Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy


Abstract
:1. Introduction
2. Epigenetic Mechanisms in Cancer
2.1. DNA Methylation
2.2. Covalent Histone Modification
2.3. Chromatin Remodeling and Associated Proteins
2.4. Non-Coding RNA
3. Epigenetic Modifications Associated with Tumor Progression and Drug Resistance in HNSCC
3.1. DNA Methylation Signature in HNSCC
3.2. Histone Acetylation and Chromatin Modifications in HNSCC
3.3. Non-Coding RNAs in HNSCC
4. Epigenetic Modifications of Cancer Stem Cells
5. Epigenetic Drugs and HNSCC Therapy
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kim, J.; Guan, J.; Chang, I.; Chen, X.; Han, D.; Wang, C.Y. PS-341 and histone deacetylase inhibitor synergistically induce apoptosis in head and neck squamous cell carcinoma cells. Mol. Cancer Ther. 2010, 9, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Pullos, A.N.; Castilho, R.M.; Squarize, C.H. HPV Infection of the Head and Neck Region and Its Stem Cells. J. Dent. Res. 2015, 94, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Mascolo, M.; Siano, M.; Ilardi, G.; Russo, D.; Merolla, F.; De Rosa, G.; Staibano, S. Epigenetic disregulation in oral cancer. Int. J. Mol. Sci. 2012, 13, 2331–2353. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P.; Bray, F.; Parkin, D.M. Estimates of the world-wide prevalence of cancer for 25 sites in the adult population. Int. J. Cancer 2002, 97, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Amornphimoltham, P.; Squarize, C.H.; Castilho, R.M.; Patel, V.; Gutkind, J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009, 45, 324–334. [Google Scholar] [CrossRef] [PubMed]
- United States Public Health Service, Office of the Surgeon General. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; U.S. Department of Health and Human Services, Public Health Service, Office of the Surgeon General: Rockville, MD, USA, 2014; Volume 2.
- Le, J.M.; Squarize, C.H.; Castilho, R.M. Histone modifications: Targeting head and neck cancer stem cells. World J. Stem Cells 2014, 6, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.; Wang, C.Y. Inhibition of HDAC6 Protein Enhances Bortezomib-induced Apoptosis in Head and Neck Squamous Cell Carcinoma (HNSCC) by Reducing Autophagy. J. Biol. Chem. 2016, 291, 18199–18209. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.O.; Abrahao, A.C.; Rosselli-Murai, L.K.; Giudice, F.S.; Zagni, C.; Leopoldino, A.M.; Squarize, C.H.; Castilho, R.M. NFkappaB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC). FEBS Open Bio 2014, 4, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. 1942. In. J. Epidemiol. 2012, 41, 10–13. [Google Scholar]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ling, S.; Rettig, E.; Sobel, R.; Tan, M.; Fertig, E.J.; Considine, M.; El-Naggar, A.K.; Brait, M.; Fakhry, C.; et al. Epigenetic screening of salivary gland mucoepidermoid carcinoma identifies hypomethylation of CLIC3 as a common alteration. Oral Oncol. 2015, 51, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenet. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Eliot, M.; Butler, R.A.; Cheong, A.; Zhang, X.; McClean, M.D.; Koestler, D.C.; Kelsey, K.T. CpG island methylation profile in non-invasive oral rinse samples is predictive of oral and pharyngeal carcinoma. Clin. Epigenet. 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capella, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell. Mol. Life Sci. 2011, 68, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Li, D.Q.; Muller, S.; Knapp, S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016, 35, 4423–4436. [Google Scholar] [CrossRef] [PubMed]
- Stricker, S.H.; Koferle, A.; Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 2017, 18, 51–66. [Google Scholar] [CrossRef] [PubMed]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone acetylation regulates intracellular pH. Mol. Cell 2013, 49, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef] [PubMed]
- Gegonne, A.; Weissman, J.D.; Singer, D.S. TAFII55 binding to TAFII250 inhibits its acetyltransferase activity. Proc. Natl. Acad. Sci. USA 2001, 98, 12432–12437. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, Part I: Covalent histone modifications. Trends Mol. Med. 2007, 13, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: Targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, F.L.; Cheng, Z.L.; Lei, Q.Y. Impact of acetylation on tumor metabolism. Mol. Cell. Oncol. 2014, 1, e963452. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Gorrini, C.; Amati, B. Tip60 in DNA damage response and growth control: Many tricks in one HAT. Trends Cell Biol. 2006, 16, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Xie, H.J.; Chang, Y.G.; Kim, M.G.; Park, H.; Lee, J.Y.; et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J. Cell. Biochem. 2012, 113, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell. Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Bedi, U.; Mishra, V.K.; Wasilewski, D.; Scheel, C.; Johnsen, S.A. Epigenetic plasticity: A central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 2014, 5, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Dorrance, A.M.; Liu, S.; Yuan, W.; Becknell, B.; Arnoczky, K.J.; Guimond, M.; Strout, M.P.; Feng, L.; Nakamura, T.; Yu, L.; et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J. Clin. Investig. 2006, 116, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Jene-Sanz, A.; Lopez-Bigas, N. The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 2013, 14, r106. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fang, J. Epigenetic regulation of epithelial-mesenchymal transition. Cell. Mol. Life Sci. 2016, 73, 4493–4515. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Nunez, F.J.; Mendez, F.; Brosnan-Cashman, J.A.; Meeker, A.K.; Lowenstein, P.R.; Castro, M.G. Mutated Chromatin Regulatory Factors as Tumor Drivers in Cancer. Cancer Res. 2017, 77, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Hodges, C.; Calarco, J.P.; Braun, S.M.; Ku, W.L.; Kadoch, C.; Zhao, K.; Crabtree, G.R. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017, 49, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Sheu, J.J.; Guan, B.; Jinawath, N.; Markowski, P.; Wang, T.L.; Shih Ie, M. Functional analysis of 11q13.5 amplicon identifies Rsf-1 (HBXAP) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer Res. 2009, 69, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Huang, H.Y.; Lee, S.W.; Lin, C.Y.; Sheu, M.J.; Chang, S.L.; Wu, L.C.; Shiue, Y.L.; Wu, W.R.; Lin, C.M.; et al. Associations of Rsf-1 overexpression with poor therapeutic response and worse survival in patients with nasopharyngeal carcinoma. J. Clin. Pathol. 2012, 65, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.I.; Ahn, J.H.; Lee, K.T.; Shih Ie, M.; Choi, J.H. RSF1 is a positive regulator of NF-kappaB-induced gene expression required for ovarian cancer chemoresistance. Cancer Res. 2014, 74, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Willingham, A.T.; Gingeras, T.R. TUF love for “junk” DNA. Cell 2006, 125, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F. Non-coding RNAs, epigenetics and complexity. Gene 2008, 410, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of Noncoding RNAs in Cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, B.; Benedetti, E.; Cimini, A.; Giordano, A. MicroRNAs: A Puzzling Tool in Cancer Diagnostics and Therapy. Anticancer Res. 2016, 36, 5571–5575. [Google Scholar] [CrossRef] [PubMed]
- Gangaraju, V.K.; Lin, H. MicroRNAs: Key regulators of stem cells. Nat. Rev. Mol. Cell Biol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Srivastava, D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell 2010, 7, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Gasche, J.A.; Goel, A. Epigenetic mechanisms in oral carcinogenesis. Future Oncol. 2012, 8, 1407–1425. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Jacks, T. MicroRNAs and cancer: Short RNAs go a long way. Cell 2009, 136, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, Y.; Lv, P.; Li, L. The role of miR-21 in proliferation and invasion capacity of human tongue squamous cell carcinoma in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 4555–4563. [Google Scholar] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Ayers, D.; Vandesompele, J. Influence of microRNAs and Long Non-Coding RNAs in Cancer Chemoresistance. Genes 2017, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Gorenchtein, M.; Poh, C.F.; Saini, R.; Garnis, C. MicroRNAs in an oral cancer context—From basic biology to clinical utility. J. Dent. Res. 2012, 91, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Kutay, H.; Nasser, M.W.; Nuovo, G.J.; Wang, B.; Majumder, S.; Liu, C.G.; Volinia, S.; Croce, C.M.; Schmittgen, T.D.; et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008, 68, 5049–5058. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Iyer, K.V.; Kumar, A.; Shivashankar, G.V. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11349–11354. [Google Scholar] [CrossRef] [PubMed]
- Giudice, F.S.; Pinto, D.S., Jr.; Nor, J.E.; Squarize, C.H.; Castilho, R.M. Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial-mesenchyme transition of head and neck cancer. PLoS ONE 2013, 8, e58672. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.O.; Guimaraes, D.M.; Martins, M.D.; Martins, M.A.T.; Warner, K.A.; Nor, J.E.; Castilho, R.M.; Squarize, C.H. Unlocking the chromatin of adenoid cystic carcinomas using HDAC inhibitors sensitize cancer stem cells to cisplatin and induces tumor senescence. Stem Cell Res. 2017, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Sun, C.X.; Tran, P.; Punyadeera, C. Salivary epigenetic biomarkers in head and neck squamous cell carcinomas. Biomark. Med. 2016, 10, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.; Yamada, Y.; Hatano, Y.; Miyazaki, Y.; Mori, H.; Shibata, T.; Hara, A. Global DNA hypomethylation suppresses squamous carcinogenesis in the tongue and esophagus. Cancer Sci. 2009, 100, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Subbalekha, K.; Pimkhaokham, A.; Pavasant, P.; Chindavijak, S.; Phokaew, C.; Shuangshoti, S.; Matangkasombut, O.; Mutirangura, A. Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol. 2009, 45, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.J.; Melchers, L.J.; Mastik, M.F.; Slagter-Menkema, L.; Groen, H.J.; van der Laan, B.F.; van Criekinge, W.; de Meyer, T.; Denil, S.; Wisman, G.B.; et al. Identification and validation of WISP1 as an epigenetic regulator of metastasis in oral squamous cell carcinoma. Genes Chromosomes Cancer 2016, 55, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Uzawa, K.; Shibahara, T.; Yokoe, H.; Noma, H.; Tanzawa, H. Expression of an inhibitor of apoptosis, survivin, in oral carcinogenesis. J. Dent. Res. 2003, 82, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Bebek, G.; Bennett, K.L.; Funchain, P.; Campbell, R.; Seth, R.; Scharpf, J.; Burkey, B.; Eng, C. Microbiomic subprofiles and MDR1 promoter methylation in head and neck squamous cell carcinoma. Hum. Mol. Genet. 2012, 21, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.H.; Horng, C.T.; Lee, C.F.; Chiang, N.N.; Tsai, F.J.; Lu, C.C.; Chiang, J.H.; Hsu, Y.M.; Yang, J.S.; Chen, F.A. Epigallocatechin gallate sensitizes cisplatin-resistant oral cancer CAR cell apoptosis and autophagy through stimulating AKT/STAT3 pathway and suppressing multidrug resistance 1 signaling. Environ. Toxicol. 2017, 32, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Arantes, L.M.; de Carvalho, A.C.; Melendez, M.E.; Carvalho, A.L.; Goloni-Bertollo, E.M. Methylation as a biomarker for head and neck cancer. Oral Oncol. 2014, 50, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Jithesh, P.V.; Risk, J.M.; Schache, A.G.; Dhanda, J.; Lane, B.; Liloglou, T.; Shaw, R.J. The epigenetic landscape of oral squamous cell carcinoma. Br. J. Cancer 2013, 108, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Peer, A.; Nair, S.; Chaturvedi, R.K. Epigenetics: A possible answer to the undeciphered etiopathogenesis and behavior of oral lesions. J. Oral Maxillofac. Pathol. 2016, 20, 122–128. [Google Scholar] [PubMed]
- Worsham, M.J.; Stephen, J.K.; Chen, K.M.; Havard, S.; Shah, V.; Gardner, G.; Schweitzer, V.G. Delineating an epigenetic continuum in head and neck cancer. Cancer Lett. 2014, 342, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Towle, R.; Truong, D.; Hogg, K.; Robinson, W.P.; Poh, C.F.; Garnis, C. Global analysis of DNA methylation changes during progression of oral cancer. Oral Oncol. 2013, 49, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.L.; Jeronimo, C.; Kim, M.M.; Henrique, R.; Zhang, Z.; Hoque, M.O.; Chang, S.; Brait, M.; Nayak, C.S.; Jiang, W.W.; et al. Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Righini, C.A.; de Fraipont, F.; Timsit, J.F.; Faure, C.; Brambilla, E.; Reyt, E.; Favrot, M.C. Tumor-specific methylation in saliva: A promising biomarker for early detection of head and neck cancer recurrence. Clin. Cancer Res. 2007, 13, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Coombes, M.M.; Briggs, K.L.; Bone, J.R.; Clayman, G.L.; El-Naggar, A.K.; Dent, S.Y. Resetting the histone code at CDKN2A in HNSCC by inhibition of DNA methylation. Oncogene 2003, 22, 8902–8911. [Google Scholar] [CrossRef] [PubMed]
- Glazer, C.A.; Chang, S.S.; Ha, P.K.; Califano, J.A. Applying the molecular biology and epigenetics of head and neck cancer in everyday clinical practice. Oral Oncol. 2009, 45, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.; Seikaly, H.; Biron, V.L. Epigenetics of oropharyngeal squamous cell carcinoma: Opportunities for novel chemotherapeutic targets. J. Otolaryngol. Head Neck Surg. 2017, 46, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Herbstman, J.; Goldman, L.R. Epigenomic biomonitors: Global DNA hypomethylation as a biodosimeter of life-long environmental exposures. Epigenomics 2011, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Butler, R.A.; Eliot, M.; Pawlita, M.; Maccani, J.Z.; McClean, M.D.; Kelsey, K.T. Novel DNA methylation targets in oral rinse samples predict survival of patients with oral squamous cell carcinoma. Oral Oncol. 2014, 50, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Svinkina, T.; Gu, H.; Silva, J.C.; Mertins, P.; Qiao, J.; Fereshetian, S.; Jaffe, J.D.; Kuhn, E.; Udeshi, N.D.; Carr, S.A. Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Anti-acetyl-lysine Antibodies and an Optimized Proteomic Workflow. Mol. Cell. Proteom. 2015, 14, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, D.M.; Almeida, L.O.; Martins, M.D.; Warner, K.A.; Silva, A.R.; Vargas, P.A.; Nunes, F.D.; Squarize, C.H.; Nor, J.E.; Castilho, R.M. Sensitizing mucoepidermoid carcinomas to chemotherapy by targeted disruption of cancer stem cells. Oncotarget 2016, 7, 42447–42460. [Google Scholar] [CrossRef] [PubMed]
- Webber, L.P.; Wagner, V.P.; Curra, M.; Vargas, P.A.; Meurer, L.; Carrard, V.C.; Squarize, C.H.; Castilho, R.M.; Martins, M.D. Hypoacetylation of acetyl-histone H3 (H3K9ac) as marker of poor prognosis in oral cancer. Histopathology 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Kao, S.Y.; Wang, H.J.; Yang, M.H. Histone modification patterns correlate with patient outcome in oral squamous cell carcinoma. Cancer 2013, 119, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, B.; Raut, S.K.; Panda, N.K.; Rattan, V.; Radotra, B.D.; Khullar, M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol. Cell. Biochem. 2016, 415, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Uzawa, K.; Onda, T.; Shiiba, M.; Yokoe, H.; Shibahara, T.; Tanzawa, H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006, 29, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Chiang, C.P.; Hung, H.C.; Lin, C.Y.; Deng, Y.T.; Kuo, M.Y. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009, 45, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Yoon, J.H. Histone deacetylase 7 silencing induces apoptosis and autophagy in salivary mucoepidermoid carcinoma cells. J. Oral Pathol. Med. 2017, 46, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Yoon, J.H. Histone deacetylase 8 as a novel therapeutic target in oral squamous cell carcinoma. Oncol. Rep. 2017, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Li, X.; Kubota, A.; Kikuchi, K.; Kameda, Y.; Zheng, H.; Miyagi, Y.; Aoki, I.; Takano, Y. SIRT1 expression is associated with good prognosis for head and neck squamous cell carcinoma patients. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 115, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Lin, P.M.; Lin, S.F.; Hsu, C.H.; Lin, H.C.; Hu, M.L.; Hsu, C.M.; Yang, M.Y. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 2013, 34, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.F.; Hu, L.; Zhuo, W.; Zhang, C.M.; Zhou, H.H.; Fan, L. Epigenetic alternations and cancer chemotherapy response. Cancer Chemother. Pharmacol. 2016, 77, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Binda, O.; Champagne, K.S.; Kuo, A.J.; Johnson, K.; Chang, H.Y.; Simon, M.D.; Kutateladze, T.G.; Gozani, O. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol. Cell 2009, 33, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Wu, L.C.; Bruick, R.K. The candidate tumor suppressor ING4 represses activation of the hypoxia inducible factor (HIF). Proc. Natl. Acad. Sci. USA 2005, 102, 7481–7486. [Google Scholar] [CrossRef] [PubMed]
- Shiseki, M.; Nagashima, M.; Pedeux, R.M.; Kitahama-Shiseki, M.; Miura, K.; Okamura, S.; Onogi, H.; Higashimoto, Y.; Appella, E.; Yokota, J.; et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003, 63, 2373–2378. [Google Scholar] [PubMed]
- Li, X.H.; Kikuchi, K.; Zheng, Y.; Noguchi, A.; Takahashi, H.; Nishida, T.; Masuda, S.; Yang, X.H.; Takano, Y. Downregulation and translocation of nuclear ING4 is correlated with tumorigenesis and progression of head and neck squamous cell carcinoma. Oral Oncol. 2011, 47, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.M.; Li, C.F.; Huang, H.Y.; Lai, M.T.; Chen, C.M.; Chiu, I.W.; Wang, T.L.; Tsai, F.J.; Shih Ie, M.; Sheu, J.J. Overexpression of a chromatin remodeling factor, RSF-1/HBXAP, correlates with aggressive oral squamous cell carcinoma. Am. J. Pathol. 2011, 178, 2407–2415. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, Q.; Xia, S.; Xia, B.; Zhang, Y.; Deng, X.; Su, W.; Huang, J. MTA1 overexpression induces cisplatin resistance in nasopharyngeal carcinoma by promoting cancer stem cells properties. Mol. Cells 2014, 37, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Kothandapani, A.; Gopalakrishnan, K.; Kahali, B.; Reisman, D.; Patrick, S.M. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp. Cell Res. 2012, 318, 1973–1986. [Google Scholar] [CrossRef] [PubMed]
- Marzook, H.; Deivendran, S.; Kumar, R.; Pillai, M.R. Role of MTA1 in head and neck cancers. Cancer Metastasis Rev. 2014, 33, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Kozaki, K.; Imoto, I.; Mogi, S.; Omura, K.; Inazawa, J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008, 68, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.F.; Huang, Y.P.; Zheng, Y.F.; Lyu, M.Y.; Wei, S.B.; Meng, Z.; Gan, Y.H. miR-29b suppresses proliferation, migration, and invasion of tongue squamous cell carcinoma through PTEN-AKT signaling pathway by targeting Sp1. Oral Oncol. 2014, 50, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.F.; Wei, S.B.; Mitchelson, K.; Gao, Y.; Zheng, Y.F.; Meng, Z.; Gan, Y.H.; Yu, G.Y. miR-34a inhibits migration and invasion of tongue squamous cell carcinoma via targeting MMP9 and MMP14. PLoS ONE 2014, 9, e108435. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.P.; Tomenson, M.; Cervigne, N.K.; Machado, J.; Jurisica, I.; Pintilie, M.; Sukhai, M.A.; Perez-Ordonez, B.; Grenman, R.; Gilbert, R.W.; et al. Programmed cell death 4 loss increases tumor cell invasion and is regulated by miR-21 in oral squamous cell carcinoma. Mol. Cancer 2010, 9, 238. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Z. MicroRNA expression and its implications for diagnosis and therapy of tongue squamous cell carcinoma. J. Cell Mol. Med. 2016, 20, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, X.; Chen, Z.; Jin, Y.; Heidbreder, C.E.; Kolokythas, A.; Wang, A.; Dai, Y.; Zhou, X. MicroRNA-7 targets IGF1R (insulin-like growth factor 1 receptor) in tongue squamous cell carcinoma cells. Biochem. J. 2010, 432, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.; Huang, Z.; Huang, Z.; He, Z.; You, S. miR-22 regulates cell invasion, migration and proliferation in vitro through inhibiting CD147 expression in tongue squamous cell carcinoma. Arch. Oral Biol. 2016, 66, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Wong, G.; Shiina, M. Up-regulation of Histone Methyltransferase, DOT1L, by Matrix Hyaluronan Promotes MicroRNA-10 Expression Leading to Tumor Cell Invasion and Chemoresistance in Cancer Stem Cells from Head and Neck Squamous Cell Carcinoma. J. Biol. Chem. 2016, 291, 10571–10585. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Huang, H.; Hou, J.; Zhang, B.; Wang, A. miR-181a-Twist1 pathway in the chemoresistance of tongue squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2013, 441, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Zhang, H.; Du, Y.; Tan, P. miR-23a promotes cisplatin chemoresistance and protects against cisplatin-induced apoptosis in tongue squamous cell carcinoma cells through Twist. Oncol. Rep. 2015, 33, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Cao, P.; He, D.; Han, S.; Zhou, J.; Tan, G.; Li, W.; Yu, F.; Yu, J.; Li, Z.; et al. MiR-634 sensitizes nasopharyngeal carcinoma cells to paclitaxel and inhibits cell growth both in vitro and in vivo. Int. J. Clin. Exp. Pathol. 2014, 7, 6784–6791. [Google Scholar] [PubMed]
- Yang, G.D.; Huang, T.J.; Peng, L.X.; Yang, C.F.; Liu, R.Y.; Huang, H.B.; Chu, Q.Q.; Yang, H.J.; Huang, J.L.; Zhu, Z.Y.; et al. Epstein-Barr Virus_Encoded LMP1 upregulates microRNA-21 to promote the resistance of nasopharyngeal carcinoma cells to cisplatin-induced Apoptosis by suppressing PDCD4 and Fas-L. PLoS ONE 2013, 8, e78355. [Google Scholar] [CrossRef] [PubMed]
- Hebert, C.; Norris, K.; Scheper, M.A.; Nikitakis, N.; Sauk, J.J. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 2007, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yao, Y.; Liu, B.; Lin, Z.; Lin, L.; Yang, M.; Zhang, W.; Chen, W.; Pan, C.; Liu, Q.; et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 2012, 31, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.P.; Das, D.N.; Panda, P.K.; Mukhopadhyay, S.; Sinha, N.; Praharaj, P.P.; Agarwal, R.; Bhutia, S.K. Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral Oncol. 2016, 62, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Cote, S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin. Investig. Drugs 2015, 24, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Meeran, S.M. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim. Biophys. Acta 2014, 1840, 3494–3502. [Google Scholar] [CrossRef] [PubMed]
- Allegra, E.; Trapasso, S.; Pisani, D.; Puzzo, L. The role of BMI1 as a biomarker of cancer stem cells in head and neck cancer: A review. Oncology 2014, 86, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Ongkeko, W.M. ABCG2: The key to chemoresistance in cancer stem cells? Expert Opin. Drug Metab. Toxicol. 2009, 5, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.; Krimmel, M.; Polligkeit, J.; Alexander, D.; Munz, A.; Kluba, S.; Keutel, C.; Hoffmann, J.; Reinert, S.; Hoefert, S. ABCB5 expression and cancer stem cell hypothesis in oral squamous cell carcinoma. Eur. J. Cancer 2012, 48, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Medema, J.P. Cancer stem cells—Important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Paduch, R. Theories of cancer origin. Eur. J. Cancer Prev. 2015, 24, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.P.; Martins, M.A.; Martins, M.D.; Warner, K.A.; Webber, L.P.; Squarize, C.H.; Nor, J.E.; Castilho, R.M. Overcoming adaptive resistance in mucoepidermoid carcinoma through inhibition of the IKK-beta/IkappaBalpha/NFkappaB axis. Oncotarget 2016, 7, 73032–73044. [Google Scholar] [PubMed]
- Matsumoto, S.; Tanaka, T.; Kurokawa, H.; Matsuno, K.; Hayashida, Y.; Takahashi, T. Effect of copper and role of the copper transporters ATP7A and CTR1 in intracellular accumulation of cisplatin. Anticancer Res. 2007, 27, 2209–2216. [Google Scholar] [PubMed]
- Singh, A.; Wu, H.; Zhang, P.; Happel, C.; Ma, J.; Biswal, S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol. Cancer Ther. 2010, 9, 2365–2376. [Google Scholar] [CrossRef] [PubMed]
- Harper, L.J.; Costea, D.E.; Gammon, L.; Fazil, B.; Biddle, A.; Mackenzie, I.C. Normal and malignant epithelial cells with stem-like properties have an extended G2 cell cycle phase that is associated with apoptotic resistance. BMC Cancer 2010, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.S.; Lan, H.Y.; Huang, C.H.; Tai, S.K.; Chang, S.Y.; Tsai, T.L.; Chang, C.C.; Tzeng, C.H.; Wu, K.J.; Kao, J.Y.; et al. Regulation of excision repair cross-complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin. Cancer Res. 2010, 16, 4561–4571. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ferdous, T.; Ueyama, Y. Establishment of 5-fluorouracil-resistant oral squamous cell carcinoma cell lines with epithelial to mesenchymal transition changes. Int. J. Oncol. 2014, 44, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ye, D.; Guo, W.; Yu, W.; He, Y.; Hu, J.; Wang, Y.; Zhang, L.; Liao, Y.; Song, H.; et al. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 6887–6901. [Google Scholar] [CrossRef] [PubMed]
- Nor, C.; Zhang, Z.; Warner, K.A.; Bernardi, L.; Visioli, F.; Helman, J.I.; Roesler, R.; Nor, J.E. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia 2014, 16, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Chen, Z.; To, K.K.; Fang, X.; Wang, F.; Cheng, B.; Fu, L. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem. Pharmacol. 2017, 124, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Johnson, N.W.; Chen, X.; Li, R.; Tao, Q.; Gao, J. Disclosure of a stem cell phenotype in an oral squamous cell carcinoma cell line induced by BMP-4 via an epithelial-mesenchymal transition. Oncol. Rep. 2011, 26, 455–461. [Google Scholar] [PubMed]
- Zhao, L.; Ren, Y.; Tang, H.; Wang, W.; He, Q.; Sun, J.; Zhou, X.; Wang, A. Deregulation of the miR-222-ABCG2 regulatory module in tongue squamous cell carcinoma contributes to chemoresistance and enhanced migratory/invasive potential. Oncotarget 2015, 6, 44538–44550. [Google Scholar] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Hatta, M.; Naganuma, K.; Kato, K.; Yamazaki, J. 3-Deazaneplanocin A suppresses aggressive phenotype-related gene expression in an oral squamous cell carcinoma cell line. Biochem. Biophys. Res. Commun. 2015, 468, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. DNA methylation as a therapeutic target in cancer. Clin. Cancer Res. 2007, 13, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. Cancer prevention: Epigenetics steps up to the plate. Cancer Prev. Res. 2008, 1, 219–222. [Google Scholar] [CrossRef] [PubMed]
- McGregor, F.; Muntoni, A.; Fleming, J.; Brown, J.; Felix, D.H.; MacDonald, D.G.; Parkinson, E.K.; Harrison, P.R. Molecular changes associated with oral dysplasia progression and acquisition of immortality: Potential for its reversal by 5-azacytidine. Cancer Res. 2002, 62, 4757–4766. [Google Scholar] [PubMed]
- Tang, X.H.; Albert, M.; Scognamiglio, T.; Gudas, L.J. A DNA methyltransferase inhibitor and all-trans retinoic acid reduce oral cavity carcinogenesis induced by the carcinogen 4-nitroquinoline 1-oxide. Cancer Prev. Res. 2009, 2, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Viet, C.T.; Dang, D.; Achdjian, S.; Ye, Y.; Katz, S.G.; Schmidt, B.L. Decitabine rescues cisplatin resistance in head and neck squamous cell carcinoma. PLoS ONE 2014, 9, e112880. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Shinohara, F.; Nishimura, K.; Echigo, S.; Rikiishi, H. Epigenetic regulation of chemosensitivity to 5-fluorouracil and cisplatin by zebularine in oral squamous cell carcinoma. Int. J. Oncol. 2007, 31, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.D.; Castilho, R.M. Histones: Controlling Tumor Signaling Circuitry. J. Carcinog. Mutagen. 2013, 1 (Suppl. S5), 1–12. [Google Scholar] [PubMed]
- Monneret, C. Histone deacetylase inhibitors. Eur. J. Med. Chem. 2005, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gan, C.P.; Hamid, S.; Hor, S.Y.; Zain, R.B.; Ismail, S.M.; Wan Mustafa, W.M.; Teo, S.H.; Saunders, N.; Cheong, S.C. Valproic acid: Growth inhibition of head and neck cancer by induction of terminal differentiation and senescence. Head Neck 2012, 34, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Gillenwater, A.M.; Zhong, M.; Lotan, R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Mol. Cancer Ther. 2007, 6, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Whang, Y.M.; Choi, E.J.; Seo, J.H.; Kim, J.S.; Yoo, Y.D.; Kim, Y.H. Hyperacetylation enhances the growth-inhibitory effect of all-trans retinoic acid by the restoration of retinoic acid receptor beta expression in head and neck squamous carcinoma (HNSCC) cells. Cancer Chemother. Pharmacol. 2005, 56, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Rikiishi, H.; Shinohara, F.; Sato, T.; Sato, Y.; Suzuki, M.; Echigo, S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int. J. Oncol. 2007, 30, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Suzuki, M.; Sato, Y.; Echigo, S.; Rikiishi, H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int. J. Oncol. 2006, 28, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Islam, M.; Dutta, S.; Roy, S.; Pan, Q.; Teknos, T.N. Suberoylanilide hydroxamic acid inhibits growth of head and neck cancer cell lines by reactivation of tumor suppressor microRNAs. Oral Oncol. 2016, 56, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jung, M.; Dritschilo, A.; Jung, M. Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors. Radiat. Res. 2004, 161, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.B.; Lima, J.P.; Cohen, E.E. Novel targeted therapies in head and neck cancer. Expert Opin. Investig. Drugs 2012, 21, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Smits, K.M.; Melotte, V.; Niessen, H.E.; Dubois, L.; Oberije, C.; Troost, E.G.; Starmans, M.H.; Boutros, P.C.; Vooijs, M.; van Engeland, M.; et al. Epigenetics in radiotherapy: Where are we heading? Radiother. Oncol. 2014, 111, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lee, S.O.; Liang, L.; Luo, J.; Huang, C.K.; Li, L.; Niu, Y.; Chang, C. Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/progenitor cells with 5-aza-2′-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J. Biol. Chem. 2012, 287, 39954–39966. [Google Scholar] [CrossRef] [PubMed]
- Zagonel, V.; Lo Re, G.; Marotta, G.; Babare, R.; Sardeo, G.; Gattei, V.; De Angelis, V.; Monfardini, S.; Pinto, A. 5-Aza-2′-deoxycytidine (Decitabine) induces trilineage response in unfavourable myelodysplastic syndromes. Leukemia 1993, 7 (Suppl. S1), 30–35. [Google Scholar] [PubMed]
- Chikamatsu, K.; Ishii, H.; Murata, T.; Sakakura, K.; Shino, M.; Toyoda, M.; Takahashi, K.; Masuyama, K. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013, 104, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
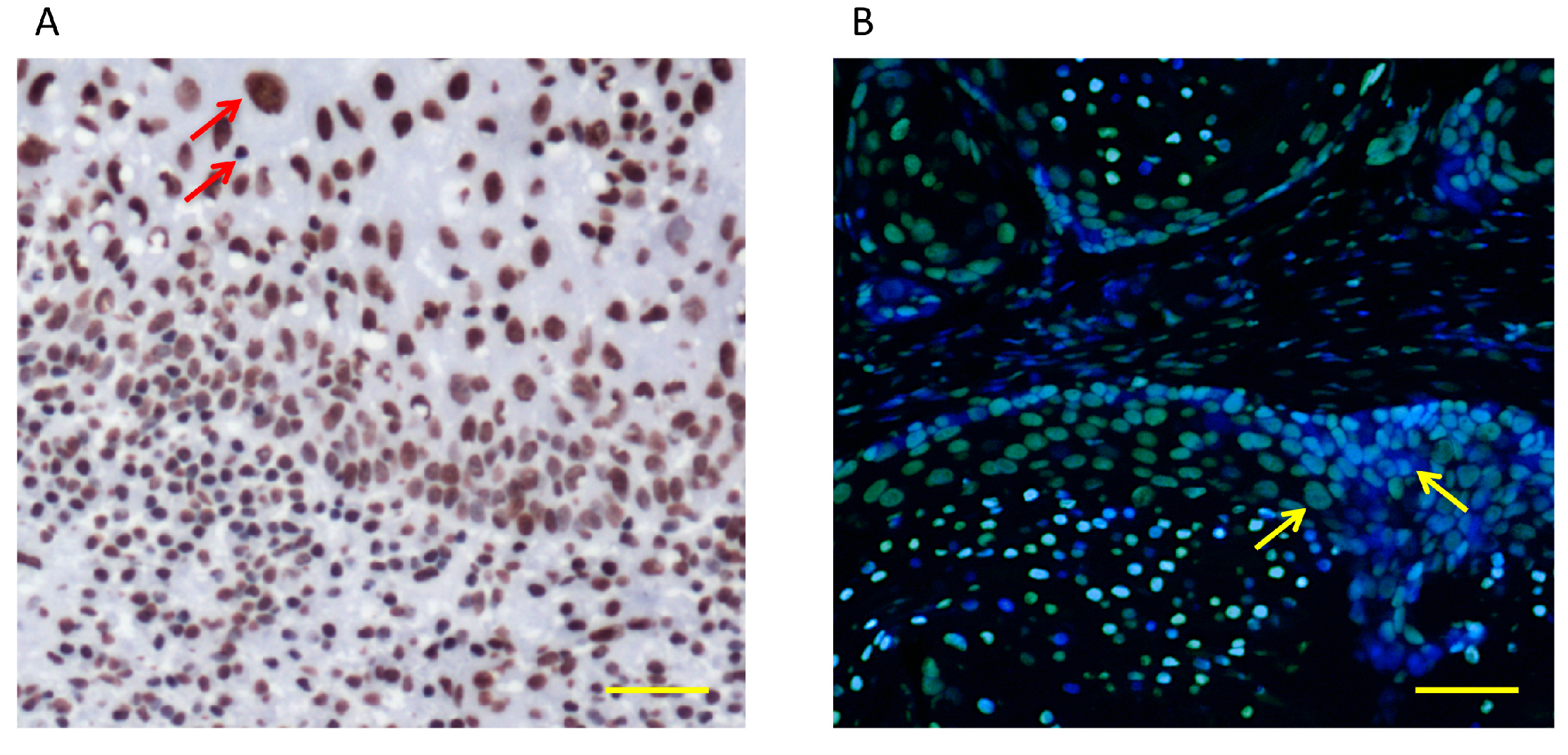

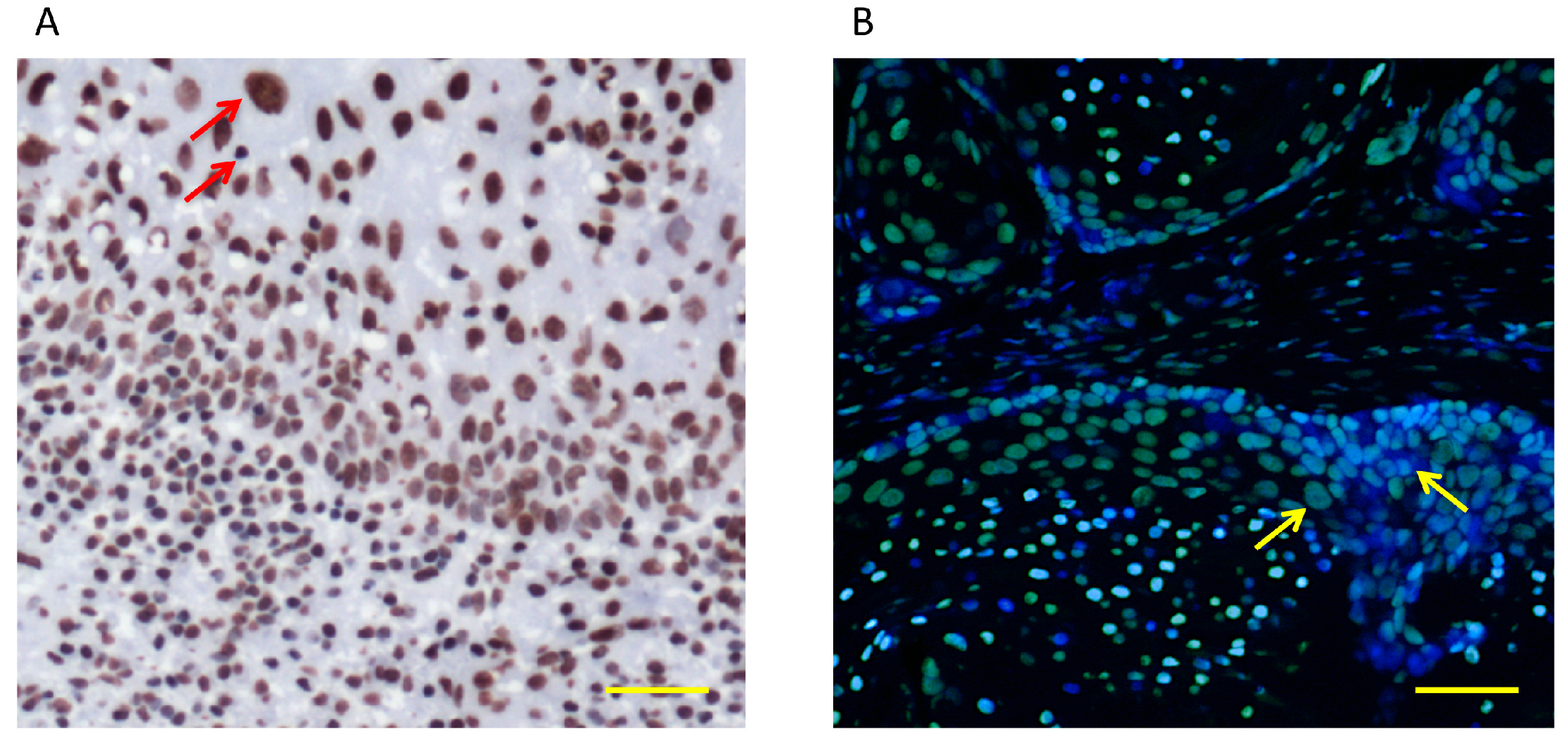{kind=link}
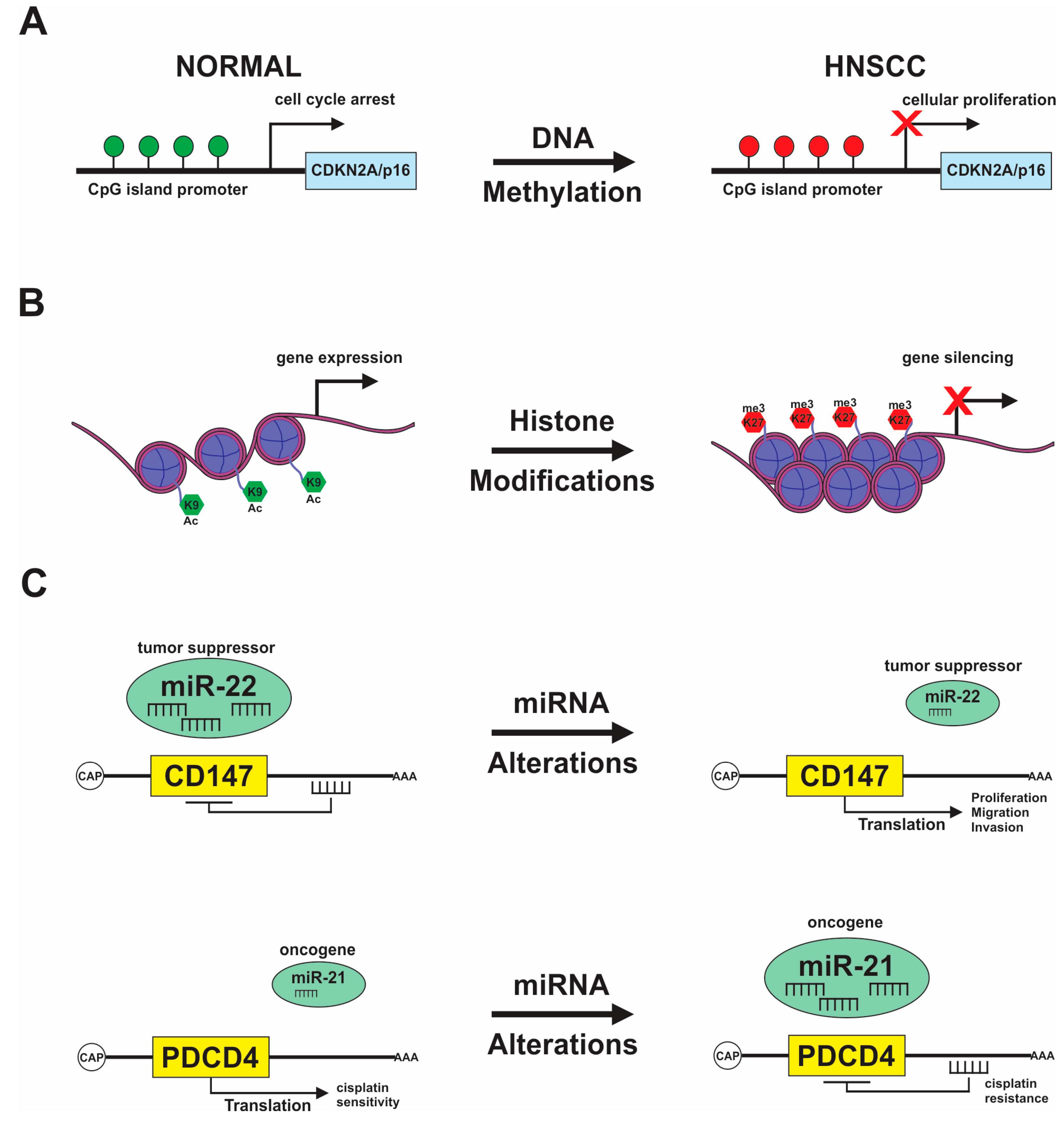
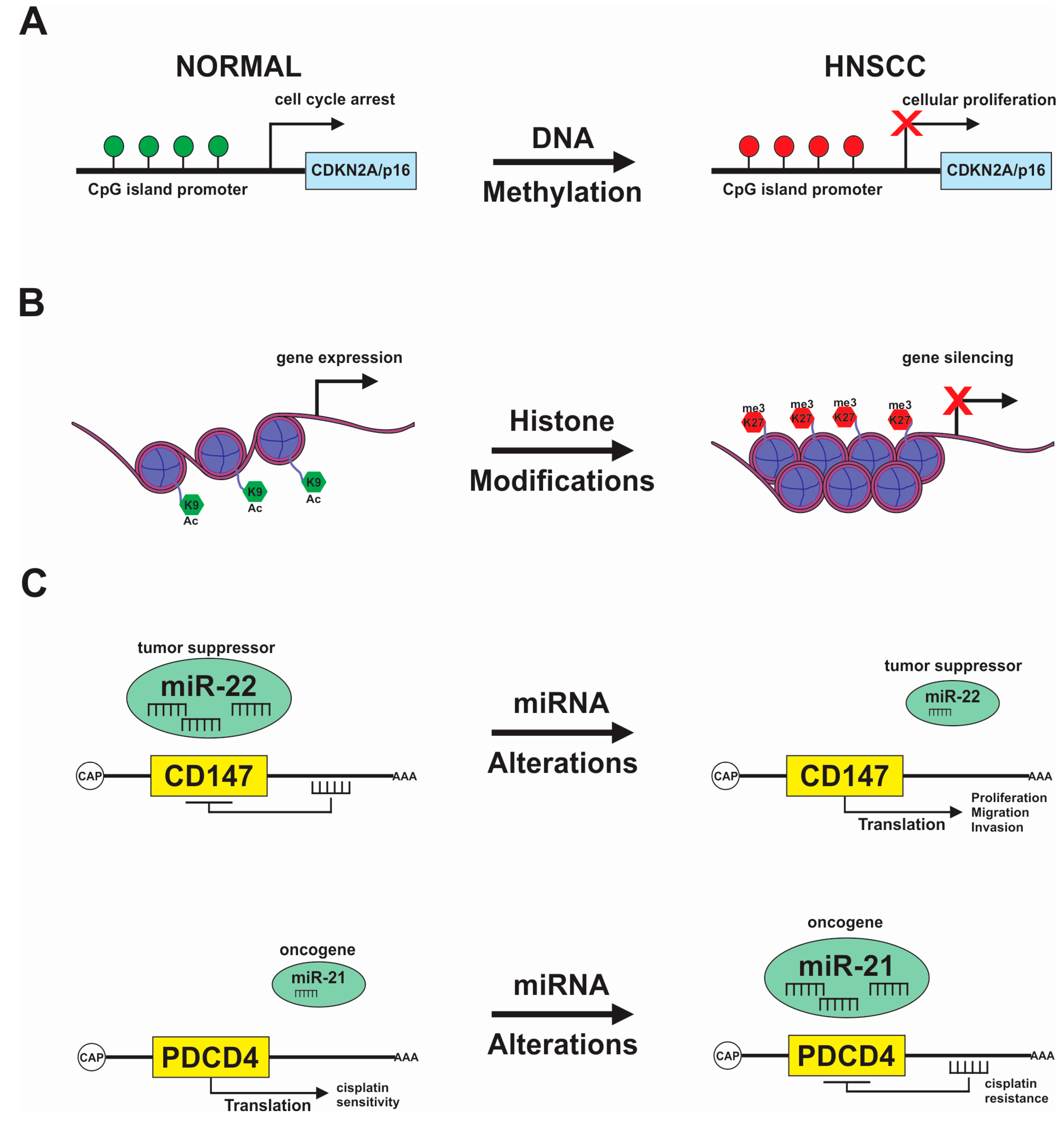{kind=link}
| Gene ID | HGNC ID | Function | Methylation Status | References |
|---|---|---|---|---|
| CDKN2A (p16; p14ARF) | HGNC:1787 | cell cycle arrest/apoptosis/senescence | hypermethylated | [5,77,89,98,99,100,101,102,103,104] |
| MGMT | HGNC:7059 | DNA damage repair | hypermethylated | [5,77,89,98,100,101,102,103,104] |
| DAPK | HGNC:2674 | apoptosis | hypermethylated | [5,89,98,100,101,102,103,104] |
| APC | HGNC:583 | cellular adhesion/migration | hypermethylated | [5,77,98,100,101,102] |
| RASSF1 | HGNC:9882 | cell cycle arrest/ cytoskeleton organization | hypermethylated | [5,77,89,101,103,104] |
| CDH1 | HGNC:1748 | cellular adhesion | hypermethylated | [77,100,102,103,104] |
| HOXA9 | HGNC:5109 | cell differentiation | hypermethylated | [91,98,105,106] |
| MLH1 | HGNC:7127 | DNA damage repair | hypermethylated | [5,77,101] |
| CDKN2B (p15) | HGNC:1788 | cell cycle arrest | hypermethylated | [5,77,100,101] |
| TIMP3 | HGNC:11822 | extracellular matrix degradation | hypermethylated | [89,98,103,104] |
| ATM | HGNC:795 | DNA damage repair | hypermethylated | [5,107] |
| MINT31 | N/A | chromatin remodeling | hypermethylated | [89,98,100] |
| CALCA | HGNC:1437 | cellular metabolism/ inflammatory response | hypermethylated | [99,103,106] |
| NPY | HGNC:7955 | cell proliferation | hypermethylated | [105,106] |
| HS3ST2 | HGNC:5195 | circadian rhythm | hypermethylated | [105,106] |
| ADGRE3 (EMR3) | HGNC:23647 | cellular surface receptor /inflammatory response | hypomethylated | [105] |
| PI3 | HGNC:8947 | inflammatory response | hypomethylated | [105] |
| AIM2 | HGNC:357 | apoptosis/ inflammatory response | hypomethylated | [105] |
| SPP1 | HGNC:11255 | cellular adhesion/ inflammatory response | hypomethylated | [105] |
| ID | Expression | Drug Resistance | Reference |
|---|---|---|---|
| miR-21 | increased | cisplatin | [148] |
| miR-634 | increased | paclitaxel | [149] |
| miR-181a | increased | cisplatin | [150] |
| miR-23a | increased | cisplatin | [151] |
| miR-10b | increased | cisplatin | [152] |
| miR-98 | increased | doxorubicin | [153] |
| miR-214 | increased | cisplatin | [140] |
| miR-200b | decreased | cisplatin | [154] |
| miR-15b | decreased | cisplatin | [154] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int. J. Mol. Sci. 2017, 18, 1506. https://doi.org/10.3390/ijms18071506
Castilho RM, Squarize CH, Almeida LO. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. International Journal of Molecular Sciences. 2017; 18(7):1506. https://doi.org/10.3390/ijms18071506
Chicago/Turabian StyleCastilho, Rogerio M., Cristiane H. Squarize, and Luciana O. Almeida. 2017. "Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy" International Journal of Molecular Sciences 18, no. 7: 1506. https://doi.org/10.3390/ijms18071506
APA StyleCastilho, R. M., Squarize, C. H., & Almeida, L. O. (2017). Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. International Journal of Molecular Sciences, 18(7), 1506. https://doi.org/10.3390/ijms18071506