Endocannabinod Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations


Abstract
:1. Introduction: Autism
2. Autism and Inflammatory State
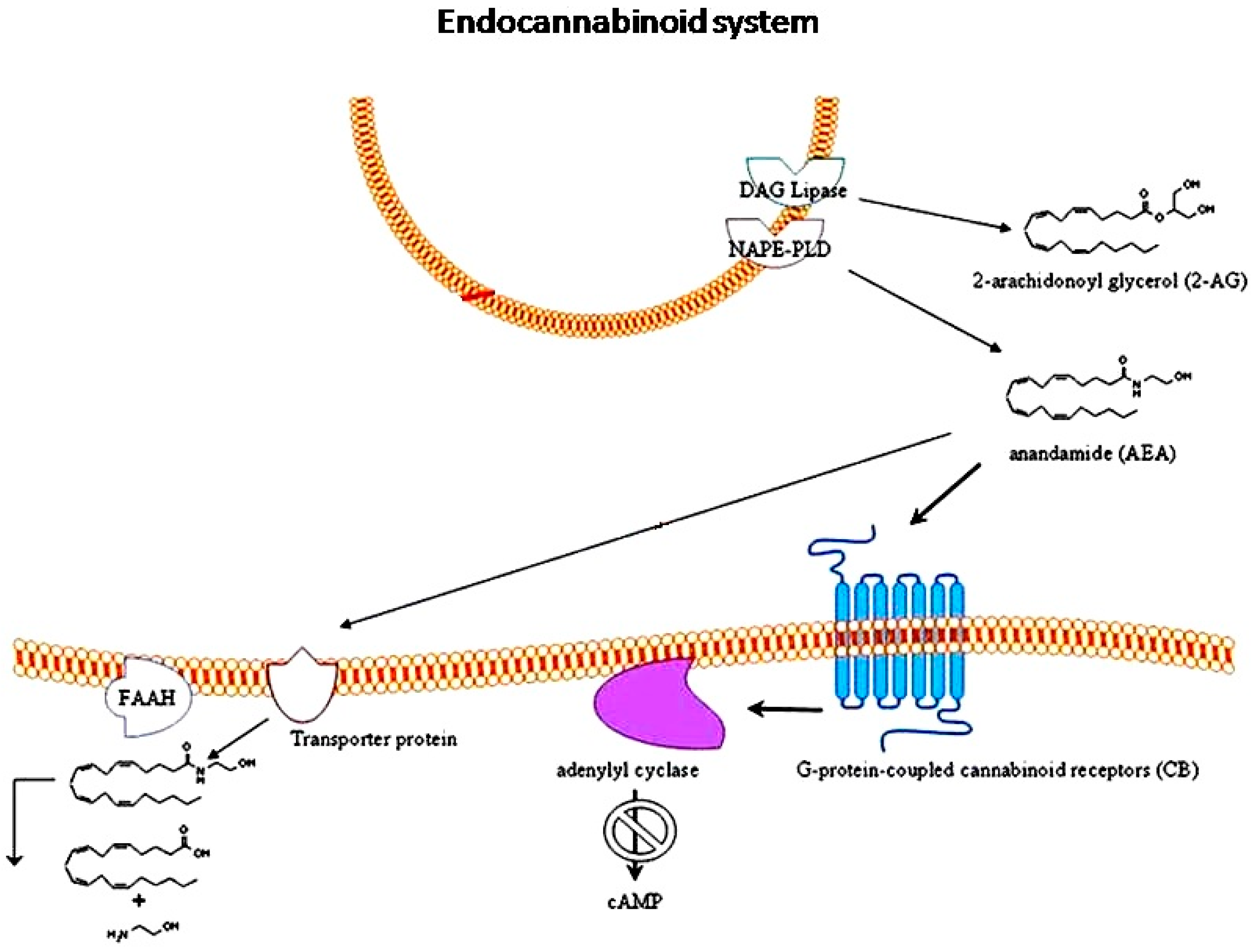
3. Endocannabinoid System
4. EC System in Neuropsychiatric Disorders
5. ECs and Autism
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Autism Spectrum Disorder. Available online: https://www.nimh.nih.gov/health/topics/autism-spectrum-disorders-asd/index.shtml (accessed on 23 June 2017).
- American Psychiatric Association. “Autism Spectrum Disorder, 299.00 (F84.0)”. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013; pp. 50–59, ISBN-13: 978-0890425558. [Google Scholar]
- Maresca, R.; de Magistris, L. Autism: What is it? In Translational Approach to Autism Spectrum Disorder, 1st ed.; Robinson-Agramonte, M., Ed.; Springer International Publishing: Basel, Switzerland, 2015; Volume 1, pp. 1–11. [Google Scholar]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Goines, P.E.; Ashwood, P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol. Teratol. 2013, 36, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Tonhajzerova, I.; Ondrejka, I.; Mestanik, M.; Mikolka, P.; Hrtanek, I.; Mestanikova, A.; Bujnakova, I.; Mokra, D. Inflammatory activity in autism spectrum disorder. Adv. Exp. Med. Biol. 2015, 861, 93–98. [Google Scholar] [PubMed]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, X.; Zhong, Y. Inflammatory cytokines: Potential biomarkers of immunologic dysfunction in autism spectrum disorders. Mediators Inflamm. 2015, 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.P.; Singh, V.K.; Averett, R.E.; Odell, J.D.; Maciulis, A.; Burger, R.A.; Daniels, W.W.; Warren, W.L. Immunogenetic studies in autism and related disorders. Mol. Chem. Neuropathol. 1996, 28, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Geng, L.; Ruby, A.; Zimmerman-Bier, B. Dysregulated innate immune responses in young children with autism spectrum disorders: Their relationship to gastrointestinal symptoms and dietary intervention. Neuropsychobiology 2005, 51, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Wills, S.; van de Water, J. The immune response in autism: A new frontier for autism research. J. Leukoc. Biol. 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Molloy, C.A.; Morrow, A.L.; Meinzen-Derr, J.; Schleifer, K.; Dienger, K.; Manning-Courtney, P.; Altaye, M.; Wills-Karp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, A.M.; Lit, L.; Onore, C.E.; Gregg, J.P.; Hansen, R.L.; Pessah, I.N.; Hertz-Picciotto, I.; van de Water, J.A.; Sharp, F.R.; Ashwood, P. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav. Immun. 2009, 23, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmun. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Harada, M.; Kamimura, D.; Park, J.H.; Kawano, F.; Yull, F.E.; Kawamoto, T.; Iwakura, Y.; Betz, U.A.; Marquez, G.; et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 2012, 148, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Aube, B.; Levesque, S.A.; Pare, A.; Chamma, E.; Kebir, H.; Gorina, R.; Lecuyer, M.A.; Alvarez, J.I.; de Koninck, Y.; Engelhardt, B.; et al. Neutrophils mediate blood-spinal cord barrier disruption in demyelinating neuroinflammatory diseases. J. Immunol. 2014, 193, 2438–2454. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, A.M.; Onore, C.E.; van de Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Warren, R.P.; Odell, J.D.; Warren, W.L.; Cole, P. Antibodies to myelin basic protein in children with autistic behavior. Brain Behav. Immun. 1993, 7, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Chez, M.G.; Pestronk, A.; Arnold, S.T.; Mehta, S.; Deuel, R.K. Serum autoantibodies to brain in Landau-Kleffner variant, autism, and other neurologic disorders. J. Pediatr. 1999, 134, 607–613. [Google Scholar] [CrossRef]
- Ashwood, P.; van de Water, J. Is autism an autoimmune disease? Autoimmun. Rev. 2004, 3, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Cohly, H.H.; Panja, A. Immunological findings in autism. Int. Rev. Neurobiol. 2005, 71, 317–341. [Google Scholar] [PubMed]
- Kawashti, M.I.; Amin, O.R.; Rowehy, N.G. Possible immunological disorders in autism: Concomitant autoimmunity and immune tolerance. Egypt J. Immunol. 2006, 13, 99–104. [Google Scholar] [PubMed]
- Wills, S.; Cabanlit, M.; Bennett, J.; Ashwood, P.; Amaral, D.; van de Water, J. Autoantibodies in autism spectrum disorders (ASD). Ann. N. Y. Acad. Sci. 2007, 1107, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Ashwood, P.; Braunschweig, D.; Cabanlit, M.; van de Water, J.; Amaral, D.G. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav. Immun. 2008, 22, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.; Haapanen, L.; Boyce, R.; Duncanson, P.; Braunschweig, D.; Delwiche, L.; Hansen, R.; Hertz-Picciotto, I.; Ashwood, P.; van de Water, J. Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav. Immun. 2011, 25, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Inga Jácome, M.C.; Morales Chacòn, L.M.; Vera Cuesta, H.; MaragotoRizo, C.; WhilbySantiesteban, M.; Ramos Hernandez, L.; NorisGarcía, E.; González Fraguela, M.E.; Fernandez Verdecia, C.I.; Vegas Hurtado, Y.; et al. Peripheral inflammatory markers contributing to comorbidities in autism. Behav. Sci. 2016, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, G.A.; Al-Ayadhi, L.Y. The relationship between the increased frequency of serum antineuronal antibodies and the severity of autism in children. Eur. J. Paediatr. Neurol. 2012, 16, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Aktas, O.; Smorodchenko, A.; Brocke, S.; Infante-Duarte, C.; Schulze, T.U.; Vogt, J.; Prozorovski, T.; Meier, S.; Osmanova, V.; Pohl, E.; et al. Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 2005, 46, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Garay, P.A.; McAllister, A.K. Novel roles for immune molecules in neural development: Implications for neurodevelopmental disorders. Front. Synaptic Neurosci. 2010, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Sapone, A.; Giordano, C.; Cirillo, A.; de Novellis, V.; de Magistris, L.; Rossi, F.; Fasano, A.; Maione, S.; Antonucci, N. The expression of caspases is enhanced in peripheral blood mononuclear cells of autism spectrum disorder patients. J. Autism Dev. Disord. 2012, 42, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Endocannabinoids and their pharmacological actions. Handb. Exp. Pharmacol. 2015, 231, 1–37. [Google Scholar] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International union of basic and clinical pharmacology. LXXIX. cannabinoidreceptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Zelasko, S.; Arnold, W.R.; Das, A. Endocannabinoid metabolism by cytochrome P450 monooxygenases. Prostaglandins Other Lipid Mediat. 2015, 116–117, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Keereetaweep, J.; Chapman, K.D. Lipidomicanalysis of endocannabinoid signaling: Targeted metabolite identification and quantification. Neural Plast. 2016, 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Mallipeddi, S.; Janero, D.R.; Zvonok, N.; Makriyannis, A. Functional selectivity at G-protein coupled receptors: Advancing cannabinoid receptors as drug targets. Biochem. Pharmacol. 2017, 128, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Di Marzo, V. Short- and long-term plasticity of the endocannabinoid system in neuropsychiatric and neurological disorders. Pharmacol. Res. 2007, 56, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Briley, E.M.; Axelrod, J.; Simpson, J.T.; Mackie, K.; Devane, W.A. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. USA 1993, 90, 7656–7660. [Google Scholar] [CrossRef] [PubMed]
- Vogel, Z.; Barg, J.; Levy, R.; Saya, D.; Heldman, E.; Mechoulam, R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J. Neurochem. 1993, 61, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; Mechoulam, R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.H.; Bayewitch, M.; Avidor-Reiss, T.; Levy, R.; Vogel, Z. Cannabinoid receptor activation differentially regulates the various adenylyl cyclase isozymes. J. Neurochem. 1998, 71, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Taskén, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Sapone, A.; Giordano, C.; Cirillo, A.; de Magistris, L.; Rossi, F.; Fasano, A.; Bradstreet, J.J.; Maione, S.; Antonucci, N. Cannabinoid receptor type 2, but not type 1, is up-regulated in peripheral blood mononuclear cells of children affected by autistic disorders. J. Autism Dev. Disord. 2013, 43, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Tsuboi, K.; Ueda, N. Enzymatic formation of anandamide. Vitam. Horm. 2009, 81, 1–24. [Google Scholar] [PubMed]
- Magotti, P.; Bauer, I.; Igarashi, M.; Babagoli, M.; Marotta, R.; Piomelli, D.; Garau, G. Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: Regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 2015, 23, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Siniscalco, D.; Trovato, A.E.; Comelli, F.; Sotgiu, M.L.; Colleoni, M.; Maione, S.; Rossi, F.; Giagnoni, G. AM404, an inhibitor of anandamide uptake, prevents pain behaviour and modulates cytokine and apoptotic pathways in a rat model of neuropathic pain. Br. J. Pharmacol. 2006, 148, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D. Endocannabinoidsystem as novel therapeutic target for autism treatment. Autism Open Access 2014, 4, e122. [Google Scholar] [CrossRef]
- Keimpema, E.; Calvigioni, D.; Harkany, T. Endocannabinoid signals in the developmental programming of delayed-onset neuropsychiatric and metabolic illnesses. Biochem. Soc. Trans. 2013, 41, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Keimpema, E.; Barabas, K.; Morozov, Y.M.; Tortoriello, G.; Torii, M.; Cameron, G.; Yanagawa, Y.; Watanabe, M.; Mackie, K.; Harkany, T. Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. J. Neurosci. 2010, 30, 13992–14007. [Google Scholar] [CrossRef] [PubMed]
- Morena, M.; de Castro, V.; Gray, J.M.; Palmery, M.; Trezza, V.; Roozendaal, B.; Hill, M.N.; Campolongo, P. Training-associated emotional arousal shapes endocannabinoid modulation of spatial memory retrieval in rats. J. Neurosci. 2015, 35, 13962–13974. [Google Scholar] [CrossRef] [PubMed]
- Khaspekov, L.G.; BrenzVerca, M.S.; Frumkina, L.E.; Hermann, H.; Marsicano, G.; Lutz, B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 2004, 19, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoidsshape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Derkinderen, P.; Valjent, E.; Toutant, M.; Corvol, J.C.; Enslen, H.; Ledent, C.; Trzaskos, J.; Caboche, J.; Girault, J.A. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J. Neurosci. 2003, 23, 2371–2382. [Google Scholar] [PubMed]
- Williams, E.J.; Walsh, F.S.; Doherty, P. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J. Cell Biol. 2003, 160, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Leonoudakis, D.; Abood, M.E.; Beattie, E.C. Cannabinoid receptor activation reduces TNFα-induced surface localization of AMPAR-type glutamate receptors and excitotoxicity. Neuropharmacology 2010, 58, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Van der Stelt, M.; Di Marzo, V. The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: Implications for neurological and psychiatric disorders. Eur. J. Pharmacol. 2003, 480, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Hernández, M.; Ramos, J.A. Cannabinoid-dopamine interaction in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 2010, 16, e72–e91. [Google Scholar] [CrossRef] [PubMed]
- Croxford, J.L. Therapeutic potential of cannabinoids in CNS disease. CNS Drugs 2003, 17, 179–202. [Google Scholar] [CrossRef] [PubMed]
- Carrier, E.J.; Patel, S.; Hillard, C.J. Endocannabinoids in neuroimmunology and stress. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Lunn, C.A.; Reich, E.P.; Bober, L. Targeting the CB2 receptor for immune modulation. Expert Opin. Ther. Targets 2006, 10, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, A.J.; Platt, B. Cannabinoids: Mechanisms and therapeutic applications in the CNS. Curr. Med. Chem. 2003, 10, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Ecker, C.; Bookheimer, S.Y.; Murphy, D.G. Neuroimaging in autism spectrum disorder: Brain structure and function across the lifespan. Lancet Neurol. 2015, 14, 1121–1134. [Google Scholar] [CrossRef]
- Harkany, T.; Mackie, K.; Doherty, P. Wiring and firing neuronal networks: Endocannabinoids take center stage. Curr. Opin. Neurobiol. 2008, 18, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Fride, E.; Gobshtis, F.E.; Dahan, H.; Weller, A.; Giuffrida, A.; Ben-Shabat, S. The endocannabinoid system during development: Emphasis on perinatal events and delayed effects. Vitam. Horm. 2009, 81, 139–158. [Google Scholar] [PubMed]
- McFadden, K.; Minshew, N.J. Evidence for dysregulation of axonal growth and guidance in the etiology of ASD. Front. Hum. Neurosci. 2013, 7, 671. [Google Scholar] [CrossRef] [PubMed]
- Lunn, C.A.; Reich, E.-P.; Fine, J.S.; Lavey, B.; Kozlowski, J.A.; Hipkin, R.W.; Lundell, D.J.; Bober, L. Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br. J. Pharmacol. 2008, 153, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Haskó, G.; Liaudet, L.; Huffman, J.W.; Csiszar, A.; Ungvari, Z.; Mackie, K.; Chatterjee, S.; et al. CB2-receptor stimulation attenuates TNF-α-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte endothelial adhesion. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2210–H2218. [Google Scholar] [CrossRef] [PubMed]
- Servadio, M.; Melancia, F.; Manduca, A.; di Masi, A.; Schiavi, S.; Cartocci, V.; Pallottini, V.; Campolongo, P.; Ascenzi, P.; Trezza, V. Targeting anandamide metabolism rescues core and associated autistic-like symptoms in rats prenatally exposed to valproic acid. Transl. Psychiatry 2016, 6, e902. [Google Scholar] [CrossRef] [PubMed]
- Doenni, V.M.; Gray, J.M.; Song, C.M.; Patel, S.; Hill, M.N.; Pittman, Q.J. Deficient adolescent social behavior following early-life inflammation is ameliorated by augmentation of anandamide signaling. Brain Behav. Immun. 2016, 58, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.M.; Gilmartin, A.; Roche, M. Pharmacological inhibition of fatty acid amide hydrolase attenuates social behavioural deficits in male rats prenatally exposed to valproic acid. Pharmacol. Res. 2016, 113, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.M.; Downey, L.; Conboy, M.; Finn, D.P.; Roche, M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Földy, C.; Malenka, R.; Südhof, T. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 2013, 78, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Bradstreet, J.J.; Cirillo, A.; Antonucci, N. The in vitroGcMAF effects on endocannabinoid system transcriptionomics, receptor formation, and cell activity of autism-derived macrophages. J. Neuroinflamm. 2014, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Chen, L.; Chen, X.; He, X.; Yang, J.; Shi, Y.; Zhou, N. The second intracellular loop of the human cannabinoid CB2 receptor governs G protein coupling in coordination with the carboxyl terminal domain. PLoS ONE 2013, 8, e63262. [Google Scholar]
- Jonsson, K.O.; Vandevoorde, S.; Lambert, D.M.; Tiger, G.; Fowler, C.J. Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoidanandamide. Br. J. Pharmacol. 2001, 133, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, L.; Gouveia-Figueira, S.; Häggström, J.; Alhouayek, M.; Fowler, C.J. The anti-inflammatory compound palmitoylethanolamide inhibits prostaglandin and hydroxyeicosatetraenoic acid production by a macrophage cell line. Pharmacol. Res. Perspect. 2017, 5, e00300. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Borchert, P.; Hinz, B. A simple method for simultaneous determination of N-arachidonoylethanolamine, N-oleoylethanolamine, N-palmitoylethanolamine and 2-arachidonoylglycerol in human cells. Anal. Bioanal. Chem. 2015, 407, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Tsuboi, K.; Zhao, L.Y.; Okamoto, Y.; Lambert, D.M.; Ueda, N. Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim. Biophys. Acta 2005, 1736, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [PubMed]
- De Magistris, L.; Familiari, V.; Pascotto, A.; Sapone, A.; Frolli, A.; Iardino, P.; Carteni, M.; De Rosa, M.; Francavilla, R.; Riegler, G.; et al. Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 418–424. [Google Scholar] [CrossRef] [PubMed]
- De Magistris, L.; Picardi, A.; Siniscalco, D.; Riccio, M.P.; Sapone, A.; Cariello, R.; Abbadessa, S.; Medici, N.; Lammers, K.M.; Schiraldi, C.; et al. Antibodies against food antigens in patients with autistic spectrum disorders. BioMed Res. Int. 2013, 2013, 729349. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, B.; Crupi, R.; Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Beneficial effects of co-ultramicronizedpalmitoylethanolamide/luteolin in a mouse model of autism and in a case report of autism. CNS Neurosci. Ther. 2017, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, N.; Cirillo, A.; Siniscalco, D. Beneficial effects of palmitoylethanolamide on expressive language, cognition, and behaviors in autism: A report of two cases. Case Rep. Psychiatry 2015, 2015, 325061. [Google Scholar] [CrossRef] [PubMed]
- Umathe, S.N.; Manna, S.S.; Utturwar, K.S.; Jain, N.S. Endocannabinoids mediate anxiolytic-like effect of acetaminophen via CB1 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, C.I.; Pérez, M.J.; Manautou, J.E.; Mottino, A.D. Acetaminophen from liver to brain: New insights into drug pharmacological action and toxicity. Pharmacol. Res. 2016, 109, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.; DeSilva, M.; Gu, T.T.; Qiang, M.; Whang, K. Effects of the analgesic acetaminophen (Paracetamol) and its para-aminophenol metabolite on viability of mouse-cultured cortical neurons. Basic Clin. Pharmacol. Toxicol. 2012, 110, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Gould, G.G.; Seillier, A.; Weiss, G.; Giuffrida, A.; Burke, T.F.; Hensler, J.G.; Rock, C.; Tristan, A.; McMahon, L.R.; Salazar, A.; et al. Acetaminophen differentially enhances social behavior and cortical cannabinoid levels in inbred mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Viberg, H.; Eriksson, P.; Gordh, T.; Fredriksson, A. Paracetamol (acetaminophen) administration during neonatal brain development affects cognitive function and alters its analgesic and anxiolytic response in adult male mice. Toxicol. Sci. 2014, 138, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.Z.; Kriebel, D. Prenatal and perinatal analgesic exposure and autism: An ecological link. Environ. Health 2013, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.T.; Klonoff-Cohen, H.S.; Wingard, D.L.; Akshoomoff, N.A.; Macera, C.A.; Ji, M. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: The results of a parent survey. Autism 2008, 12, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.T. Can autism be triggered by acetaminophen activation of the endocannabinoid system? Acta Neurobiol. Exp. 2010, 70, 227–231. [Google Scholar]
- Högestätt, E.D.; Jönsson, B.A.; Ermund, A.; Andersson, D.A.; Björk, H.; Alexander, J.P.; Cravatt, B.F.; Basbaum, A.I.; Zygmunt, P.M. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J. Biol. Chem. 2005, 280, 31405–31412. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, A.; Ferrari, A.; Ottani, A.; Guerzoni, S.; Tacchi, R.; Leone, S. Paracetamol: New vistas of an old drug. CNS Drug Rev. 2006, 12, 250–275. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J. Paracetamol (Acetaminophen): Mechanisms of action. Paediatr. Anaesth. 2008, 18, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Daulhac, L.; Bonnefont, J.; Ledent, C.; Etienne, M.; Chapuy, E.; Libert, F.; Eschalier, A. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain 2008, 139, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Ottani, A.; Leone, S.; Sandrini, M.; Ferrari, A.; Bertolini, A. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur. J. Pharmacol. 2006, 531, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.G.; Schultz, S.T. Similarities in features of autism and asthma and a possible link to acetaminophen use. Med. Hypotheses 2010, 74, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.; Hornik, C.D.; Bilbo, S.; Holzknecht, Z.E.; Gentry, L.; Rao, R.; Lin, S.S.; Herbert, M.R.; Nevison, C.D. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J. Int. Med. Res. 2017, 45, 407–438. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ikeda, S.; Kishimoto, S.; Gokoh, M.; Yanagimoto, S.; Waku, K.; Sugiura, T. 2-Arachidonoylglycerol, an endogenous cannabinoid receptor ligand, induces the migration of EoL-1 human eosinophilic leukemia cells and human peripheral blood eosinophils. J. Leukoc. Biol. 2004, 76, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Frei, R.B.; Luschnig, P.; Parzmair, G.P.; Peinhaupt, M.; Schranz, S.; Fauland, A.; Wheelock, C.E.; Heinemann, A.; Sturm, E.M. Cannabinoid receptor 2 augments eosinophil responsiveness and aggravates allergen-induced pulmonary inflammation in mice. Allergy 2016, 71, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Reuter, I.; Knaup, S.; Romanos, M.; Lesch, K.P.; Drepper, C.; Lillesaar, C. Developmental exposure to acetaminophen does not induce hyperactivity in zebrafish larvae. J. Neural Transm. 2016, 123, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Trezza, V.; Palmery, M.; Trabace, L.; Cuomo, V. Developmental exposure to cannabinoids causes subtle and enduring neurofunctional alterations. Int. Rev. Neurobiol. 2009, 85, 117–133. [Google Scholar] [PubMed]
- Jager, G.; Ramsey, N.F. Long-term consequences of adolescent cannabis exposure on the development of cognition, brain structure and function: An overview of animal and human research. Curr. Drug Abuse Rev. 2008, 1, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Anavi-Goffer, S.; Mulder, J. The polarized life of the endocannabinoid system in CNS development. Chembiochemistry 2009, 10, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, C.; Navarra, M.; Calapai, F.; Spagnolo, E.V.; Busardò, F.P.; Da Cas, R.; Ippolito, F.M.; Calapai, G. Neurological aspects of medical use of cannabidiol. CNS Neurol. Disord. Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Mishima, K.; Fujiwara, M. Therapeutic potential of non-psychotropic cannabidiol in ischemic stroke. Pharmaceuticals 2010, 3, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Zamberletti, E.; Parolaro, D. Endocannabinoids and mental disorders. Handb. Exp. Pharmacol. 2015, 231, 261–283. [Google Scholar] [PubMed]
- Cassano, T.; Calcagnini, S.; Pace, L.; de Marco, F.; Romano, A.; Gaetani, S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol. Ther. 2012, 133, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Solimini, R.; Rotolo, M.C.; Pichini, S.; Pacifici, R. Neurological disorders in medical use of cannabis: An update. CNS Neurol. Disord. Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| First Author | Year | Title | Reference |
|---|---|---|---|
| Schultz | 2008 | Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: The results of a parent survey | [97] |
| Schultz | 2010 | Can Autism Be Triggered by Acetaminophen Activation of the Endocannabinoid System? | [98] |
| Becker | 2010 | Similarities in features of autism and asthma and a possible link to acetaminophen use. | [104] |
| Bauer | 2013 | Prenatal and perinatal analgesic exposure and autism: An ecological link | [96] |
| McFadden | 2013 | Evidence for dysregulation of axonal growth and guidance in the etiology of ASD. | [71] |
| Siniscalco | 2013 | Cannabinoid receptor type 2, but not type 1, is up-regulated in peripheral blood mononuclear cells of children affected by autistic disorders. | [47] |
| Siniscalco | 2014 | The in vitro GcMAF effects on endocannabinoid system transcriptionomics, receptor formation, and cell activity of autism-derived macrophages. | [80] |
| Parker | 2017 | The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. | [105] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brigida, A.L.; Schultz, S.; Cascone, M.; Antonucci, N.; Siniscalco, D. Endocannabinod Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations. Int. J. Mol. Sci. 2017, 18, 1425. https://doi.org/10.3390/ijms18071425
Brigida AL, Schultz S, Cascone M, Antonucci N, Siniscalco D. Endocannabinod Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations. International Journal of Molecular Sciences. 2017; 18(7):1425. https://doi.org/10.3390/ijms18071425
Chicago/Turabian StyleBrigida, Anna Lisa, Stephen Schultz, Mariana Cascone, Nicola Antonucci, and Dario Siniscalco. 2017. "Endocannabinod Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations" International Journal of Molecular Sciences 18, no. 7: 1425. https://doi.org/10.3390/ijms18071425
APA StyleBrigida, A. L., Schultz, S., Cascone, M., Antonucci, N., & Siniscalco, D. (2017). Endocannabinod Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations. International Journal of Molecular Sciences, 18(7), 1425. https://doi.org/10.3390/ijms18071425