Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine

 ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Glutamate and Migraine
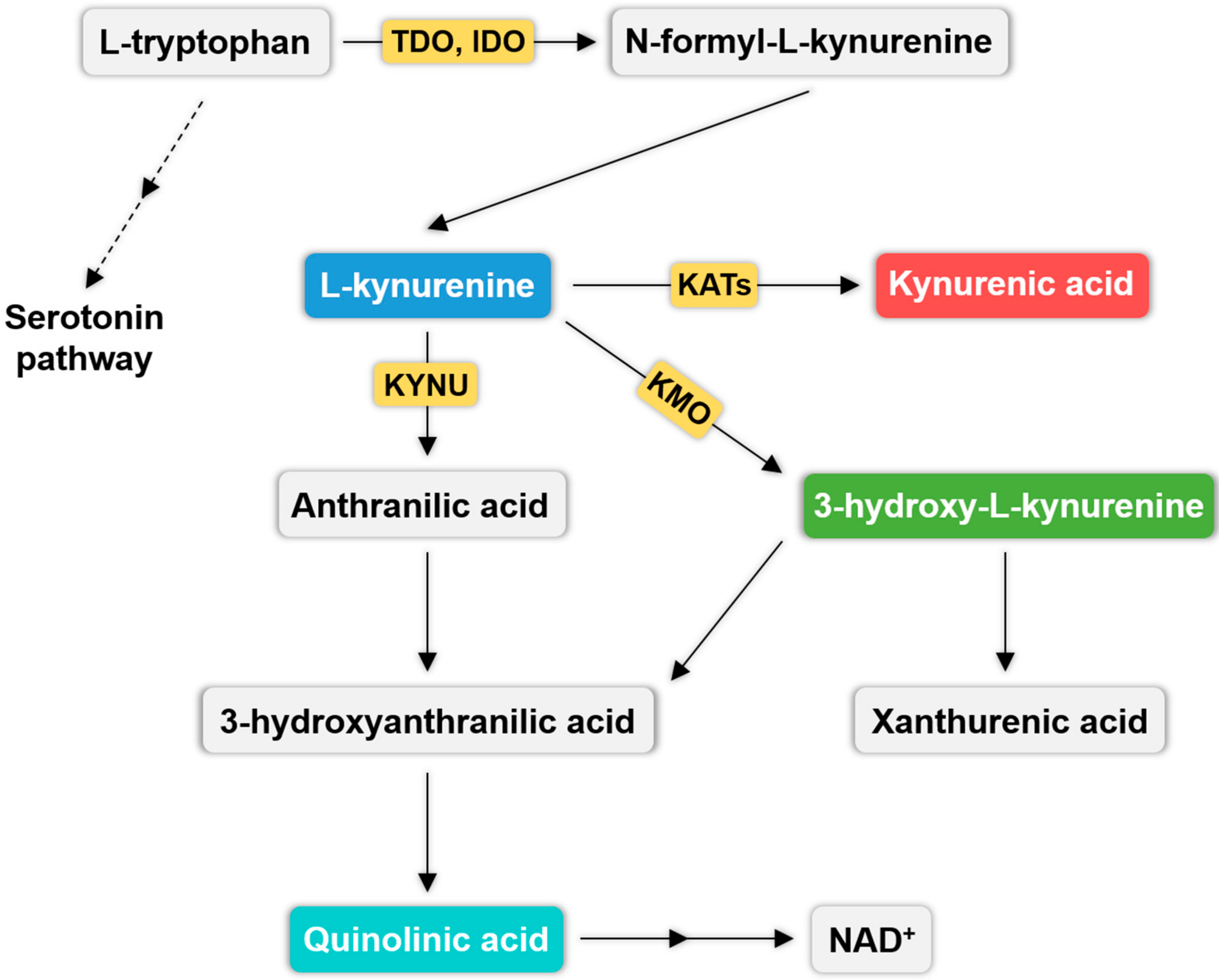
3. Kynurenine Pathway (KP) and Migraine
4. The Endocannabinoid System and Migraine
4.1. The Endocannabinoid System
4.2. The Role of Endocannabinoids in Migraine
5. Cannabinoid and Glutamate Receptors in the Trigeminal System
6. Known and Potential Functional Interactions betweenthe Endocannabinoid and Kynurenine System: Possible Pharmacological Targets against Migraine
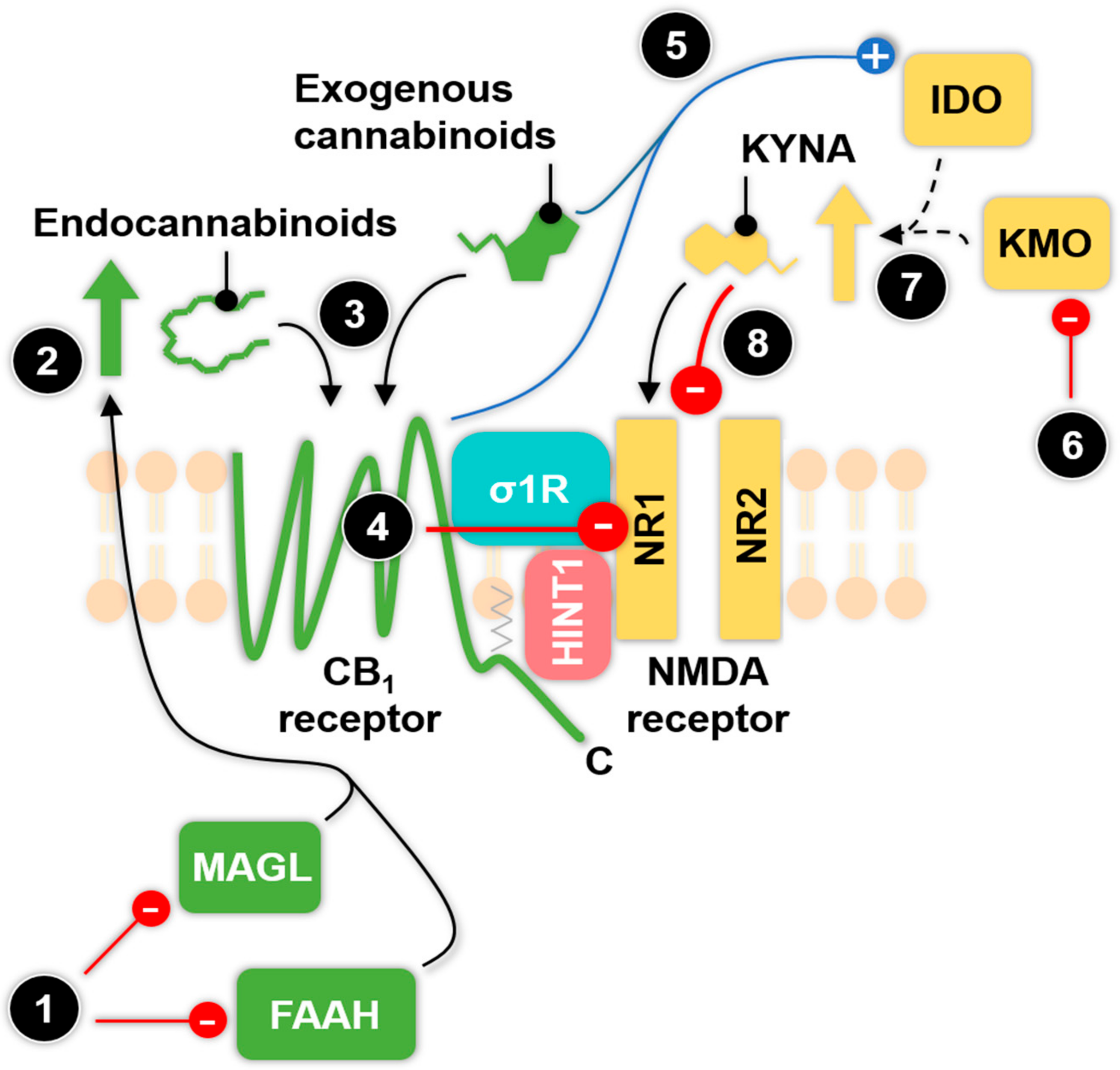
6.1. The Relationship between Enzymes of the KP and Exogenous Cannabinoids
6.2. The Type 1 Cannabinoid Receptor-N-methyl-d-aspartate (CB1-NMDA) Receptor Complex
6.3. Possible Interaction between the μ Opioid, the CB1 and the NMDA Receptor
6.4. The Interaction between the CB1 and the α7nACh Receptor
6.5. GPR35: A Possible Interactional Partner for Cannabinoid Receptors
7. Final Remarks and Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| 3-HA | 3-hydroxyanthranilic acid |
| 3-HK | 3-hydroxy-l-kynurenine |
| AEA | anandamide |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| ANA | anthranilic acid |
| cAMP | cyclic adenosine monophosphate |
| CB1 | type 1 cannabinoid receptor |
| CB2 | type 2 cannabinoid receptor |
| CFA | complete Freund’s Adjuvant |
| CGRP | calcitonin gene-related peptide |
| CSD | cortical spreading depression |
| DRG | dorsal root ganglion |
| FAAH | fatty acid amide hydrolase |
| GABA | γ-Aminobutyric acid |
| GPCR | G-protein coupled receptor |
| GPR35 | G protein coupled receptor 35 |
| HINT1 | histidine triad nucleotide-binding protein 1 |
| IDO | indoleamine 2,3-dioxygenase |
| KA1 | N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride |
| KA2 | N-(2-N-pyrrolidinylethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride |
| KATII | kynurenine aminotransferase II |
| KMO | kynurenine 3-monooxygenase |
| KP | kynurenine pathway |
| KYNA | kynurenic acid |
| KYNU | l-kynurenine hydrolase |
| L-KYN | l-kynurenine |
| MAGL | monoacylglycerol lipase |
| MAP | mitogen-activated protein kinases |
| MOR | μ opioid receptor |
| NAD+ | nicotinamide adenine dinucleotid |
| NMDA | N-methyl-d-aspartate |
| NO | nitric oxide |
| NTG | nitroglycerin |
| NAc | nucleus accumbens |
| PAG | periaqueaductal grey |
| QUIN | quinolinic acid |
| TDO | tryptophan 2,3-dioxygenase |
| THC | Δ9-tetrahydrocannabinol |
| Trp | tryptophan |
| α7nAChR | α7 nicotinic acetylcholine receptor |
| σ1R | σ receptor type 1 |
References
- Rodríguez de Fonseca, F.; Del Arco, I.; Bermudez-Silva, F.J.; Bilbao, A.; Cippitelli, A.; Navarro, M. The endocannabinoid system: Physiology and pharmacology. Alcohol Alcohol. 2005, 40, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Bifulco, M.; De Petrocellis, L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Clinical endocannabinoid deficiency (CECD): Can this concept explain therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuro Endocrinol. Lett. 2004, 25, 31–39. [Google Scholar] [PubMed]
- Smitherman, T.A.; Burch, R.; Sheikh, H.; Loder, E. The prevalence, impact, and treatment of migraine and severe headaches in the United States: A review of statistics from national surveillance studies. Headache 2013, 53, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Gustavsson, A.; Svensson, M.; Wittchen, H.U.; Jonsson, B. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2012, 19, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Vikelis, M.; Mitsikostas, D.D. The role of glutamate and its receptors in migraine. CNS Neurol. Disord. Drug Targets 2007, 6, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Majlath, Z.; Balog, A.; Tajti, J. Drug targets of migraine and neuropathy: Treatment of hyperexcitability. CNS Neurol. Disord. Drug Targets 2015, 14, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Grotenhermen, F.; Muller-Vahl, K. The therapeutic potential of cannabis and cannabinoids. Dtsch. Arztebl. Int. 2012, 109, 495–501. [Google Scholar] [PubMed]
- Greenamyre, J.T.; Porter, R.H. Anatomy and physiology of glutamate in the CNS. Neurology 1994, 44, S7–S13. [Google Scholar] [PubMed]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [PubMed]
- Ziff, E.B. Recent excitement in the ionotropic glutamate receptor field. Ann. N. Y. Acad. Sci. 1999, 868, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Brauner-Osborne, H.; Egebjerg, J.; Nielsen, E.O.; Madsen, U.; Krogsgaard-Larsen, P. Ligands for glutamate receptors: Design and therapeutic prospects. J. Med. Chem. 2000, 43, 2609–2645. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Sharpe, L.G. Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science 1969, 166, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 1985, 58, 293–297. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Ong, W.Y.; Horrocks, L.A. Biochemical aspects of neurodegeneration in human brain: Involvement of neural membrane phospholipids and phospholipases A2. Neurochem. Res. 2004, 29, 1961–1977. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, C.F.; Griffiths, L.R. The biology of the glutamatergic system and potential role in migraine. Int. J. Biomed. Sci. 2013, 9, 1–8. [Google Scholar] [PubMed]
- Cananzi, A.R.; D’Andrea, G.; Perini, F.; Zamberlan, F.; Welch, K.M. Platelet and plasma levels of glutamate and glutamine in migraine with and without aura. Cephalalgia 1995, 15, 132–135. [Google Scholar] [CrossRef] [PubMed]
- D’Eufemia, P.; Finocchiaro, R.; Lendvai, D.; Celli, M.; Viozzi, L.; Troiani, P.; Turri, E.; Giardini, O. Erythrocyte and plasma levels of glutamate and aspartate in children affected by migraine. Cephalalgia 1997, 17, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Castillo, J.; Rodriguez, J.R.; Leira, R.; Noya, M. Neuroexcitatory amino acid levels in plasma and cerebrospinal fluid during migraine attacks. Cephalalgia 1993, 13, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L.; Villalon, C.M.; MaassenVanDenBrink, A. Basic mechanisms of migraine and its acute treatment. Pharmacol. Ther. 2012, 136, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.P.; Woolf, C.J. Noxious stimuli induce an N-methyl-d-aspartate receptor-dependent hypersensitivity of the flexion withdrawal reflex to touch: Implications for the treatment of mechanical allodynia. Pain 1995, 61, 383–390. [Google Scholar] [CrossRef]
- Hughes, D.I.; Scott, D.T.; Todd, A.J.; Riddell, J.S. Lack of evidence for sprouting of abeta afferents into the superficial laminas of the spinal cord dorsal horn after nerve section. J. Neurosci. 2003, 23, 9491–9499. [Google Scholar] [PubMed]
- Soliman, A.C.; Yu, J.S.; Coderre, T.J. Mglu and NMDA receptor contributions to capsaicin-induced thermal and mechanical hypersensitivity. Neuropharmacology 2005, 48, 325–332. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, G.; Cananzi, A.R.; Joseph, R.; Morra, M.; Zamberlan, F.; Ferro Milone, F.; Grunfeld, S.; Welch, K.M. Platelet glycine, glutamate and aspartate in primary headache. Cephalalgia 1991, 11, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Rothrock, J.F.; Mar, K.R.; Yaksh, T.L.; Golbeck, A.; Moore, A.C. Cerebrospinal fluid analyses in migraine patients and controls. Cephalalgia 1995, 15, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Sullivan, S.G.; Stern, A. Oxidative reactivity of the tryptophan metabolites 3-hydroxyanthranilate, cinnabarinate, quinolinate and picolinate. Biochem. Pharmacol. 1987, 36, 211–217. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenate and FG9041 have both competitive and non-competitive antagonist actions at excitatory amino acid receptors. Eur. J. Pharmacol. 1988, 151, 313–315. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. Actions of kynurenic acid and quinolinic acid in the rat hippocampus in vivo. Exp. Neurol. 1985, 88, 570–579. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F.; George, S.R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Williams, D.J.; Puhl, H.L.; Ikeda, S.R. Inhibition of n-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J. Pharmacol. Exp. Ther. 2008, 324, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, A.E.; Milligan, G. The emerging pharmacology and function of GPR35 in the nervous system. Neuropharmacology 2017, 113, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Schwarcz, R. 3-hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–3863. [Google Scholar] [CrossRef] [PubMed]
- Behan, W.M.; McDonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.G.; Tasca, C.I.; Santos, C.E.; Wajner, M.; Souza, D.O.; Dutra-Filho, C.S. Quinolinic acid inhibits glutamate uptake into synaptic vesicles from rat brain. NeuroReport 2000, 11, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.A.; Foster, A.C.; Leeson, P.D.; Priestley, T.; Tridgett, R.; Iversen, L.L.; Woodruff, G.N. 7-chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-d-aspartate receptor complex. Proc. Natl. Acad. Sci. USA 1988, 85, 6547–6550. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Robotka, H.; Rozsa, E.; Agoston, M.; Szenasi, G.; Gigler, G.; Marosi, M.; Kis, Z.; Farkas, T.; Vecsei, L.; et al. Kynurenine diminishes the ischemia-induced histological and electrophysiological deficits in the rat hippocampus. Neurobiol. Dis. 2008, 32, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Di Clemente, L.; Coppola, G.; Magis, D.; Gerardy, P.Y.; Fumal, A.; De Pasqua, V.; Di Piero, V.; Schoenen, J. Nitroglycerin sensitises in healthy subjects CNS structures involved in migraine pathophysiology: Evidence from a study of nociceptive blink reflexes and visual evoked potentials. Pain 2009, 144, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Tassorelli, C.; Joseph, S.A. Systemic nitroglycerin induces Fos immunoreactivity in brainstem and forebrain structures of the rat. Brain Res. 1995, 682, 167–181. [Google Scholar] [CrossRef]
- Fejes-Szabo, A.; Bohar, Z.; Vamos, E.; Nagy-Grocz, G.; Tar, L.; Veres, G.; Zadori, D.; Szentirmai, M.; Tajti, J.; Szatmari, I.; et al. Pre-treatment with new kynurenic acid amide dose-dependently prevents the nitroglycerine-induced neuronal activation and sensitization in cervical part of trigemino-cervical complex. J. Neural Transm. (Vienna) 2014, 121, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Vamos, E.; Pardutz, A.; Fejes, A.; Tajti, J.; Toldi, J.; Vecsei, L. Modulatory effects of probenecid on the nitroglycerin-induced changes in the rat caudal trigeminal nucleus. Eur. J. Pharmacol. 2009, 621, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Vamos, E.; Pardutz, A.; Varga, H.; Bohar, Z.; Tajti, J.; Fulop, F.; Toldi, J.; Vecsei, L. l-kynurenine combined with probenecid and the novel synthetic kynurenic acid derivative attenuate nitroglycerin-induced nNOS in the rat caudal trigeminal nucleus. Neuropharmacology 2009, 57, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Grocz, G.; Tar, L.; Bohar, Z.; Fejes-Szabo, A.; Laborc, K.F.; Spekker, E.; Vecsei, L.; Pardutz, A. The modulatory effect of anandamide on nitroglycerin-induced sensitization in the trigeminal system of the rat. Cephalalgia 2016, 36, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Grócz, G.; Laborc, K.F.; Veres, G.; Bajtai, A.; Bohar, Z.; Zádori, D.; Fejes-Szabó, A.; Spekker, E.; Vécsei, L.; Párdutz, Á. The effect of systemic nitroglycerin administration on the kynurenine pathway in the rat. Front. Neurol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, M.; Warfvinge, K.; Kruse, L.S.; Tajti, J.; Fulop, F.; Toldi, J.; Vecsei, L.; Edvinsson, L. KYNA analogue SZR72 modifies CFA-induced dural inflammation- regarding expression of pERK1/2 and IL-1beta in the rat trigeminal ganglion. J. Headache Pain 2016, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Clavelou, P.; Pajot, J.; Dallel, R.; Raboisson, P. Application of the formalin test to the study of orofacial pain in the rat. Neurosci. Lett. 1989, 103, 349–353. [Google Scholar] [CrossRef]
- Veres, G.; Fejes-Szabo, A.; Zadori, D.; Nagy-Grocz, G.; Laszlo, A.M.; Bajtai, A.; Mandity, I.; Szentirmai, M.; Bohar, Z.; Laborc, K.; et al. A comparative assessment of two kynurenic acid analogs in the formalin model of trigeminal activation: A behavioral, immunohistochemical and pharmacokinetic study. J. Neural Transm. (Vienna) 2017, 124, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Redavide, E.; Pampalone, S.; Toldi, J.; Fulop, F.; Blandini, F.; Nappi, G.; Sandrini, G.; et al. Effects of kynurenic acid analogue 1 (KYNA-A1) in nitroglycerin-induced hyperalgesia: Targets and anti-migraine mechanisms. Cephalalgia 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Knyihar-Csillik, E.; Chadaide, Z.; Okuno, E.; Krisztin-Peva, B.; Toldi, J.; Varga, C.; Molnar, A.; Csillik, B.; Vecsei, L. Kynurenine aminotransferase in the supratentorial dura mater of the rat: Effect of stimulation of the trigeminal ganglion. Exp. Neurol. 2004, 186, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C. Cortical spreading depression triggers migraine attack: Pro. Headache 2010, 50, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M. Pathophysiology of the migraine aura. The spreading depression theory. Brain 1994, 117 Pt 1, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Knapp, L.; Szita, B.; Kocsis, K.; Vecsei, L.; Toldi, J. Nitroglycerin enhances the propagation of cortical spreading depression: Comparative studies with sumatriptan and novel kynurenic acid analogues. Drug Des. Dev. Ther. 2017, 11, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M.; Rice, M.E.; Okada, Y.; Nicholson, C. Quisqualate, kainate and NMDA can initiate spreading depression in the turtle cerebellum. Brain Res. 1988, 475, 317–327. [Google Scholar] [CrossRef]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Perugino, F.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered serum levels of kynurenine metabolites in patients affected by cluster headache. J. Headache Pain 2015, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kiyama, H.; Park, H.T.; Tohyama, M. AMPA, KA and NMDA receptors are expressed in the rat DRG neurones. Neuroreport 1993, 4, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, H.; Tonai-Kachi, H.; Ichikawa, K. GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 2008, 365, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Mecs, L.; Tuboly, G.; Nagy, E.; Benedek, G.; Horvath, G. The peripheral antinociceptive effects of endomorphin-1 and kynurenic acid in the rat inflamed joint model. Anesth. Analg. 2009, 109, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Ji, G.C.; Wu, G.C.; Zhao, Z.Q. Kynurenic acid enhances electroacupuncture analgesia in normal and carrageenan-injected rats. Brain Res. 2003, 966, 300–307. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cdna. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, U.; Marsicano, G.; Cota, D.; Lutz, B.; Pasquali, R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr. Rev. 2006, 27, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Haskó, G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2008, 153, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S163–S171. [Google Scholar] [CrossRef] [PubMed]
- Malan, T.P.; Ibrahim, M.M.; Deng, H.; Liu, Q.; Mata, H.P.; Vanderah, T.; Porreca, F.; Makriyannis, A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain 2001, 93, 239–245. [Google Scholar] [CrossRef]
- Romero, T.R.; Resende, L.C.; Guzzo, L.S.; Duarte, I.D. CB1 and CB2 cannabinoid receptor agonists induce peripheral antinociception by activation of the endogenous noradrenergic system. Anesth. Analg. 2013, 116, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Targa, M.; Corciulo, C.; Tabrizi, M.A.; Merighi, S.; Gessi, S.; Saponaro, G.; Baraldi, P.G.; Borea, P.A.; Varani, K. Antinociceptive effects of the selective CB2 agonist MT178 in inflammatory and chronic rodent pain models. Pain 2013, 154, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Mangione, A.S.; Sandrini, G.; Nappi, G.; Tassorelli, C. Activation of CB2 receptors as a potential therapeutic target for migraine: Evaluation in an animal model. J. Headache Pain 2014, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Fride, E.; Di Marzo, V. Endocannabinoids. Eur. J. Pharmacol. 1998, 359, 1–18. [Google Scholar] [CrossRef]
- Justinova, Z.; Panlilio, L.V.; Moreno-Sanz, G.; Redhi, G.H.; Auber, A.; Secci, M.E.; Mascia, P.; Bandiera, T.; Armirotti, A.; Bertorelli, R.; et al. Effects of fatty acid amide hydrolase (FAAH) inhibitors in non-human primate models of nicotine reward and relapse. Neuropsychopharmacology 2015, 40, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Haller, V.L.; Stevens, D.L.; Welch, S.P. Modulation of opioids via protection of anandamide degradation by fatty acid amide hydrolase. Eur. J. Pharmacol. 2008, 600, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Bushlin, I.; Rozenfeld, R.; Devi, L.A. Cannabinoid-opioid interactions during neuropathic pain and analgesia. Curr. Opin. Pharmacol. 2010, 10, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Kondo, H.; Nakane, S.; Kodaka, T.; Tokumura, A.; Waku, K.; Sugiura, T. 2-arachidonoylglycerol, an endogenous cannabinoid receptor agonist: Identification as one of the major species of monoacylglycerols in various rat tissues, and evidence for its generation through Ca2+-dependent and -independent mechanisms. FEBS Lett. 1998, 429, 152–156. [Google Scholar] [CrossRef]
- Karlsson, M.; Contreras, J.A.; Hellman, U.; Tornqvist, H.; Holm, C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997, 272, 27218–27223. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001, 63, 569–611. [Google Scholar] [CrossRef]
- Akerman, S.; Holland, P.R.; Goadsby, P.J. Cannabinoid (CB1) receptor activation inhibits trigeminovascular neurons. J. Pharmacol. Exp. Ther. 2007, 320, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Cupini, L.M.; Costa, C.; Sarchielli, P.; Bari, M.; Battista, N.; Eusebi, P.; Calabresi, P.; Maccarrone, M. Degradation of endocannabinoids in chronic migraine and medication overuse headache. Neurobiol. Dis. 2008, 30, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Pini, L.A.; Cupini, M.L.; Calabresi, P.; Sarchielli, P. Endocannabinoids in platelets of chronic migraine patients and medication-overuse headache patients: Relation with serotonin levels. Eur. J. Clin. Pharmacol. 2008, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Pini, L.A.; Coppola, F.; Rossi, C.; Baldi, A.; Mancini, M.L.; Calabresi, P. Endocannabinoids in chronic migraine: CSF findings suggest a system failure. Neuropsychopharmacology 2007, 32, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Cupini, L.M.; Bari, M.; Battista, N.; Argiro, G.; Finazzi-Agro, A.; Calabresi, P.; Maccarrone, M. Biochemical changes in endocannabinoid system are expressed in platelets of female but not male migraineurs. Cephalalgia 2006, 26, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Gasperi, V.; Maccarrone, M.; Tassorelli, C. The endocannabinoid system and migraine. Exp. Neurol. 2010, 224, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Gasperi, V.; Sandrini, G.; Bagetta, G.; Nappi, G.; Maccarrone, M.; Tassorelli, C. Alterations of the endocannabinoid system in an animal model of migraine: Evaluation in cerebral areas of rat. Cephalalgia 2010, 30, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Akerman, S.; Kaube, H.; Goadsby, P.J. Anandamide is able to inhibit trigeminal neurons using an in vivo model of trigeminovascular-mediated nociception. J. Pharmacol. Exp. Ther. 2004, 309, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, S.; Endo, Y.; Nakahara, H.; Yoshizawa, K.; Hashiba, Y.; Kawashiri, S.; Tanaka, A.; Nakagawa, K.; Matsuoka, Y.; Kogo, M.; et al. Inhibition of invasion and metastasis in oral cancer by targeting urokinase-type plasminogen activator receptor. Oral Oncol. 2005, 41, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, H.; Rahgozar, M.; Speckmann, E.J.; Gorji, A. Effect of cannabinoid receptor activation on spreading depression. Iran. J. Basic Med. Sci. 2012, 15, 926–936. [Google Scholar] [PubMed]
- Akerman, S.; Holland, P.R.; Lasalandra, M.P.; Goadsby, P.J. Endocannabinoids in the brainstem modulate dural trigeminovascular nociceptive traffic via CB1 and “triptan” receptors: Implications in migraine. J. Neurosci. 2013, 33, 14869–14877. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann. Neurol. 1988, 23, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Desroches, J.; Beaulieu, P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br. J. Pharmacol. 2007, 150, 693–701. [Google Scholar] [CrossRef] [PubMed]
- La Rana, G.; Russo, R.; Campolongo, P.; Bortolato, M.; Mangieri, R.A.; Cuomo, V.; Iacono, A.; Raso, G.M.; Meli, R.; Piomelli, D.; et al. Modulation of neuropathic and inflammatory pain by the endocannabinoid transport inhibitor am404 [N-(4-hydroxyphenyl)-eicosa-5,8,11,14-tetraenamide]. J. Pharmacol. Exp. Ther. 2006, 317, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Strangman, N.M.; Walker, J.M. Cannabinoid win 55,212-2 inhibits the activity-dependent facilitation of spinal nociceptive responses. J. Neurophysiol. 1999, 82, 472–477. [Google Scholar] [PubMed]
- Ahn, K.; Johnson, D.S.; Mileni, M.; Beidler, D.; Long, J.Z.; McKinney, M.K.; Weerapana, E.; Sadagopan, N.; Liimatta, M.; Smith, S.E.; et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Dragunow, M.; Faull, R.L. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Thomas, B.F.; Wei, X.; Martin, B.R. Characterization and autoradiographic localization of the cannabinoid binding site in rat brain using [3H] 11-OH-delta 9-THC-DMH. J. Pharmacol. Exp. Ther. 1992, 263, 1383–1390. [Google Scholar] [PubMed]
- Wotherspoon, G.; Fox, A.; McIntyre, P.; Colley, S.; Bevan, S.; Winter, J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience 2005, 135, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Colin-Gonzalez, A.L.; Aguilera, G.; Santamaria, A. Cannabinoids: Glutamatergic transmission and kynurenines. Adv. Neurobiol. 2016, 12, 173–198. [Google Scholar] [PubMed]
- Juhasz, G.; Lazary, J.; Chase, D.; Pegg, E.; Downey, D.; Toth, Z.G.; Stones, K.; Platt, H.; Mekli, K.; Payton, A.; et al. Variations in the cannabinoid receptor 1 gene predispose to migraine. Neurosci. Lett. 2009, 461, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Tallaksen-Greene, S.J.; Young, A.B.; Penney, J.B.; Beitz, A.J. Excitatory amino acid binding sites in the trigeminal principal sensory and spinal trigeminal nuclei of the rat. Neurosci. Lett. 1992, 141, 79–83. [Google Scholar] [CrossRef]
- Watanabe, M.; Mishina, M.; Inoue, Y. Distinct gene expression of the N-methyl-d-aspartate receptor channel subunit in peripheral neurons of the mouse sensory ganglia and adrenal gland. Neurosci. Lett. 1994, 165, 183–186. [Google Scholar] [CrossRef]
- Furuyama, T.; Kiyama, H.; Sato, K.; Park, H.T.; Maeno, H.; Takagi, H.; Tohyama, M. Region-specific expression of subunits of ionotropic glutamate receptors (AMPA-type, KA-type and NMDA receptors) in the rat spinal cord with special reference to nociception. Brain Res. Mol. Brain Res. 1993, 18, 141–151. [Google Scholar] [CrossRef]
- Lomazzo, E.; Bindila, L.; Remmers, F.; Lerner, R.; Schwitter, C.; Hoheisel, U.; Lutz, B. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology 2015, 40, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Nie, Z.; Siggins, G.R. Mu-opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J. Neurosci. 1997, 17, 11–22. [Google Scholar] [PubMed]
- Patel, S.; Hill, M.N.; Cheer, J.F.; Wotjak, C.T.; Holmes, A. The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci. Biobehav. Rev. 2017, 76, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of migraine: A disorder of sensory processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Critical enzymes involved in endocannabinoid metabolism. Protein Pept. Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892. [Google Scholar] [CrossRef] [PubMed]
- Jenny, M.; Santer, E.; Pirich, E.; Schennach, H.; Fuchs, D. Delta9-tetrahydrocannabinol and cannabidiol modulate mitogen-induced tryptophan degradation and neopterin formation in peripheral blood mononuclear cells in vitro. J. Neuroimmunol. 2009, 207, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Aloyo, V.J.; Berg, K.A.; Clarke, W.P.; Spampinato, U.; Harvey, J.A. Inverse agonism at serotonin and cannabinoid receptors. Prog. Mol. Biol. Transl. Sci. 2010, 91, 1–40. [Google Scholar] [PubMed]
- Devlin, M.G.; Christopoulos, A. Modulation of cannabinoid agonist binding by 5-HT in the rat cerebellum. J. Neurochem. 2002, 80, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Justinova, Z.; Mascia, P.; Wu, H.Q.; Secci, M.E.; Redhi, G.H.; Panlilio, L.V.; Scherma, M.; Barnes, C.; Parashos, A.; Zara, T.; et al. Reducing cannabinoid abuse and preventing relapse by enhancing endogenous brain levels of kynurenic acid. Nat. Neurosci. 2013, 16, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Merlos, M.; Garzón-Niño, J. Endocannabinoid control of glutamate NMDA receptors: The therapeutic potential and consequences of dysfunction. Oncotarget 2016, 7, 55840–55862. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, J.; Urban, L.; Capogna, M.; Bevan, S.; Nagy, I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience 2000, 100, 685–688. [Google Scholar] [CrossRef]
- Turski, M.P.; Turska, M.; Paluszkiewicz, P.; Parada-Turska, J.; Oxenkrug, G.F. Kynurenic acid in the digestive system-new facts, new challenges. Int. J. Tryptophan Res. 2013, 6, 47–55. [Google Scholar] [PubMed]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Krystal, J.H. NMDA receptor regulation of memory and behavior in humans. Hippocampus 2001, 11, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.; McKillop-Smith, S.; Ross, N.L.; Pertwee, R.G.; Hampson, R.E.; Platt, B.; Riedel, G. Hippocampal endocannabinoids inhibit spatial learning and limit spatial memory in rats. Psychopharmacology 2008, 198, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Wise, L.E.; Iredale, P.A.; Lichtman, A.H. The cannabinoid CB (1) receptor antagonist CE prolongs spatial memory duration in a rat delayed radial arm maze memory task. Eur. J. Pharmacol. 2008, 590, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.M.; Carelli, R.M.; Dykstra, L.A.; Suchey, T.L.; Everett, C.V. Effects of the competitive N-methyl-d-aspartate receptor antagonist, ly235959 [(−)-6-phosphonomethyl-deca-hydroisoquinoline-3-carboxylic acid], on responding for cocaine under both fixed and progressive ratio schedules of reinforcement. J. Pharmacol. Exp. Ther. 2005, 315, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.M.; Uban, K.A.; Atwood, E.M.; Albeck, D.S.; Yamamoto, D.J. Continuous intracerebroventricular infusion of the competitive NMDA receptor antagonist, LY235959, facilitates escalation of cocaine self-administration and increases break point for cocaine in sprague-dawley rats. Pharmacol. Biochem. Behav. 2007, 88, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Mendizábal, V.; Touriño, C.; Robledo, P.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Lack of CB1 cannabinoid receptor impairs cocaine self-administration. Neuropsychopharmacology 2005, 30, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- De Vry, J.; Denzer, D.; Reissmueller, E.; Eijckenboom, M.; Heil, M.; Meier, H.; Mauler, F. 3-[2-cyano-3-(trifluoromethyl)phenoxy]phenyl-4,4,4-trifluoro-1-butanesulfonate (BAY 59-3074): A novel cannabinoid CB1/CB2 receptor partial agonist with antihyperalgesic and antiallodynic effects. J. Pharmacol. Exp. Ther. 2004, 310, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Pelissier, T.; Infante, C.; Constandil, L.; Espinosa, J.; Lapeyra, C.D.; Hernández, A. Antinociceptive effect and interaction of uncompetitive and competitive NMDA receptor antagonists upon capsaicin and paw pressure testing in normal and monoarthritic rats. Pain 2008, 134, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: Implications in psychosis and schizophrenia. Front. Pharmacol. 2014, 4, 169. [Google Scholar] [CrossRef] [PubMed]
- Corlew, R.; Brasier, D.J.; Feldman, D.E.; Philpot, B.D. Presynaptic NMDA receptors: Newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist 2008, 14, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.G.; Briley, E.M.; Herkenham, M. Pre- and postsynaptic distribution of cannabinoid and mu opioid receptors in rat spinal cord. Brain Res. 1999, 822, 17–25. [Google Scholar] [CrossRef]
- Marchalant, Y.; Cerbai, F.; Brothers, H.M.; Wenk, G.L. Cannabinoid receptor stimulation is anti-inflammatory and improves memory in old rats. Neurobiol. Aging 2008, 29, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Salio, C.; Fischer, J.; Franzoni, M.F.; Conrath, M. Pre- and postsynaptic localizations of the CB1 cannabinoid receptor in the dorsal horn of the rat spinal cord. Neuroscience 2002, 110, 755–764. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Vicente-Sánchez, A.; Garzón, J. Cannabinoid receptors couple to NMDA receptors to reduce the production of no and the mobilization of zinc induced by glutamate. Antioxid. Redox Signal 2013, 19, 1766–1782. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Salter, M.W. Glutamate receptor phosphorylation and trafficking in pain plasticity in spinal cord dorsal horn. Eur. J. Neurosci. 2010, 32, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Shyu, Y.J.; Suarez, C.D.; Hu, C.D. Visualization of ternary complexes in living cells by using a BiFC-based FRET assay. Nat. Protoc. 2008, 3, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Gardoni, F.; Spano, P.; Di Luca, M.; Missale, C. Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-d-aspartate receptors. J. Biol. Chem. 2003, 278, 20196–20202. [Google Scholar] [CrossRef] [PubMed]
- Perroy, J.; Raynaud, F.; Homburger, V.; Rousset, M.C.; Telley, L.; Bockaert, J.; Fagni, L. Direct interaction enables cross-talk between ionotropic and group I metabotropic glutamate receptors. J. Biol. Chem. 2008, 283, 6799–6805. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Vicente-Sánchez, A.; Berrocoso, E.; Garzón, J. The mu-opioid receptor and the NMDA receptor associate in pag neurons: Implications in pain control. Neuropsychopharmacology 2012, 37, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Herrero-Labrador, R.; Burgueño, J.; Zamanillo, D.; Garzón, J. The calcium-sensitive sigma-1 receptor prevents cannabinoids from provoking glutamate NMDA receptor hypofunction: Implications in antinociception and psychotic diseases. Int. J. Neuropsychopharmacol. 2014, 17, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Cortés-Montero, E.; Pozo-Rodrigálvarez, A.; Sánchez-Blázquez, P.; Garzón-Niño, J. The ON:Off switch, σ1R-HINT1 protein, controls GPCR-NMDA receptor cross-regulation: Implications in neurological disorders. Oncotarget 2015, 6, 35458–35477. [Google Scholar] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Herrero-Labrador, R.; Martínez-Murillo, R.; Merlos, M.; Vela, J.M.; Garzón, J. The σ1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxid. Redox Signal 2015, 22, 799–818. [Google Scholar] [CrossRef] [PubMed]
- Khaspekov, L.G.; Brenz Verca, M.S.; Frumkina, L.E.; Hermann, H.; Marsicano, G.; Lutz, B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 2004, 19, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bhat, M.; Bowen, W.D.; Cheng, J. Signaling pathways from cannabinoid receptor-1 activation to inhibition of N-methyl-d-aspartic acid mediated calcium influx and neurotoxicity in dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 2009, 331, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Sánchez, A.; Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Mol. Brain 2013, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Yang, H.; Zhang, J.; Chen, C. Reduced expression of glutamate receptors and phosphorylation of CREB are responsible for in vivo Delta9-THC exposure-impaired hippocampal synaptic plasticity. J. Neurochem. 2010, 112, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Schachter, S.C. The NMDA receptor complex as a therapeutic target in epilepsy: A review. Epilepsy Behav. 2011, 22, 617–640. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, L.; Hall, W.; Lynskey, M. Testing hypotheses about the relationship between cannabis use and psychosis. Drug Alcohol Depend. 2003, 71, 37–48. [Google Scholar] [CrossRef]
- Fernandez-Espejo, E.; Viveros, M.P.; Núñez, L.; Ellenbroek, B.A.; Rodriguez de Fonseca, F. Role of cannabis and endocannabinoids in the genesis of schizophrenia. Psychopharmacology 2009, 206, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; Vawter, M.P.; Ginsberg, S.D. Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology 2009, 34, 18–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, X.; O’Neill, F.A.; Walsh, D.; Kendler, K.S.; Chen, X. Is the histidine triad nucleotide-binding protein 1 (HINT1) gene a candidate for schizophrenia? Schizophr. Res. 2008, 106, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nunokawa, A.; Kaneko, N.; Shibuya, M.; Egawa, J.; Someya, T. Supportive evidence for the association between the Gln2Pro polymorphism in the SIGMAR1 gene and schizophrenia in the Japanese population: A case-control study and an updated meta-analysis. Schizophr. Res. 2012, 141, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Abadias, M.; Escriche, M.; Vaqué, A.; Sust, M.; Encina, G. Safety, tolerability and pharmacokinetics of single and multiple doses of a novel sigma-1 receptor antagonist in three randomized phase I studies. Br. J. Clin. Pharmacol. 2013, 75, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.S.; Gibel, A.; Shleifer, T.; Boguslavsky, I.; Zayed, A.; Maayan, R.; Weizman, A.; Lerner, V. Pregnenolone and dehydroepiandrosterone as an adjunctive treatment in schizophrenia and schizoaffective disorder: An 8-week, double-blind, randomized, controlled, 2-center, parallel-group trial. J. Clin. Psychiatry 2010, 71, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Horvath, G.; Kekesi, G.; Tuboly, G.; Benedek, G. Antinociceptive interactions of triple and quadruple combinations of endogenous ligands at the spinal level. Brain Res. 2007, 1155, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Kekesi, G.; Joo, G.; Csullog, E.; Dobos, I.; Klimscha, W.; Toth, K.; Benedek, G.; Horvath, G. The antinociceptive effect of intrathecal kynurenic acid and its interaction with endomorphin-1 in rats. Eur. J. Pharmacol. 2002, 445, 93–96. [Google Scholar] [CrossRef]
- Marek, P.; Ben-Eliyahu, S.; Gold, M.; Liebeskind, J.C. Excitatory amino acid antagonists (kynurenic acid and MK-801) attenuate the development of morphine tolerance in the rat. Brain Res. 1991, 547, 77–81. [Google Scholar] [CrossRef]
- Morgan, M.M.; Bobeck, E.N.; Ingram, S.L. Glutamate modulation of antinociception, but not tolerance, produced by morphine microinjection into the periaqueductal gray of the rat. Brain Res. 2009, 1295, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Safrany-Fark, A.; Petrovszki, Z.; Kekesi, G.; Keresztes, C.; Benedek, G.; Horvath, G. Telemetry monitoring for non-invasive assessment of changes in core temperature after spinal drug administration in freely moving rats. J. Pharmacol. Toxicol. Methods 2015, 72, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Zador, F.; Samavati, R.; Szlavicz, E.; Tuka, B.; Bojnik, E.; Fulop, F.; Toldi, J.; Vecsei, L.; Borsodi, A. Inhibition of opioid receptor mediated g-protein activity after chronic administration of kynurenic acid and its derivative without direct binding to opioid receptors. CNS Neurol. Disord. Drug Targets 2014, 13, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Samavati, R.; Zádor, F.; Szűcs, E.; Tuka, B.; Martos, D.; Veres, G.; Gáspár, R.; Mándity, I.M.; Fülöp, F.; Vécsei, L.; et al. Kynurenic acid and its analogue can alter the opioid receptor g-protein signaling after acute treatment via NMDA receptor in rat cortex and striatum. J. Neurol. Sci. 2017, 376, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, D.L. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 2004, 74, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Desroches, J.; Beaulieu, P. Opioids and cannabinoids interactions: Involvement in pain management. Curr. Drug Targets 2010, 11, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.; Valverde, O. Participation of the opioid system in cannabinoid-induced antinociception and emotional-like responses. Eur. Neuropsychopharmacol. 2003, 13, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Mao, J. NMDA and opioid receptors: Their interactions in antinociception, tolerance and neuroplasticity. Brain Res. Brain Res. Rev. 1999, 30, 289–304. [Google Scholar] [CrossRef]
- Robledo, P.; Berrendero, F.; Ozaita, A.; Maldonado, R. Advances in the field of cannabinoid—Opioid cross-talk. Addict. Biol. 2008, 13, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Scavone, J.L.; Sterling, R.C.; Van Bockstaele, E.J. Cannabinoid and opioid interactions: Implications for opiate dependence and withdrawal. Neuroscience 2013, 248, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Zádor, F.; Wollemann, M. Receptome: Interactions between three pain-related receptors or the “triumvirate” of cannabinoid, opioid and TRPV1 receptors. Pharmacol. Res. 2015, 102, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Scherma, M.; Muntoni, A.L.; Melis, M.; Fattore, L.; Fadda, P.; Fratta, W.; Pistis, M. Interactions between the endocannabinoid and nicotinic cholinergic systems: Preclinical evidence and therapeutic perspectives. Psychopharmacology 2016, 233, 1765–1777. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [PubMed]
- Albuquerque, E.X.; Schwarcz, R. Kynurenic acid as an antagonist of α7 nicotinic acetylcholine receptors in the brain: Facts and challenges. Biochem. Pharmacol. 2013, 85, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Dobelis, P.; Staley, K.J.; Cooper, D.C. Lack of modulation of nicotinic acetylcholine alpha-7 receptor currents by kynurenic acid in adult hippocampal interneurons. PLoS ONE 2012, 7, e41108. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.H.; Fricker, A.C.; Weil, A.; Kew, J.N. Electrophysiological characterisation of the actions of kynurenic acid at ligand-gated ion channels. Neuropharmacology 2009, 57, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Bertrand, D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I.; Moretti, M.; Pedrazzi, P.; Pucci, L.; Zoli, M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef] [PubMed]
- Wonnacott, S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997, 20, 92–98. [Google Scholar] [CrossRef]
- Solinas, M.; Scherma, M.; Fattore, L.; Stroik, J.; Wertheim, C.; Tanda, G.; Fratta, W.; Goldberg, S.R. Nicotinic alpha 7 receptors as a new target for treatment of cannabis abuse. J. Neurosci. 2007, 27, 5615–5620. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.; Brea, J.; Smith, N.J.; Hudson, B.D.; Reilly, G.; Bryant, N.J.; Castro, M.; Loza, M.I.; Milligan, G. Identification of novel species-selective agonists of the G-protein-coupled receptor GPR35 that promote recruitment of β-arrestin-2 and activate Gα13. Biochem. J. 2010, 432, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.; Alvarez-Curto, E.; Campbell, K.; de Munnik, S.; Canals, M.; Schlyer, S.; Milligan, G. Agonist activation of the g protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα₁₃ and β-arrestin-2. Br. J. Pharmacol. 2011, 162, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Fallarini, S.; Magliulo, L.; Paoletti, T.; de Lalla, C.; Lombardi, G. Expression of functional GPR35 in human iNKT cells. Biochem. Biophys. Res. Commun. 2010, 398, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, A.E.; Lappin, J.E.; Taylor, D.L.; Nicklin, S.A.; Milligan, G. GPR35 as a novel therapeutic target. Front. Endocrinol. (Lausanne) 2011, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRS, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Hohmann, A.G.; Martin, W.J.; Strangman, N.M.; Huang, S.M.; Tsou, K. The neurobiology of cannabinoid analgesia. Life Sci. 1999, 65, 665–673. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tonai-Kachi, H.; Shinjo, K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006, 580, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Naftali, T.; Bar-Lev Schleider, L.; Dotan, I.; Lansky, E.P.; Sklerovsky Benjaminov, F.; Konikoff, F.M. Cannabis induces a clinical response in patients with Crohn’s disease: A prospective placebo-controlled study. Clin. Gastroenterol. Hepatol. 2013, 11, 1276–1280.e1. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Baldassano, R.N.; Griffiths, A.; Russell, R.K.; Annese, V.; Dubinsky, M.; Kugathasan, S.; Bradfield, J.P.; Walters, T.D.; Sleiman, P.; et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009, 41, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.C.; Ahluwalia, N.; Anderson, T.J.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 2009, 284, 19189–19195. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, V.; Evangelista, D.; Chiurchiù, V.; Florenzano, F.; Savini, I.; Oddi, S.; Avigliano, L.; Catani, M.V.; Maccarrone, M. 2-arachidonoylglycerol modulates human endothelial cell/leukocyte interactions by controlling selectin expression through CB1 and CB2 receptors. Int. J. Biochem. Cell Biol. 2014, 51, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem. Pharmacol. 2014, 92, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Kianian, M.; Al-Banna, N.A.; Kelly, M.E.; Lehmann, C. Inhibition of endocannabinoid degradation in experimental endotoxemia reduces leukocyte adhesion and improves capillary perfusion in the gut. J. Basic Clin. Physiol. Pharmacol. 2013, 24, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Kianian, M.; Zhou, J.; Küster, I.; Kuschnereit, R.; Whynot, S.; Hung, O.; Shukla, R.; Johnston, B.; Cerny, V.; et al. Cannabinoid receptor 2 activation reduces intestinal leukocyte recruitment and systemic inflammatory mediator release in acute experimental sepsis. Crit. Care 2012, 16, R47. [Google Scholar] [CrossRef] [PubMed]
- Lunn, C.A.; Fine, J.S.; Rojas-Triana, A.; Jackson, J.V.; Fan, X.; Kung, T.T.; Gonsiorek, W.; Schwarz, M.A.; Lavey, B.; Kozlowski, J.A.; et al. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J. Pharmacol. Exp. Ther. 2006, 316, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Burger, F.; Mach, F.; Steffens, S. CB2 cannabinoid receptor agonist JWH-015 modulates human monocyte migration through defined intracellular signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1145–H1155. [Google Scholar] [CrossRef] [PubMed]
- Murikinati, S.; Jüttler, E.; Keinert, T.; Ridder, D.A.; Muhammad, S.; Waibler, Z.; Ledent, C.; Zimmer, A.; Kalinke, U.; Schwaninger, M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010, 24, 788–798. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy-Grócz, G.; Zádor, F.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, Á.; Vécsei, L. Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine. Int. J. Mol. Sci. 2017, 18, 1617. https://doi.org/10.3390/ijms18081617
Nagy-Grócz G, Zádor F, Dvorácskó S, Bohár Z, Benyhe S, Tömböly C, Párdutz Á, Vécsei L. Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine. International Journal of Molecular Sciences. 2017; 18(8):1617. https://doi.org/10.3390/ijms18081617
Chicago/Turabian StyleNagy-Grócz, Gábor, Ferenc Zádor, Szabolcs Dvorácskó, Zsuzsanna Bohár, Sándor Benyhe, Csaba Tömböly, Árpád Párdutz, and László Vécsei. 2017. "Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine" International Journal of Molecular Sciences 18, no. 8: 1617. https://doi.org/10.3390/ijms18081617
APA StyleNagy-Grócz, G., Zádor, F., Dvorácskó, S., Bohár, Z., Benyhe, S., Tömböly, C., Párdutz, Á., & Vécsei, L. (2017). Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine. International Journal of Molecular Sciences, 18(8), 1617. https://doi.org/10.3390/ijms18081617