Roche). Curr. Opin. Investig. Drugs 2004, 5, 657–667. [Google Scholar] [PubMed]
- Nettles, J.H.; Li, H.; Cornett, B.; Krahn, J.M.; Snyder, J.P.; Downing, K.H. The binding mode of epothilone A on alpha; beta-tubulin by electron crystallography. Science 2004, 305, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.; Wang, S.; Li, M.; Nan, L.; Rhie, J.K.; Covey, J.M.; Zhang, R.; Hill, D.L. Preclinical pharmacology of epothilone D; a novel tubulin-stabilizing antitumor agent. Cancer Chemother. Pharmacol. 2005, 56, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.M.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, A.; Salin, P.; Schweitzer, A.; Bégou, M.; Pachoud, B.; Brun, P.; Gory-Fauré, S.; Kujala, P.; Suaud-Chagny, M.F.; Höfle, G.; et al. Microtubule stabilizer ameliorates synaptic function and behavior in a mouse model for schizophrenia. Biol. Psychiatry 2006, 60, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Spivak-Pohis, I.; Gozes, I. ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J. Mol. Neurosci. 2008, 35, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I. Activity-dependent neuroprotective protein (ADNP): From autism to Alzheimer’s disease. Springerplus 2015, 4 (Suppl. 1), L37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wittmann, T. +TIPs: SxIPping along microtubule ends. Trends Cell Biol. 2012, 22, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Quraishe, S.; Cowan, C.M.; Mudher, A. NAP (davunetide) rescues neuronal dysfunction in a Drosophila model of tauopathy. Mol. Psychiatry 2013, 18, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Divinski, I.; Holtser-Cochav, M.; Vulih-Schultzman, I.; Steingart, R.A.; Gozes, I. Peptide neuroprotection through specific interaction with brain tubulin. J. Neurochem. 2006, 98, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Vaisburd, S.; Shemer, Z.; Yeheskel, A.; Giladi, E.; Gozes, I. Risperidone and NAP protect cognition and normalize gene expression in a schizophrenia mouse model. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Buchanan, R.W.; Keefe, R.S.; Kern, R.; McMahon, R.P.; Green, M.F.; Lieberman, J.; Goff, D.C.; Csernansky, J.G.; McEvoy, J.P.; et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 2012, 136, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Merenlender-Wagner, A.; Shemer, Z.; Touloumi, O.; Lagoudaki, R.; Giladi, E.; Andrieux, A.; Grigoriadis, N.C.; Gozes, I. New horizons in schizophrenia treatment: Autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 2014, 10, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Zemlyak, I.; Manley, N.; Vulih-Shultzman, I.; Cutler, A.B.; Graber, K.; Sapolsky, R.M.; Gozes, I. The microtubule interacting drug candidate NAP protects against kainic acid toxicity in a rat model of epilepsy. J. Neurochem. 2009, 111, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Toso, L.; Cameroni, I.; Roberson, R.; Abebe, D.; Bissell, S.; Spong, C.Y. Prevention of developmental delays in a Down syndrome mouse model. Obstet. Gynecol. 2008, 112, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; McGurk, S.R. The effects of clozapine; risperidone; and olanzapine on cognitive function in schizophrenia. Schizophr. Bull. 1999, 25, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Politte, L.C.; Henry, C.A.; McDougle, C.J. Psychopharmacological interventions in autism spectrum disorder. Harv. Rev. Psychiatry 2014, 22, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Delotterie, D.; Ruiz, G.; Brocard, J.; Schweitzer, A.; Roucard, C.; Roche, Y.; Suaud-Chagny, M.F.; Bressand, K.; Andrieux, A. Chronic administration of atypical antipsychotics improves behavioral and synaptic defects of STOP null mice. Psychopharmacology 2010, 208, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Altun, A.; Ugur-Altun, B. Melatonin: Therapeutic and clinical utilization. Int. J. Clin. Pract. 2007, 61, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Benítez-King, G. Melatonin as a cytoskeletal modulator: Implications for cell physiology and disease. J. Pineal. Res. 2006, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, J.; Maldonado, V.; Ortega, A. Effect of melatonin on beta-tubulin and MAP2 expression in NIE-115 cells. Neurochem. Res. 1996, 21, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Gómez, B.; Velázquez-Paniagua, M.; Cisneros, L.O.; Reyes-Vázquez, C.; Jiménez-Trejo, F.; Reyes, M.E.; Mendoza-Torreblanca, J.; Gutiérrez-Ospina, G. Melatonin attenuates the decrement of dendritic protein MAP-2 immuno-staining in the hippocampal CA1 and CA3 fields of the aging male rat. Neurosci. Lett. 2008, 448, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Benitez-King, G.; Ramírez-Rodríguez, G.; Ortíz, L.; Meza, I. The neuronal cytoskeleton as a potential therapeutical target in neurodegenerative diseases and schizophrenia. Curr. Drug. Targets CNS Neurol. Disord. 2004, 3, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Baulieu, E.E. 3β-Methoxy-pregnenolone (MAP4343) as an innovative therapeutic approach for depressive disorders. Proc. Natl. Acad. Sci. USA 2012, 109, 1713–1718, Erratum in 2012, 109, 4708. [Google Scholar] [CrossRef] [PubMed]
- Parésys, L.; Hoffmann, K.; Froger, N.; Bianchi, M.; Villey, I.; Baulieu, E.E.; Fuchs, E. Effects of the Synthetic Neurosteroid: 3β-Methoxypregnenolone (MAP4343) on Behavioral and Physiological Alterations Provoked by Chronic Psychosocial Stress in Tree Shrews. Int. J. Neuropsychopharmacol. 2016, 19, pyv119. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Fellous, A.; Baulieu, E.E.; Robel, P. Pregnenolone binds to microtubule-associated protein 2 and stimulates microtubule assembly. Proc. Natl. Acad. Sci. USA 2000, 97, 3579–3584, Erratum in 2000, 97, 9819. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.G.; Owen, R.; Gordon-Weeks, P.R. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J. Cell Sci. 1999, 112 Pt 19, 3373–3384. [Google Scholar] [PubMed]
- Yamada, M.; Yoshida, Y.; Mori, D.; Takitoh, T.; Kengaku, M.; Umeshima, H.; Takao, K.; Miyakawa, T.; Sato, M.; Sorimachi, H.; et al. Inhibition of calpain increases LIS1 expression and partially rescues in vivo phenotypes in a mouse model of lissencephaly. Nat. Med. 2009, 15, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Toba, S.; Tamura, Y.; Kumamoto, K.; Yamada, M.; Takao, K.; Hattori, S.; Miyakawa, T.; Kataoka, Y.; Azuma, M.; Hayasaka, K.; et al. Post-natal treatment by a blood-brain-barrier permeable calpain inhibitor, SNJ1945 rescued defective function in lissencephaly. Sci. Rep. 2013, 3, 1224. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.; Smith, A.B. Non-Naturally Occurring Small Molecule Microtubule-Stabilizing Agents: A Potential Tactic for CNS-Directed Therapies. ACS Chem. Neurosci. 2017, 8, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ayral-Kaloustian, S.; Nguyen, T.; Afragola, J.; Hernandez, R.; Lucas, J.M.; Gibbons, J.; Beyer, C. Synthesis and SAR of [1,2,4]triazolo[1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition. J. Med. Chem. 2007, 50, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ayral-Kaloustian, S.; Nguyen, T.; Hernandez, R.; Lucas, J.; Discafani, C.; Beyer, C. Synthesis and SAR of 6-chloro-4-fluoroalkylamino-2-heteroaryl-5-(substituted)phenylpyrimidines as anti-cancer agents. Bioorg. Med. Chem. 2009, 17, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lou, K.; Yao, Y.; Hoye, A.T.; James, M.J.; Cornec, A.S.; Hyde, E.; Gay, B.; Lee, V.M.; Trojanowski, J.Q.; Smith, A.B.; et al. Brain-penetrant, orally bioavailable microtubule-stabilizing small molecules are potential candidate therapeutics for Alzheimer’s disease and related tauopathies. J. Med. Chem. 2014, 57, 6116–6127. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Genes/Protein | Effects | Ref. |
|---|---|---|
| ADNP | ADNP mut cause intellectual disability, facial dysmorphisms | [66,67,68,69] |
| ADNP−/− mice are not viable due to failure of neural tube closure | ||
| ADNP−/+ mice present tauopathy and neuronal cell death, deficit in social behavior and object recognition | ||
| ADNP knock down in cells provokes decreased number of neurites and decreased number and size of embryoid bodies | ||
| Slit/Robo | Increased expression of Slit 1, decreased expression of ABL1 and Cdc42 are associated to alteration in neurite branching and axon pathfinding | [70,71] |
| JAKMIP1 | JAKMIP1 overexpression causes the formation of tight and stable MTs boundles in human cell lines | [72,73] |
| JAKMIP1−/− mice present ASD-like behaviors | ||
| STOP/MAP6 | STOP/MAP6−/− mice present synaptic abnormalities and ASD-like behavioral deficits (impairments in maternal care, social behavior and reduced cognitive performance) | [74,75,76,77,78] |
| Reduced plasma levels of STOP/MAP6 protein in ASD patients | ||
| KIRREL3/MAP1B | KIRREL3/MAP1B interaction involved in ASD pathogenesis | [79,80,81,82] |
| AUTS2/Rac1 | AUTS2 mut neurons display abnormal morphology during migration and reduced activity of JNK | [83,84,85,86] |
| YWHAE | YWHAE duplication associated to ASD | [87,88] |
| YWHAE regulator of neurite formation acting on Dcx | ||
| MARKs | Dysregulation of MARKs has been linked to ASD | [89,90,91] |
| MARKs regulate MTs dynamics during cell polarity, migration and vesicular transport | ||
| KATNAL2 | KATNAL2 de novo mut associated to ASD in human | [92,93] |
| KATNAL2−/− mice present reduced neurite branching and length |
| Genes/Protein | Effects | Ref. |
|---|---|---|
| α-tubulin and β-tubulin | Altered cytoskeletal organization | [100,101,102] |
| DISC-1 | Mutation S704C confers susceptibility to schizophrenia in humans | [110,111] |
| ULK4 | Ulk4−/− mice showed low levels of acetylated α-tubulin | [103] |
| ULK4 gene deletions have been found in schizophrenic patients | ||
| TTLL 11 | Balanced chromosomal translocation combined with chromosomal micro-duplication is associated with increased schizophrenia susceptibility | [104,105] |
| MAP1B | Low immunoreactivity in hippocampal subiculum associated with altered cyto-architecture and neurotransmission deficits in individuals with schizophrenia | [81,82] |
| MAP2 | Low immunoreactivity in brains of individuals with schizophrenia | [107,108] |
| MAP6 | Neuronal transport defects | [78,109] |
| Deletion of gene causes altered mood and cognitive performance in mice models of schizophrenia | ||
| ADNP | ADNP protein deregulated in postmortem hippocampi of schizophrenia patients | [112,113] |
| ADNP involved in autophagy regulation |
| Genes/Protein | Effects | Ref. |
|---|---|---|
| LIS1 | Loss of lissencephaly-1 (LIS1) protein is a major cause of lissencephaly | [143,144,145] |
| Mutations on LIS1 gene cause alterations in neuronal migration, in MTs network organization and intracellular transport | ||
| DCX | DCX gene mutations cause neuronal migration abnormalities by altering MTs dynamics | [146,147,148,149,150] |
| DCX gene has been associated with lissencephaly and subcortical band heterotopia | ||
| TUBA1A | Mutations in the TUBA1A gene have been associates with type I lissencephaly | [151,152] |
| TUBB2B | Mutations in TUBB2B gene cause asymmetrical bilateral polygyria | [153] |
| RELN | RELN gene mutations cause loss of reelin protein, leading to alterations in neuronal migration and positioning in the developing brain | [53] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonini, S.A.; Mastinu, A.; Ferrari-Toninelli, G.; Memo, M. Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders. Int. J. Mol. Sci. 2017, 18, 1627. https://doi.org/10.3390/ijms18081627
Bonini SA, Mastinu A, Ferrari-Toninelli G, Memo M. Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders. International Journal of Molecular Sciences. 2017; 18(8):1627. https://doi.org/10.3390/ijms18081627
Chicago/Turabian StyleBonini, Sara Anna, Andrea Mastinu, Giulia Ferrari-Toninelli, and Maurizio Memo. 2017. "Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders" International Journal of Molecular Sciences 18, no. 8: 1627. https://doi.org/10.3390/ijms18081627
APA StyleBonini, S. A., Mastinu, A., Ferrari-Toninelli, G., & Memo, M. (2017). Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders. International Journal of Molecular Sciences, 18(8), 1627. https://doi.org/10.3390/ijms18081627