Mechanism Investigation of Rifampicin-Induced Liver Injury Using Comparative Toxicoproteomics in Mice

 ,
, 
 and
and
Abstract
:1. Introduction
2. Results
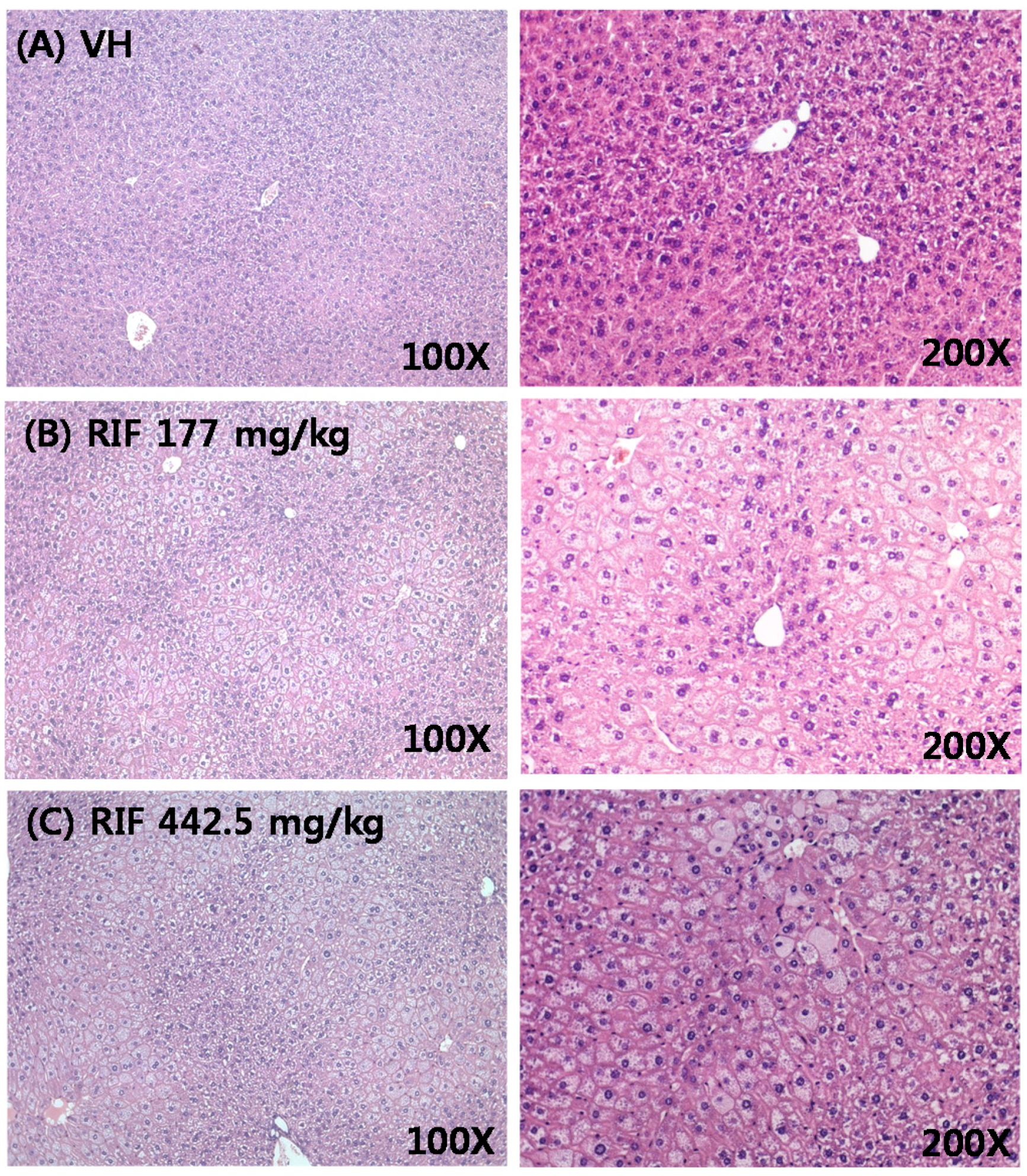
2.1. Rifampicin-Induced Hepatotoxicity
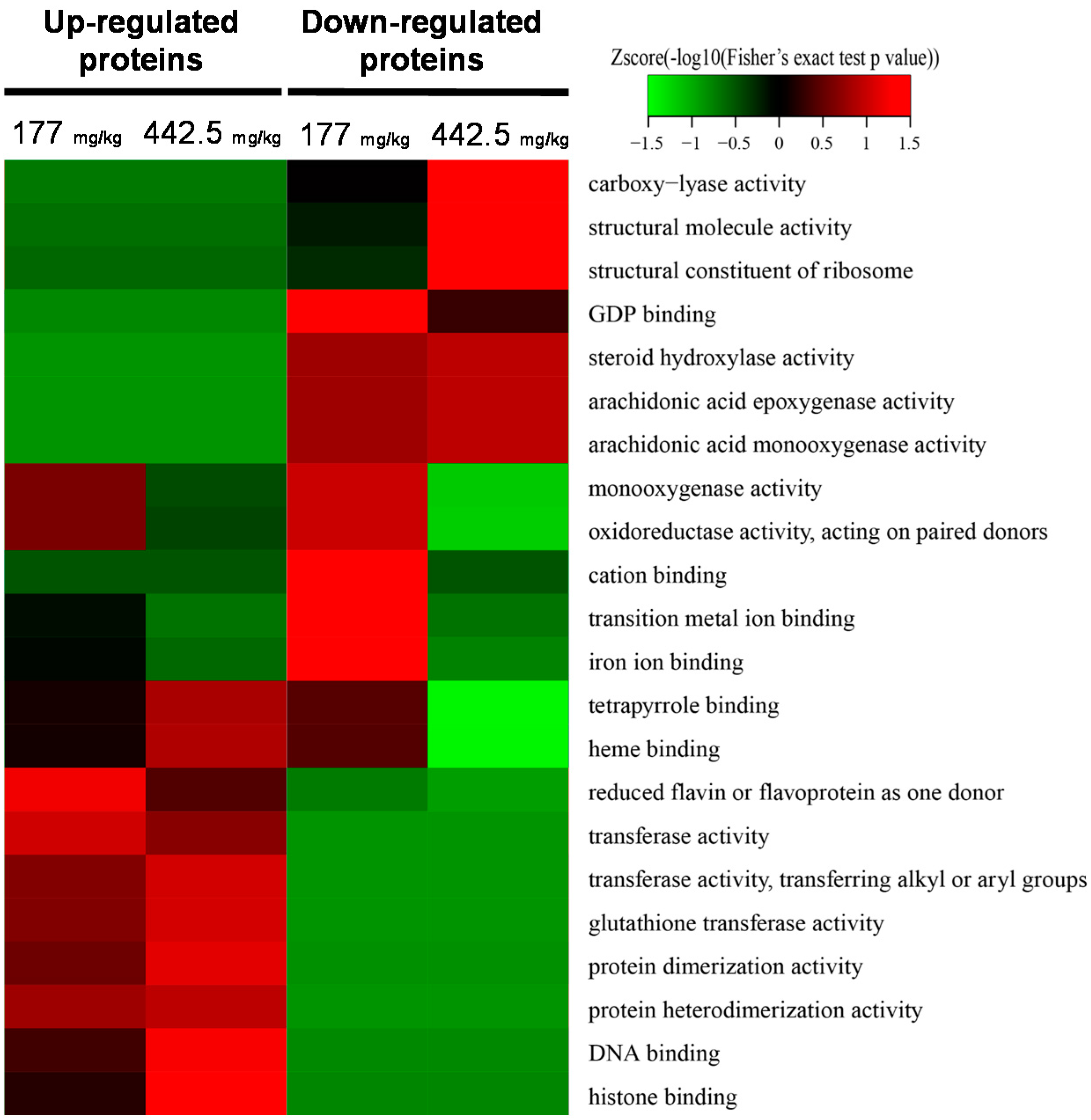
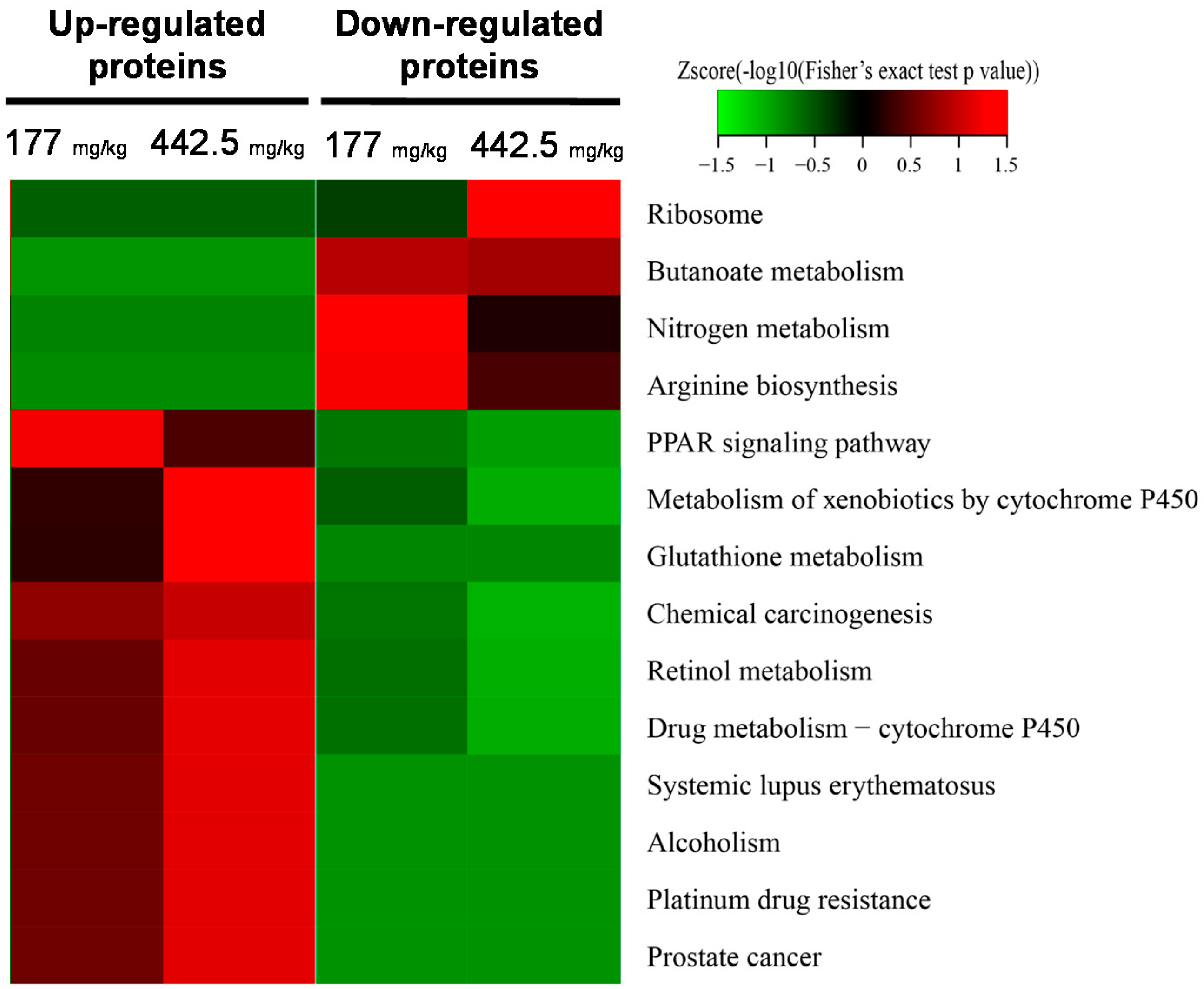
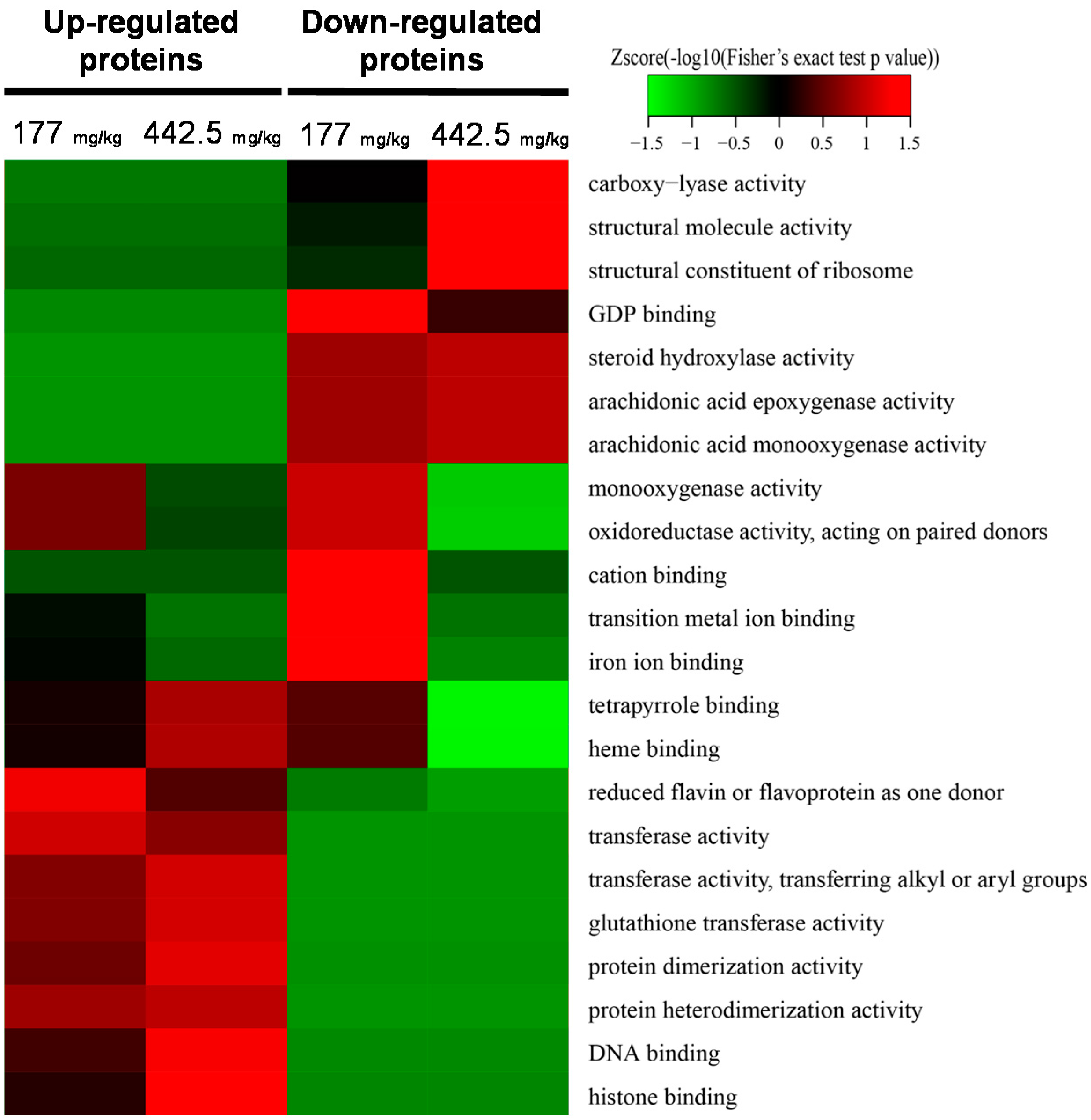
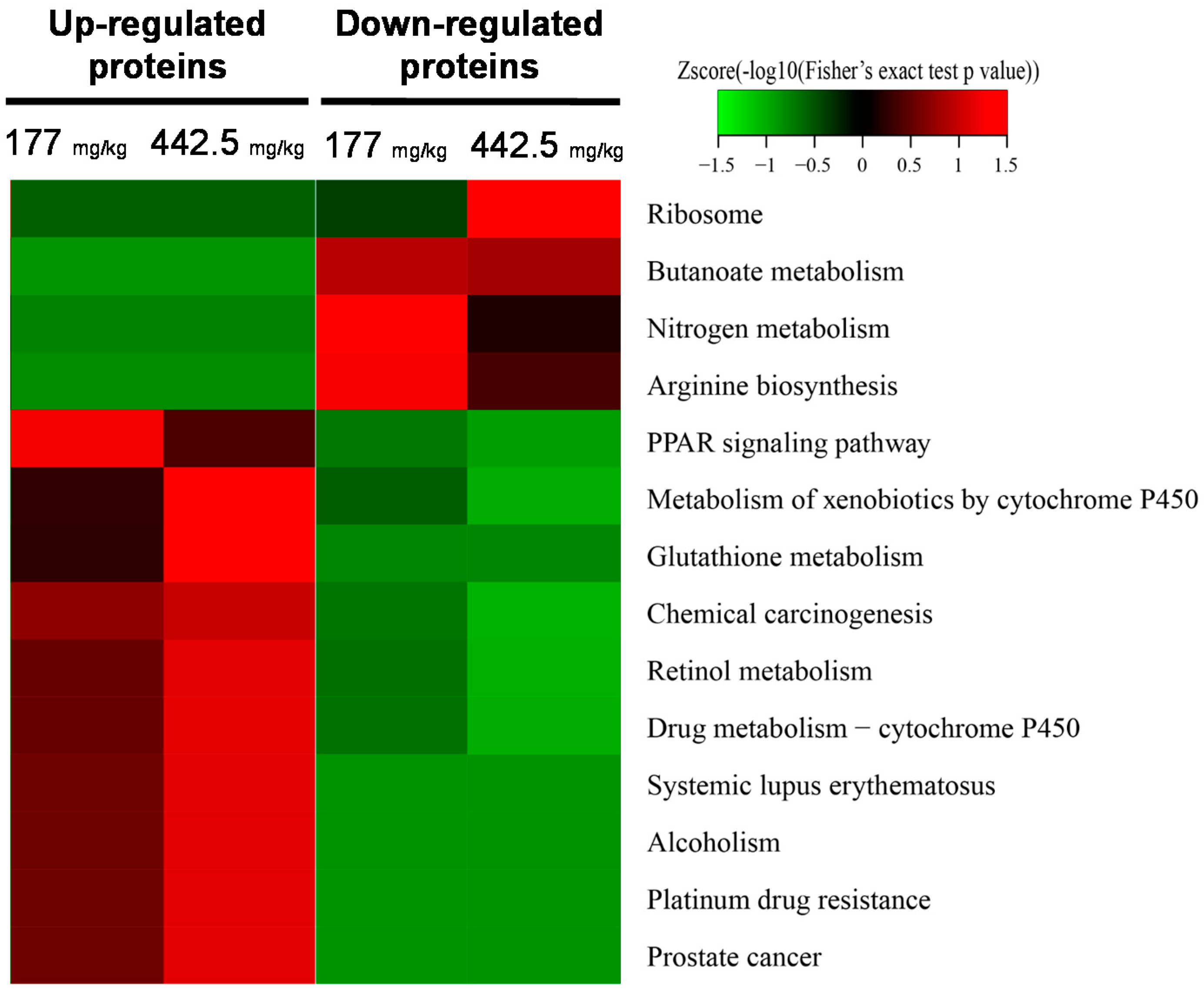
2.2. Quantitative Proteomic Analysis of RIF-Administered Liver Proteins
3. Discussion
4. Materials and Methods
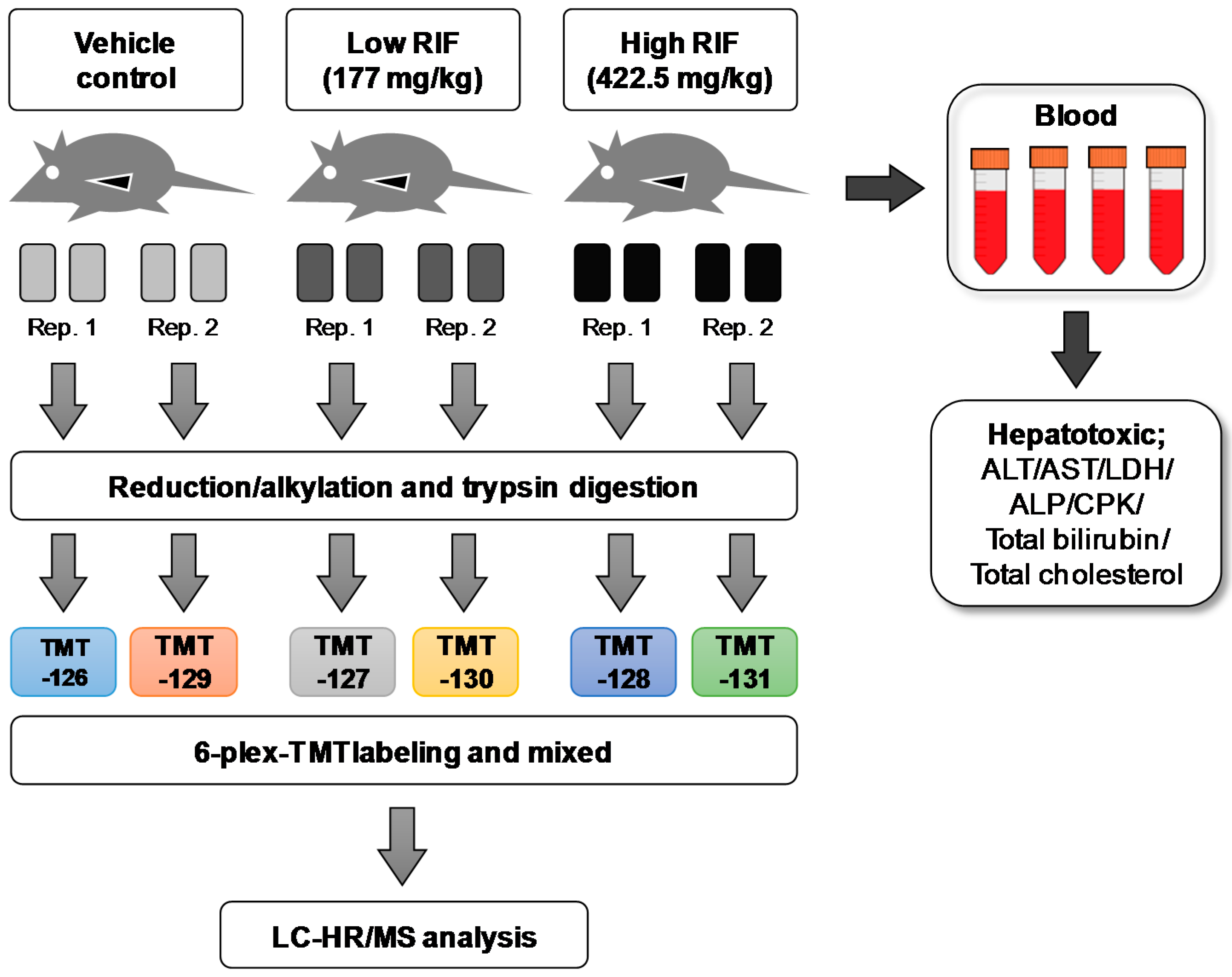
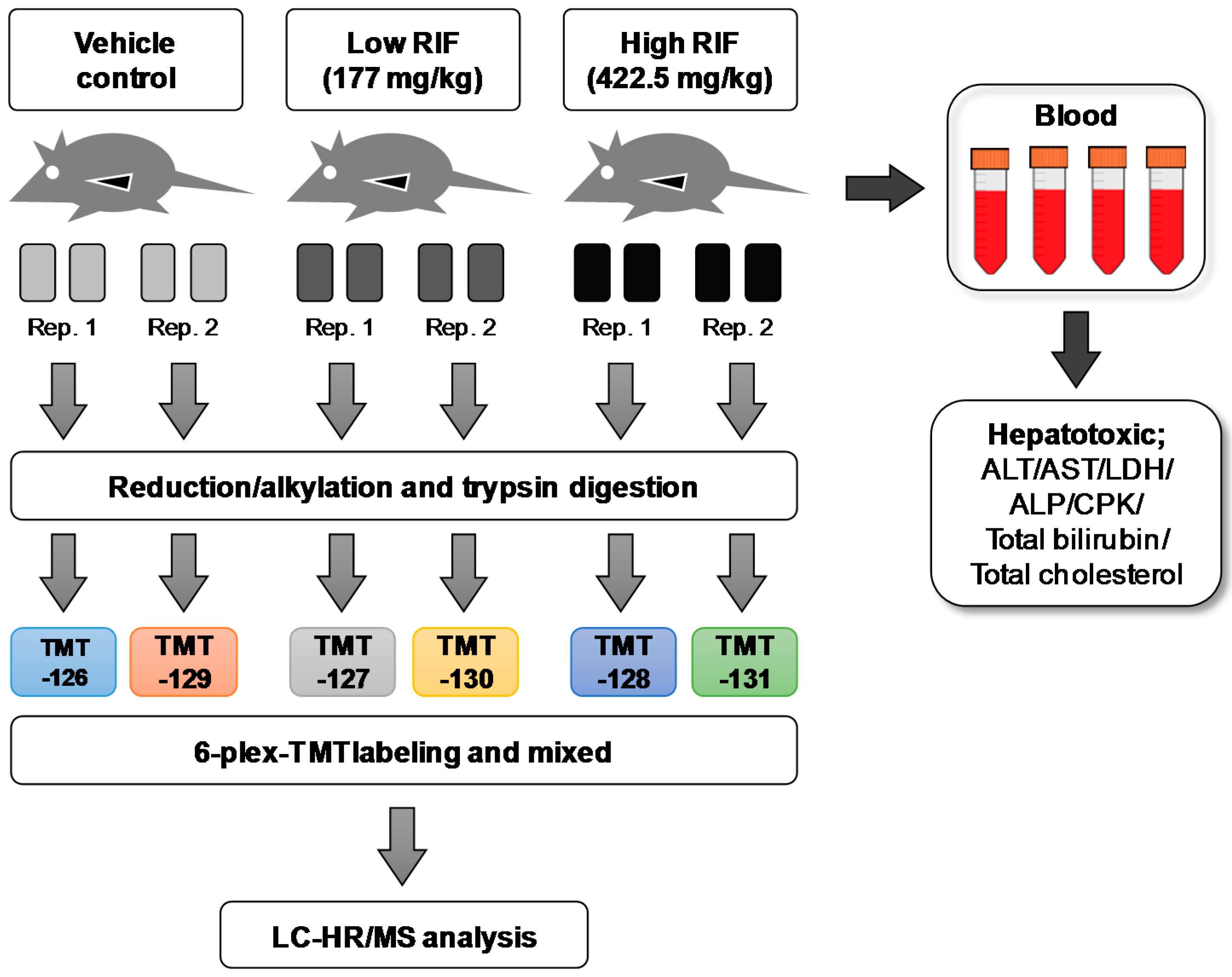
4.1. Experimental Design and Animal Treatment
4.2. Hepatotoxic Parameters and Histology
4.3. Liver Sample Preparation for Proteomic Analysis
4.4. NanoLC-Mass Spectrometry
4.5. Protein Identification and Quantification
4.6. Bioinformatics
4.7. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Holt, M.; Ju, C. Drug-induced liver injury. Handb. Exp. Pharmacol. 2010, 196, 3–27. [Google Scholar]
- Stirnimann, G.; Kessebohm, K.; Lauterburg, B. Liver injury caused by drugs: An update. Swiss Méd. Wkly. 2010, 140, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, L.; Liao, Z.; He, X.; Zhou, Y.; Guo, H. Epidemiology of drug-induced liver injury in China: A systematic analysis of the Chinese literature including 21,789 patients. Eur. J. Gastroen. Hepat. 2013, 25, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Lucena, M.I.; Fernández, M.C.; Pelaez, G.; Pachkoria, K.; García-Ruiz, E.; García-Muñoz, B.; González-Grande, R.; Pizarro, A.; Durán, J.A. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology 2005, 129, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Davern, T.; Serrano, J.; Yang, H.; Rochon, J.; Drug-Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008, 135, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Tuberculosis, Fact Sheet No. 104, Reviewed March 2017. Available online: http://www.who.int/mediacentre/factsheets/fs104/en/ (accessed on 30 June 2017).
- Singh, J.; Arora, A.; Garg, P.; Thakur, V.; Pande, J.; Tandon, R. Antituberculosis treatment-induced hepatotoxicity: Role of predictive factors. Postgrad. Méd. J. 1995, 71, 359–362. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Treatment of Tuberculosis Guidelines, 4th ed.; World Health Organization: Geneva, Switzerland, 2010; p. 46. ISBN 978-92-4-154783-3. [Google Scholar]
- Girling, D. The hepatic toxicity of antituberculosis regimens containing isoniazid, rifampicin and pyrazinamide. Tubercle 1977, 59, 13–32. [Google Scholar] [CrossRef]
- Steel, M.A.; Burk, R.F.; DesPrez, R.M. Toxic hepatitis with isoniazid and rifampin. Chest 1991, 99, 467–471. [Google Scholar] [CrossRef]
- Girling, D. Adverse reactions to rifampicin in antituberculosis regimens. J. Antimicrob. Chemoth. 1977, 3, 115–132. [Google Scholar] [CrossRef]
- Capelle, P.; Dhumeaux, D.; Mora, M.; Feldmann, G.; Berthelot, P. Effect of rifampicin on liver function in man. Gut 1972, 13, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Westphal, J.; Vetter, D.; Brogard, J. Hepatic side-effects of antibiotics. J. Antimicrob. Chemother. 1994, 33, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Zhang, C.; Zhang, D.G.; Li, L.; Chen, X.; Xu, D.X. Rifampicin-induced hepatic lipid accumulation: Association with up-regulation of peroxisome proliferator-activated receptor gamma in mouse liver. PLoS ONE 2016, 11, e0165787. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.T.; Chen, T. PXR variants: The impact on drug metabolism and therapeutic responses. Acta Pharm. Sin. B 2016, 6, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Hong, M.; Choi, H.G.; Kim, J.A.; Lee, S. Investigation of selective inhibitory effects of glycyrol on human CYP 1A1 and 2C9. Xenobiotica 2016, 46, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.F.; Zhao, J.Q.; Bhadauria, M.; Nirala, S.K. Baicalin prevents cadmium induced hepatic cytotoxicity, oxidative stress and histomorphometric alterations. Exp. Toxicol. Pathol. 2013, 65, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Devarbhavi, H.; Singh, R.; Patil, M.; Sheth, K.; Adarsh, C.K.; Balaraju, G. Outcome and determinants of mortality in 269 patients with combination anti-tuberculosis drug-induced liver injury. J. Gastroenterol. Hepatol. 2013, 28, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Santra, A.; Bhattacharjee, K.; Ghatak, S.; Saha, D.R.; Dhali, G.K. Mitochondrial oxidative stress and permeability transition in isoniazid and rifampicin induced liver injury in mice. J. Hepatol. 2006, 45, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Batt, A.M.; Ferrari, L. Manifestations of chemically induced liver damage. Clin. Chem. 1995, 41, 1882–1887. [Google Scholar] [PubMed]
- Yan, J.; Xie, W. A brief history of the discovery of PXR and CAR as xenobiotic receptors. Acta Pharm. Sin. B 2016, 6, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Garty, N.B.; Blanchette-Mackie, E.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar] [PubMed]
- Londos, C.; Brasaemle, D.L.; Schultz, C.J.; Segrest, J.P.; Kimmel, A.R. Perilipins, ADRP, and other proteins that associate with intracellular neutral lipid droplets in animal cells. Semin. Cell Dev. Biol. 1999, 10, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Subramanian, V.; Garcia, A.; Marcinkiewicz, A.; Rothenberg, A. Perilipin a and the control of triacylglycerol metabolism. Mol. Cell. Biochem. 2009, 326, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Schoonjans, K.; Ulven, S.M.; Weedon-Fekjaer, M.S.; Bentzen, T.G.; Koutnikova, H.; Auwerx, J.; Nebb, H.I. Adipose tissue expression of the lipid droplet–associating proteins S3–12 and perilipin is controlled by peroxisome proliferator—Activated receptor-γ. Diabetes 2004, 53, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Straub, B.K.; Stoeffel, P.; Heid, H.; Zimbelmann, R.; Schirmacher, P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 2008, 47, 1936–1946. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J. Atheroscler. Thromb. 2010, 16, 893–901. [Google Scholar] [CrossRef]
- Enriquez-Cortina, C.; Almonte-Becerril, M.; Clavijo-Cornejo, D.; Palestino-Domínguez, M.; Bello-Monroy, O.; Nuño, N.; López, A.; Bucio, L.; Souza, V.; Hernández-Pando, R.; et al. Hepatocyte growth factor protects against isoniazid/rifampicin-induced oxidative liver damage. Toxicol. Sci. 2013, 135, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Tostmann, A.; Boeree, M.J.; Aarnoutse, R.E.; de Lange, W.C.; van der Ven, A.J.; Dekhuijzen, R. Antituberculosis drug-induced hepatotoxicity: Concise up-to-date review. J. Gastroenterol. Hepatol. 2008, 23, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.V.; Ramzan, I.; Murray, M. Impaired microsomal oxidation of the atypical antipsychotic agent clozapine in hepatic steatosis. J. Pharmacol. Exp. Ther. 2007, 322, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.; Jimenez, N.; Serralta, A.; Mir, J.; Castell, J.; Gómez-Lechón, M. Effects of steatosis on drug-metabolizing capability of primary human hepatocytes. Toxicol. In Vitro 2007, 21, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, N.J.; Gómez Lechón, M.J.; Houston, J.B.; Hallifax, D.; Brown, H.S.; Maurel, P.; Kenna, J.G.; Gustavsson, L.; Lohmann, C.; Skonberg, C. Primary hepatocytes: Current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab. Rev. 2007, 39, 159–234. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Bailey, D.; Thomson, S.; Askari, A.; Faulkner, A.; Wheeler-Jones, C. Lipid-metabolizing CYPs in the regulation and dysregulation of metabolism. Annu. Rev. Nutr. 2014, 34, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Taniguchi, K.; Seki, T.; Ishimoto, T.; Sato-Watanabe, M.; Yasuda, Y.; Doi, M.; Kametani, S.; Tomishima, Y.; Ueki, T.; et al. Het0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br. J. Pharmacol. 2001, 133, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Sacerdoti, D.; Balazy, M.; Angeli, P.; Gatta, A.; McGiff, J.C. Eicosanoid excretion in hepatic cirrhosis. Predominance of 20-HETE. J. Clin. Investig. 1997, 100, 1264. [Google Scholar] [CrossRef] [PubMed]
- Woolsey, S.J.; Beaton, M.D.; Choi, Y.H.; Dresser, G.K.; Gryn, S.E.; Kim, R.B.; Tirona, R.G. Relationships between endogenous plasma biomarkers of constitutive cytochrome P450 3A activity and single-time-point oral midazolam microdose phenotype in healthy subjects. Basic Clin. Pharmacol. Toxicol. 2016, 118, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Marde Arrhen, Y.; Nylen, H.; Lovgren-Sandblom, A.; Kanebratt, K.P.; Wide, K.; Diczfalusy, U. A comparison of 4β-hydroxycholesterol: Cholesterol and 6β-hydroxycortisol: Cortisol as markers of CYP3A4 induction. Br. J. Clin. Pharmacol. 2013, 75, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Diczfalusy, U.; Nylen, H.; Elander, P.; Bertilsson, L. 4β-Hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br. J. Clin. Pharmacol. 2011, 71, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R.; Chen, S.; Karin, M.; Tukey, R.H. Reduced expression of UGT1A1 in intestines of humanized UGT1 mice via inactivation of NF-κB leads to hyperbilirubinemia. Gastroenterology 2012, 142, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yueh, M.F.; Evans, R.M.; Tukey, R.H. Pregnane-X-receptor controls hepatic glucuronidation during pregnancy and neonatal development in humanized UGT1 mice. Hepatology 2012, 56, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Galal, A.M.; Walker, L.A.; Khan, I.A. Induction of gst and related events by dietary phytochemicals: Sources, chemistry, and possible contribution to chemoprevention. Curr. Top. Med. Chem. 2015, 14, 2802–2821. [Google Scholar] [CrossRef] [PubMed]
- Tasduq, S.A.; Kaiser, P.; Sharma, S.C.; Johri, R.K. Potentiation of isoniazid-induced liver toxicity by rifampicin in a combinational therapy of antitubercular drugs (rifampicin, isoniazid and pyrazinamide) in Wistar rats: A toxicity profile study. Hepatol. Res. 2007, 37, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 Update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (mg/kg) | Body Weight (g) | Liver Weight (g) | Weight Ratio (Liver/Body Weight) | ||
|---|---|---|---|---|---|
| 0 Day | 7 Days | 14 Days | 14 Days | ||
| 0 | 26.4 ± .2 | 30.1 ± 2.1 | 33.1 ± 2.8 | 1.9 ± 0.23 | 5.8 ± 0.46 |
| 177 | 26.3 ± 0.9 | 30.6 ± 1.5 | 34.5 ± 2.4 | 3.2 ± 0.62 * | 9.1 ± 1.1 * |
| 442.5 | 26.3 ± 1.0 | 30.8 ± 1.6 | 34.2 ± 2.5 | 4.5 ± 0.74 ** | 12.7 ± 1.4 ** |
| Dose (mg/kg) | ALT (IU/L) | AST (IU/L) | ALP (KA Units) | Total Bilirubin (mg/dL) | Total Triglycerides (mg/dL) | Total Cholesterol (mg/dL) |
|---|---|---|---|---|---|---|
| 0 | 85.5 ± 3.1 | 140.5 ± 3.6 | 24.2 ± 2.1 | 0.2 ± 0.1 | 43.7 ± 2.7 | 65.5 ± 5.8 |
| 177 | 185.2 ± 39.2 * | 244.3 ± 28.6 * | 29.4 ± 2.2 | 0.9 ± 0.5 | 31.8 ± 2.7 | 153.2 ± 29.4 * |
| 442.5 | 289.3 ± 55.3 * | 406.2 ± 80.6 * | 28.6 ± 7.2 | 1.8 ± 0.6 | 24.1 ± 2.6 | 224.7 ± 48.0 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Nam, W.S.; Kim, S.J.; Kwon, O.K.; Seung, E.J.; Jo, J.J.; Shresha, R.; Lee, T.H.; Jeon, T.W.; Ki, S.H.; et al. Mechanism Investigation of Rifampicin-Induced Liver Injury Using Comparative Toxicoproteomics in Mice. Int. J. Mol. Sci. 2017, 18, 1417. https://doi.org/10.3390/ijms18071417
Kim J-H, Nam WS, Kim SJ, Kwon OK, Seung EJ, Jo JJ, Shresha R, Lee TH, Jeon TW, Ki SH, et al. Mechanism Investigation of Rifampicin-Induced Liver Injury Using Comparative Toxicoproteomics in Mice. International Journal of Molecular Sciences. 2017; 18(7):1417. https://doi.org/10.3390/ijms18071417
Chicago/Turabian StyleKim, Ju-Hyun, Woong Shik Nam, Sun Joo Kim, Oh Kwang Kwon, Eun Ji Seung, Jung Jae Jo, Riya Shresha, Tae Hee Lee, Tae Won Jeon, Sung Hwan Ki, and et al. 2017. "Mechanism Investigation of Rifampicin-Induced Liver Injury Using Comparative Toxicoproteomics in Mice" International Journal of Molecular Sciences 18, no. 7: 1417. https://doi.org/10.3390/ijms18071417
APA StyleKim, J.-H., Nam, W. S., Kim, S. J., Kwon, O. K., Seung, E. J., Jo, J. J., Shresha, R., Lee, T. H., Jeon, T. W., Ki, S. H., Lee, H. S., & Lee, S. (2017). Mechanism Investigation of Rifampicin-Induced Liver Injury Using Comparative Toxicoproteomics in Mice. International Journal of Molecular Sciences, 18(7), 1417. https://doi.org/10.3390/ijms18071417