Histone Deacetylase Inhibitors as Anticancer Drugs


Abstract
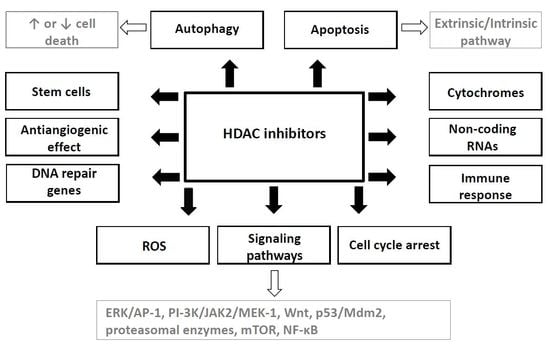
:
1. Introduction
2. Acetylation and Deacetylation
3. HDAC Inhibition and Its Effects
4. Mechanisms of HDAC Inhibitors Action
4.1. Cell Cycle Arrest
4.2 Apoptosis Induction
4.3. The Effects on Induction of Autophagy
4.4. The Effects on Non-Coding RNA
4.5. The Effects on Cellular Signaling Pathways
4.6. The Antiangiogenic Effect
4.7. HDAC Inhibitor-Induced Modulation of Immune Response
4.8. The Effects on Stem Cells
4.9. Other Effects of HDAC Inhibitors
5. Combination of HDAC Inhibitors with Other Anticancer Therapeutic Modalities—Preclinical Studies
6. Clinical Studies and Registered Drugs
7. Conclusions and Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, G.; Margueron, R.; Hu, G.; Stokes, D.; Wang, Y.H.; Reinberg, D. Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol. Cell 2010, 38, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Scarpati, G.D.V.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Herman, J.G.; Guo, M. Epigenome-based personalized medicine in human cancer. Epigenomics 2015, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDACl in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.M.J.; Johnstone, R.R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.-I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Allis, C.D. Histone acetylation and chromatin assembly: A single escort, multiple dances? Cell 1996, 87, 5–8. [Google Scholar] [CrossRef]
- Kazanets, A.; Shorstova, T.; Hilmi, K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yan, H.; Zhuang, S. Histone deacetylases as targets for treatment of multiple diseases. Clin. Sci. 2013, 124, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [PubMed]
- Trapp, J.; Jung, M. The role of NAD+ dependent histone deacetylases (sirtuins) in ageing. Curr. Drug Targets 2006, 7, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Wong, J. HDAC3: Taking the SMRT-N-CoRrect road to repression. Oncogene 2007, 26, 5439–5449. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Mariadason, J.M.; Corner, G.A.; Augenlicht, L.H. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: Comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000, 60, 4561–4572. [Google Scholar] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins—Novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008, 7, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Mai, A. Small-molecule inhibitors of histone deacetylase for the treatment of cancer and non-cancer diseases: A patent review (2011–2013). Expert Opin. Ther. Pat. 2014, 24, 401–415. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, W.K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig. Drugs 2007, 16, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Eckschlager, T.; Poljakova, J.; Hrabeta, J.; Adam, V.; Kizek, R.; Frei, E. The synergistic effects of DNA-targeted chemotherapeutics and histone deacetylase inhibitors as therapeutic strategies for cancer treatment. Curr. Med. Chem. 2012, 19, 4218–4238. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Hadjimichael, C.; Charmpilas, N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012, 2012, 184154. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Sung, J.; Cohen, M.; Chowdhury, W.H.; Sachs, M.D.; Li, Y.; Lakshmanan, Y.; Yung, B.Y.; Lupold, S.E.; Rodriguez, R. Valproic acid inhibits invasiveness in bladder cancer but not in prostate cancer cells. J. Pharmacol. Exp. Ther. 2006, 319, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Stockhausen, M.-T.; Sjölund, J.; Manetopoulos, C.; Axelson, H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br. J. Cancer 2005, 92, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Vrana, J.A.; Decker, R.H.; Johnson, C.R.; Wang, Z.; Jarvis, W.D.; Richon, V.M.; Ehinger, M.; Fisher, P.B.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent G1arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.; Cui, H.; Bradbury, C.M.; Cook, J.; Smart, D.D.K.; Zhao, S.; Young, L.; Brandenburg, S.A.; Hu, Y.; Bisht, K.S.; et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 2004, 6, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 2006, 26, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Mahyar-Roemer, M.; Roemer, K. p21 Waf1/Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene 2001, 20, 3387–3398. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yokozaki, H.; Kuniyasu, H.; Hayashi, K.; Naka, K.; Ono, S.; Ishikawa, T.; Tahara, E.; Yasui, W. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int. J. Cancer 2000, 88, 992–997. [Google Scholar] [CrossRef]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, D.; Donadelli, M.; Pozza, E.D.; Rinalducci, S.; Zolla, L.; Scupoli, M.T.; Righetti, P.G.; Scarpa, A.; Palmieri, M. Synergistic effect of trichostatin A and 5-aza-2′-deoxycytidine on growth inhibition of pancreatic endocrine tumour cell lines: A proteomic study. Proteomics 2009, 9, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.I.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef]
- Walton, T.J.; Li, G.; Seth, R.; McArdle, S.E.; Bishop, M.C.; Rees, R.C. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor β and induce apoptosis in prostate cancer cell-lines. Prostate 2008, 68, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Singh, M.M.; Rivera-Del Valle, N.; Manton, C.A.; Chandra, J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011, 2011, 514261. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.W.; Lee, H.W.; Han, J.W. Apicidin, a Histone Deacetylase Inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA. 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.X.; Huang, L.D.; Jiang, Y.M.; Gutkind, J.S.; Manji, H.K.; Chen, G. The Mood Stabilizer Valproic Acid Activates Mitogen-activated Protein Kinases and Promotes Neurite Growth. J. Biol. Chem. 2001, 276, 31674–31683. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Mobley, A.; Miller, C.; Boklan, J.; Chandra, J. Potentiation of reactive oxygen species is a marker for synergistic cytotoxicity of MS-275 and 5-azacytidine in leukemic cells. Leuk. Res. 2008, 32, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.-S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Shao, L.E.; Diccianni, M.B.; Tanaka, T.; Gribi, R.; Yu, A.L.; Pullen, J.D.; Camitta, B.M.; Yu, J. Thioredoxin expression in primary T-cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001, 61, 7333–7338. [Google Scholar] [PubMed]
- Cipro, Š.; Hřebačková, J.; Hraběta, J.; Poljaková, J.; Eckschlager, T. Valproic acid overcomes hypoxia-induced resistance to apoptosis. Oncol. Rep. 2012, 27, 1219–1226. [Google Scholar] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Choi, I.K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Autophagy, B.; Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyên, L.; Di Lenardo, T.; Semmes, O.J.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar]
- Park, M.A.; Reinehr, R.; Haussinger, D.; Voelkel-Johnson, C.; Ogretmen, B.; Yacoub, A.; Grant, S.; Dent, P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol. Cancer Ther. 2010, 9, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Lassalle, S.; Hofman, V.; Bordone, O.; Tanga, V.G.; Bonnetaud, C.; Moreilhon, C.; Rios, G.; Santini, J.; Barbry, P.; et al. MiR-129–5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Dimri, M.; Dimri, G.P. MicroRNA-31 is a transcriptional target of histone deacetylase inhibitors and a regulator of cellular senescence. J. Biol. Chem. 2015, 290, 10555–10567. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Hiebert, S.W.; Eischen, C.M. Myc Induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016, 76, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kikuchi, J.; Furukawa, Y. Histone deacetylase 1 enhances microRNA processing via deacetylation of DGCR8. EMBO Rep. 2012, 13, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Moran, V.A.; Perera, R.J.; Khalil, A.M. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012, 40, 6391–6400. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhong, Y.; Xie, H.; Lai, X.; Xu, M.; Nie, Y.; Liu, S.; Wan, Y.J.Y. Induction of the liver cancer-down-regulated long noncoding RNA uc002mbe.2 mediates trichostatin-induced apoptosis of liver cancer cells. Biochem. Pharmacol. 2013, 85, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelièvre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces Cancer stem cells differentiation in breast Cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef] [PubMed]
- Asghari, V.; Wang, J.F.; Reiach, J.S.; Young, L.T. Differential effects of mood stabilizers on FosrJun proteins and AP-1 DNA binding activity in human neuroblastoma SH-SY5Y cells. Mol. Brain Res. 1998, 58, 95–102. [Google Scholar] [CrossRef]
- Bassa, B.V.; Roh, D.D.; Vaziri, N.D.; Kirschenbaum, M.A.; Kamanna, V.S. Lysophosphatidylcholine activates mesangial cell PKC and MAP kinase by PLCgamma-1 and tyrosine kinase-Ras pathways. Am. J. Physiol. 1999, 277, F328–F337. [Google Scholar] [PubMed]
- Cieslik, K.; Abrams, C.S.; Wu, K.K. Up-regulation of endothelial nitric-oxide synthase promoter by the phosphatidylinositol 3-kinase γ/Janus kinase 2/MEK-1-dependent pathway. J. Biol. Chem. 2001, 276, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Shen, Y.; Schaefer, J.; Meiri, K.F. Failure to express GAP-43 during neurogenesis affects cell cycle regulation and differentiation of neural precursors and stimulates apoptosis of neurons. Mol. Cell. Neurosci. 2001, 17, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Cinatl, J. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Bug, G.; Gul, H.; Schwarz, K.; Pfeifer, H.; Kampfmann, M.; Zheng, X.; Beissert, T.; Boehrer, S.; Hoelzer, D.; Ottmann, O.G.; Ruthardt, M. Valproic acid stimulates proliferation and self-renewal of hematopoietic stem cells. Cancer Res 2005, 65, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Krämer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef] [PubMed]
- Zupkovitz, G.; Tischler, J.; Posch, M.; Sadzak, I.; Ramsauer, K.; Egger, G.; Grausenburger, R.; Schweifer, N.; Chiocca, S.; Decker, T.; et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell Biol. 2006, 26, 7913–7928. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [PubMed]
- Fulton, D.; Gratton, J.; McCabe, T.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.; Papapetropoulos, A.; Sessa, W. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [PubMed]
- Kawasaki, K.; Smith, R.S.; Hsieh, C.-M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef] [PubMed]
- Rössig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Förstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 2002, 91, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Kotchetkov, R.; Blaheta, R.; Driever, P.H.; Vogel, J.U.; Cinatl, J. Induction of differentiation and suppression of malignant phenotype of human neuroblastoma BE(2)-C cells by valproic acid: Enhancement by combination with interferon-α. Int. J. Oncol. 2002, 20, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Chelluri, R.; Caza, T.; Woodford, M.R.; Reeder, J.E.; Bratslavsky, G.; Byler, T. Valproic acid alters angiogenic and trophic gene expression in human prostate cancer models. Anticancer Res. 2016, 36, 5079–5086. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Suhan, T.; Michaelis, U.R.; Beek, K.; Rothweiler, F.; Tausch, L.; Werz, O.; Eikel, D.; Zörnig, M.; Nau, H.; et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 2006, 13, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, Y.W.; Chen, C.T.; Ho, C.M.; Su, W.H.; Jou, Y.S. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed]
- Balliu, M.; Guandalini, L.; Romanelli, M.N.; D’Amico, M.; Paoletti, F. HDAC-inhibitor (S)-8 disrupts HDAC6-PP1 complex prompting A375 melanoma cell growth arrest and apoptosis. J. Cell. Mol. Med. 2015, 19, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Lienlaf, M.; Wang, H.-W.; Perez-Villarroel, P.; Lee, C.; Woan, K.; Rock-Klotz, J.; Sahakian, E.; Woods, D.; Pinilla-Ibarz, J.; et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 2014, 193, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, M.; Gielen, P.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Malamas, A.S.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget 2016, 7, 7390–7402. [Google Scholar] [PubMed]
- Sabbatino, F.; Schwab, J.H.; Ferrone, S.; Ferrone, C.R. Evolution of studies of HLA class I antigen processing machinery (APM) components in malignant cells. Clin. Transpl. 2013, 1, 453–463. [Google Scholar]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Kortenhorst, M.S.; Wissing, M.D.; Rodríguez, R.; Kachhap, S.K.; Jans, J.J.; Van der Groep, P.; Verheul, H.M.; Gupta, A.; Aiyetan, P.O.; van der Wall, E.; et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics 2013, 8, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, A.; Ling, Q.-D.; Kumar, S.S.; Munusamy, M.A.; Alarfaj, A.A.; Chang, Y.; Kao, S.-H.; Lin, K.-C.; Wang, H.-C.; Umezawa, A. Generation of pluripotent stem cells without the use of genetic material. Lab. Investig. 2015, 95, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Burba, I.; Colombo, G.I.; Staszewsky, L.I.; De Simone, M.; Devanna, P.; Nanni, S.; Avitabile, D.; Molla, F.; Cosentino, S.; Russo, I.; et al. Histone deacetylase inhibition enhances self renewal and cardioprotection by human cord blood-derived CD34 cells. PLoS ONE 2011, 6, e22158. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Cen, J.; Li, J.; Zhao, R.; Zhu, C.; Wang, Z.; Xie, J.; Tang, W. Histone deacetylase inhibitor valproic acid (VPA) promotes the epithelial mesenchymal transition of colorectal cancer cells via up regulation of Snail. Cell Adhes. Migr. 2015, 9, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Luo, Z.; Yu, P.-J.; Xie, H.; He, Y.-W. Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol. Chem. 2016, 397, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Sasai, K.; Kimura, T.; Wang, L.; Aoyanagi, E.; Kohsaka, S.; Tanino, M.; Nishihara, H.; Tanaka, S. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008, 18, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wan, H.; Xue, C.; Jiang, T.; Qian, C.; Zhang, Y. Histone deacetylase 3 implicated in the pathogenesis of children glioma by promoting glioma cell proliferation and migration. Brain Res. 2013, 1520, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.A.; Hraběta, J.; Groh, T.; Procházka, P.; Doktorová, H.; Eckschlager, T. Valproic acid increases CD133 positive cells that show low sensitivity to cytostatics in neuroblastoma. PLoS ONE 2016, 11, e0162916. [Google Scholar] [CrossRef] [PubMed]
- Rudà, R.; Pellerino, A.; Soffietti, R. Does valproic acid affect tumor growth and improve survival in glioblastomas? CNS Oncol. 2016, 5, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Hřebačková, J.; Poljaková, J.; Eckschlager, T.; Hraběta, J.; Procházka, P.; Smutný, S.; Stiborová, M. Histone deacetylase inhibitors valproate and trichostatin A are toxic to neuroblastoma cells and modulate cytochrome P450 1A1, 1B1 and 3A4 expression in these cells. Interdiscip. Toxicol. 2009, 2, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S. Histone deacetylases 1 and 2 regulate DNA replication and DNA repair: Potential targets for genome stability-mechanism-based therapeutics for a subset of cancers. Cell Cycle 2015, 14, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.C.; Jiang, S.; Zhou, X.C.; Hou, X.; Jin, F.; Podratz, K.C.; Jiang, S.-W. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006, 5, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.M.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone deacetylase inhibitors: Current status and overview of recent clinical trials. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Liao, W.S.L.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hoshino, K.; Sanchez-Gonzalez, B.; Kantarjian, H.; Garcia-Manero, G. Antileukemia activity of the combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk. Res. 2005, 29, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Mo, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor Effects of a Combined 5-Aza-2′ Deoxycytidine and Valproic Acid Treatment on Rhabdomyosarcoma and Medulloblastoma in Ptch Mutant Mice. Cancer Res. 2009, 69, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Klisovic, M.I.; Maghraby, E.A.; Parthun, M.R.; Guimond, M.; Sklenar, A.R.; Whitman, S.P.; Chan, K.K.; Murphy, T.; Anon, J.; Archer, K.J.; et al. Depsipeptide (FR 901228) promotes histone acetylation, gene transcription, apoptosis and its activity is enhanced by DNA methyltransferase inhibitors in AML1/ETO-positive leukemic cells. Leukemia 2003, 17, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Li, L.; Zhou, W.; Wu, L.; Zhao, Y.; Wang, D.; Lu, S.; Yu, Y.; Wang, H.; McNutt, M.A.; et al. HDAC inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing removal of incorporated abases in lung cancer cells. PLoS ONE 2008, 3, e2445. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Zhang, H.; Jia, Y.; Wei, Y.; Yang, H.; Zhang, W.; Fiskus, W.; Bhalla, K.; et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood 2010, 116, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Greve, G.; Schiffmann, I.; Pfeifer, D.; Pantic, M.; Schüler, J.; Lübbert, M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer 2015, 15, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Takashina, T.; Kinoshita, I.; Kikuchi, J.; Shimizu, Y.; Sakakibara-Konishi, J.; Oizumi, S.; Nishimura, M.; Dosaka-Akita, H. Combined inhibition of EZH2 and histone deacetylases as a potential epigenetic therapy for non-small-cell lung cancer cells. Cancer Sci. 2016, 107, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.-Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.I.; Dunner, K.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Lembersky, D.; Kramer, L.; Fisher, R.I.; Friedberg, J.; Dent, P.; Grant, S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 2010, 115, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Lembersky, D.; Son, M.P.; Attkisson, E.; Dent, P.; Fisher, R.I.; Friedberg, J.W.; Grant, S. Carfilzomib interacts synergistically with histone deacetylase inhibitors in mantle cell lymphoma cells in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Groh, T.; Hrabeta, J.; Khalil, M.A.; Doktorova, H.; Eckschlager, T.; Stiborova, M. The synergistic effects of DNA-damaging drugs cisplatin and etoposide with a histone deacetylase inhibitor valproate in high-risk neuroblastoma cells. Int. J. Oncol. 2015, 47, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Luchenko, V.L.; Salcido, C.D.; Zhang, Y.; Agama, K.; Komlodi-Pasztor, E.; Murphy, R.F.; Giaccone, G.; Pommier, Y.; Bates, S.E.; Varticovski, L. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle 2011, 10, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of Histone Deacetylase Increases Cytotoxicity to Anticancer Drugs Targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Catalano, M.G.; Poli, R.; Pugliese, M.; Fortunati, N.; Boccuzzi, G. Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr. Relat. Cancer 2007, 14, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.H.; Yoon, W.S.; Park, K.Y.; Kim, S.M.; Lim, J.Y.; Woo, J.S.; Jeong, C.H.; Hou, Y.; Jeun, S.S. Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. J. Biomed. Biotechnol. 2012, 2012, 987495. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. DNA Methyltransferase and Histone Deacetylase Inhibitors in the Treatment of Myelodysplastic Syndromes. Semin. Hematol. 2008, 45, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Rudek, M.A.; Zhao, M.; He, P.; Hartke, C.; Gilbert, J.; Gore, S.D.; Carducci, M.A.; Baker, S.D. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J. Clin. Oncol. 2005, 23, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Vesole, D.H.; Jagannath, S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: A case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010, 10, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Afifi, S.; Michael, A.; Azimi, M.; Rodriguez, M.; Lendvai, N.; Landgren, O. Role of Histone Deacetylase Inhibitors in Relapsed Refractory Multiple Myeloma: A Focus on Vorinostat and Panobinostat. Pharmacotherapy 2015, 35, 1173–1188. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Tambaro, F.P.; Bekele, N.B.; Yang, H.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Kadia, T.M.; Konopleva, M.Y.; Faderl, S.; et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J. Clin. Oncol. 2012, 30, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Altman, J.K.; Licht, J.D. New strategies in acute myeloid leukemia: Redefining prognostic markers to guide therapy. Clin. Cancer Res. 2012, 18, 5163–5171. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIB Multicenter Trial of Vorinostat in Patients With Persistent, Progressive, or Treatment Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Marcucci, G. Targeting epigenetic changes in acute myeloid leukemia. Clin. Adv. Hematol. Oncol. 2005, 3, 855–865. [Google Scholar] [PubMed]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Campigotto, F.; Murphy, T.J.; Boswell, E.N.; Banwait, R.; Azab, F.; Chuma, S.; Kunsman, J.; Donovan, A.; Masood, F.; et al. Results of a phase 2 trial of the single-agent histone deacetylase inhibitor panobinostat in patients with relapsed/refractory Waldenstrom macroglobulinemia. Blood 2013, 121, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Advani, A.; Fernandez, L.; Van Der Jagt, R.; Brandwein, J.; Kambhampati, S.; Kassis, J.; Davis, M.; Bonfils, C.; Dubay, M.; et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2009, 147, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.; Fischer, T.; Cortes, J.; Garcia-Manero, G.; Beck, J.; Ravandi, F.; Masson, E.; Rae, P.; Laird, G.; Sharma, S.; et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res. 2006, 12, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Futur. Med Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- O'connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; Van Den Bent, M.J.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Scherpereel, A.; Berghmans, T.; Lafitte, J.J.; Colinet, B.; Richez, M.; Bonduelle, Y.; Meert, A.P.; Dhalluin, X.; Leclercq, N.; Paesmans, M.; et al. Valproate-doxorubicin: Promising therapy for progressing mesothelioma. A phase II study. Eur. Respir. J. 2011, 37, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Jotte, R.M.; Konduri, K.; Neubauer, M.A.; Spira, A.I.; Ruxer, R.L.; Varella-Garcia, M.; Bunn, P.A.; Hirsch, F.R. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012, 30, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic therapeutics: A new weapon in the war against cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed]
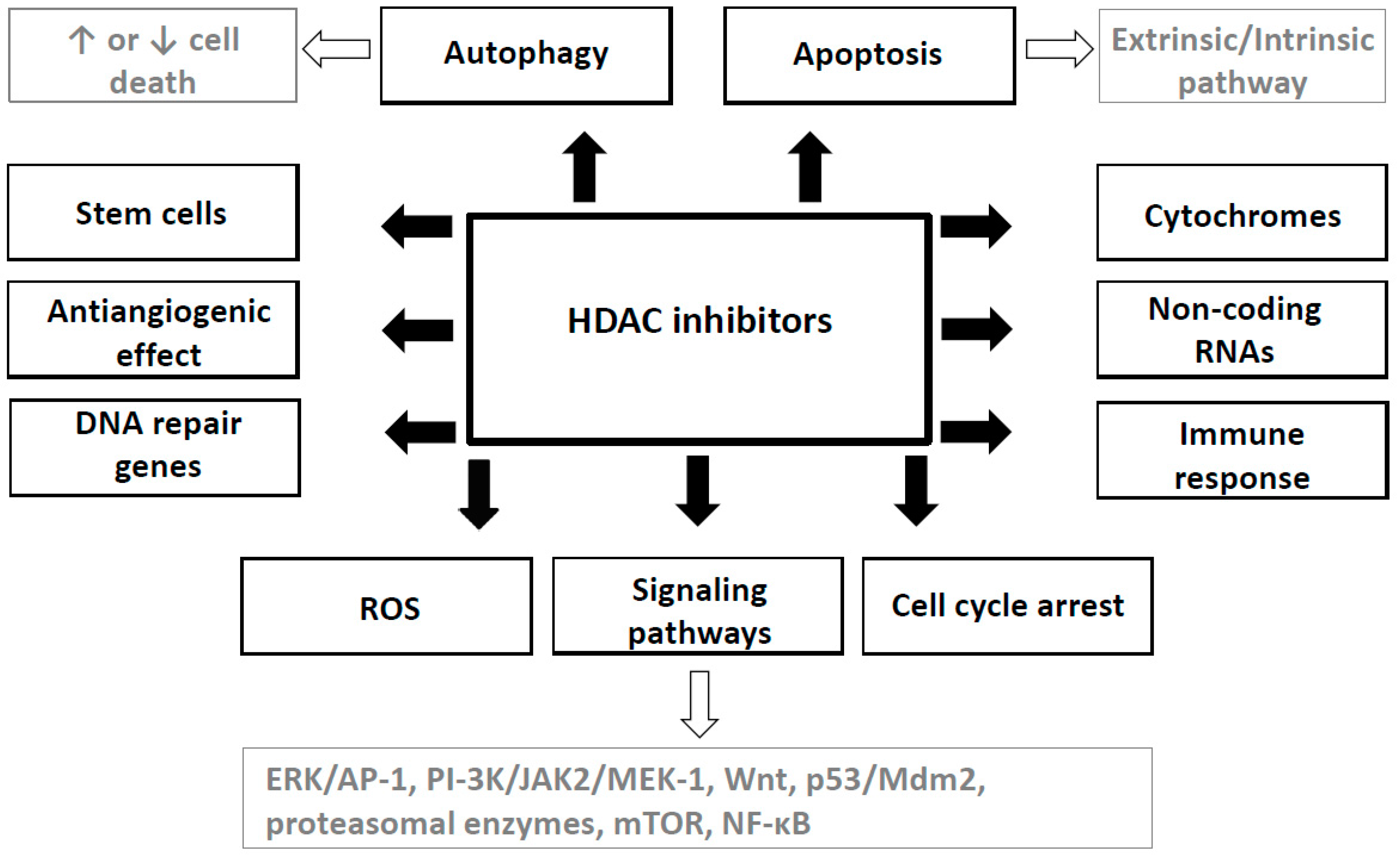

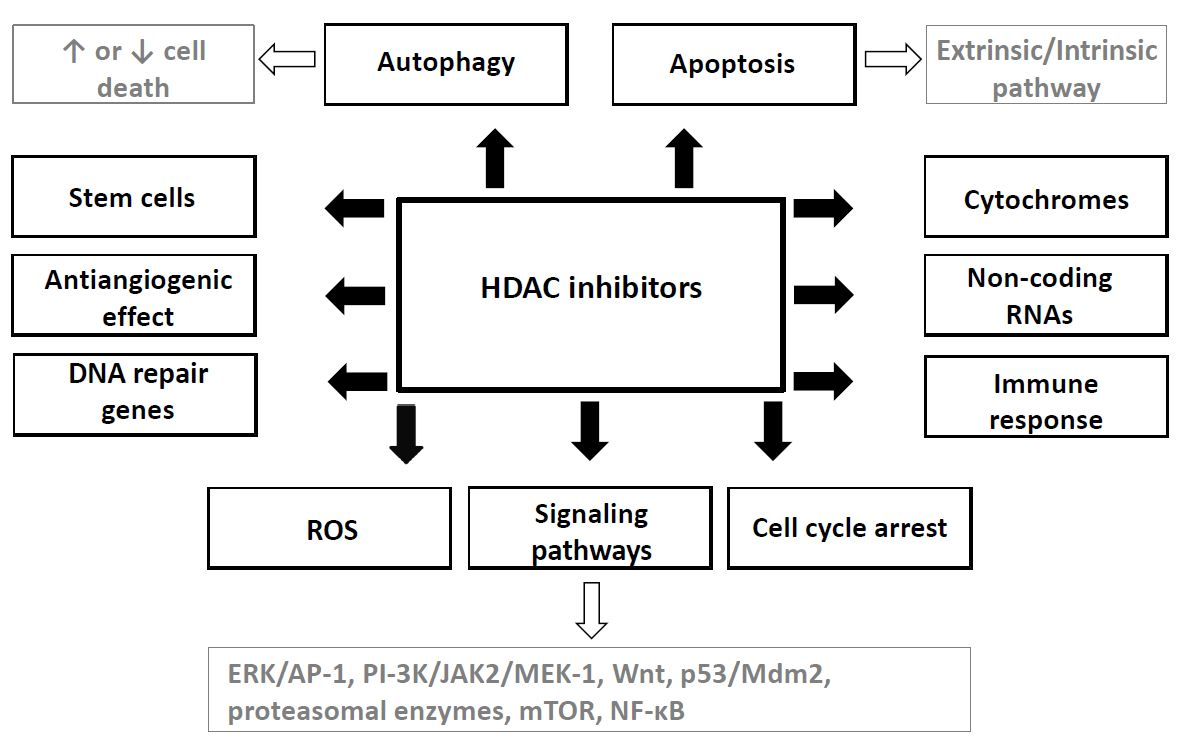{kind=link}
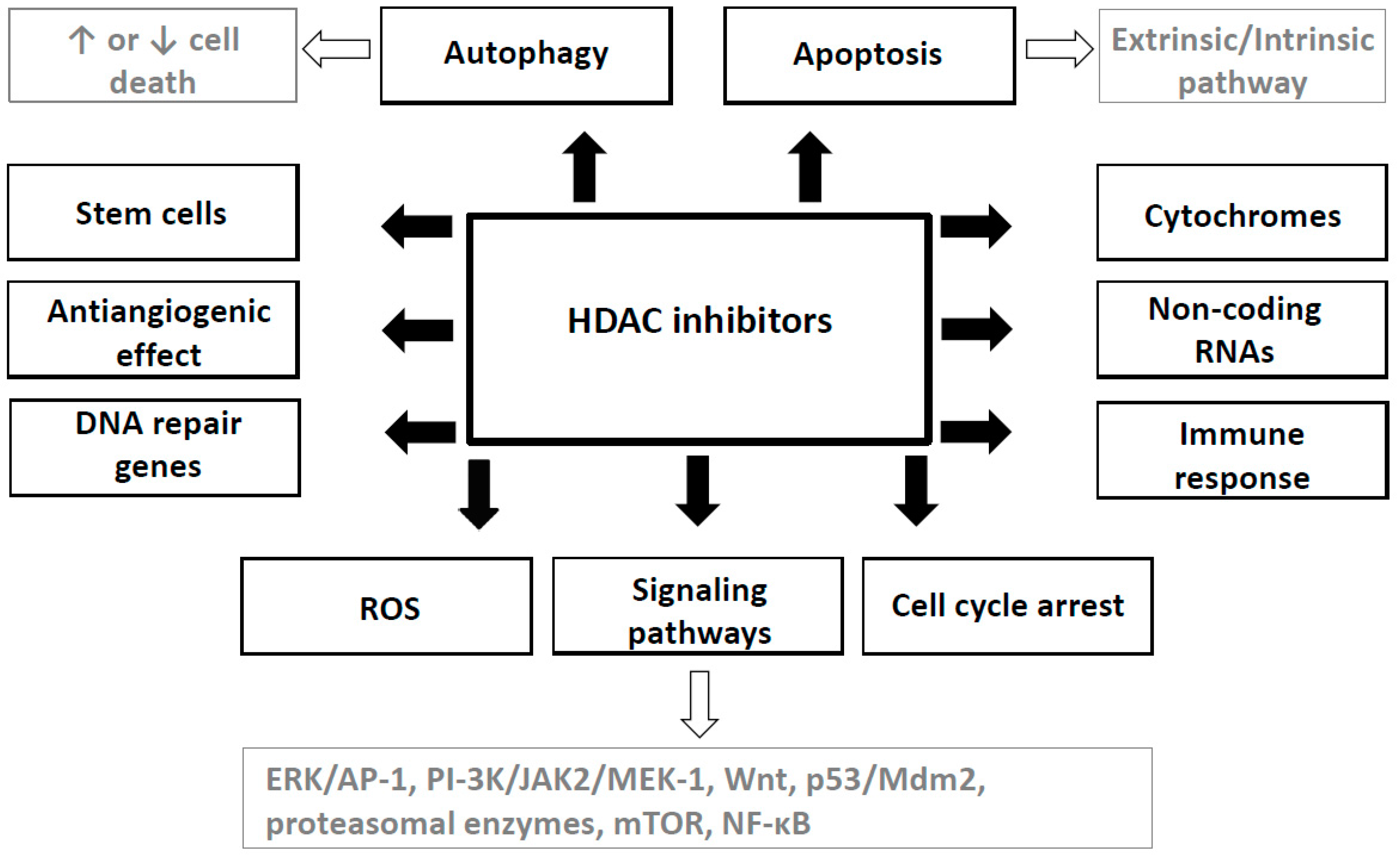{kind=link}
| Class | HDAC Inhibitor | Target HDAC Class | Clinical Status |
|---|---|---|---|
| hydroxamic acids | Trichostatin A | pan | preclinical |
| SAHA | pan | approved for cutaneous T-cell lymphoma | |
| Belinostat | pan | approved for peripheral T-cell lymphoma | |
| Panabiostat | pan | approved for multiple myeloma | |
| Givinostat | pan | phase II clinical trials—relapsed leukemia and multiple myeloma | |
| Resminostat | pan | phase I and II clinical trials—hepatocellular carcinoma | |
| Abexinostat | pan | phase II clinical trial—B-cell lymphoma | |
| Quisinostat | pan | phase I clinical trial—multiple myeloma | |
| Rocilinostat | II | phase I clinical trial—multiple myeloma | |
| Practinostat | I, II and IV | phase II clinical trial—prostate cancer | |
| CHR-3996 | I | phase I clinical trial—advanced/metastatic solid tumors refractory to standard therapy | |
| short chain fatty acids | Valproic acid | I, IIa | approved for epilepsia, bipolar disorders and migraine, phase II clinical trials—several studies |
| Butyric acid | I, II | phase II clinical trials—several studies | |
| Phenylbutyric acid | I, II | phase I clinical trials—several studies | |
| benzamides | Entinostat | I | phase II clinical trials—breast cancer, Hodgkin´s lymphoma, non-small cell lung cancer, phase III clinical trial—hormone receptor positive breast cancer |
| Tacedinaline | I | phase III clinical trial—non-small cell lung cancer and pancreatic cancer | |
| 4SC202 | I | phase I clinical trial—advanced hematological malignancies | |
| Mocetinostat | I, IV | phase II clinical trials—Hodgkin´s lymphoma | |
| cyclic tetrapeptides | Romidepsin | I | approved for cutaneous T-cell lymphoma |
| sirtuins inhibitors | Nicotinamide | all class III | phase III clinical trial—laryngeal cancer |
| Sirtinol | SIRT 1 and 2 | Preclinical | |
| Cambinol | SIRT 1 and 2 | Preclinical | |
| EX-527 | SIRT 1 and 2 | cancer preclinical, phase I and II clinical trials—Huntington disease, glaucoma |
| Mechanism | Target | Model | HDAC Inhibitor | Ref. |
|---|---|---|---|---|
| Cell cycle arrest | CDKN1A/p21 | in vitro leukemic cells U937 | SAHA | [42] |
| CDKN1A/p21 | in vitro bladder cancer cells T24 | SAHA | [43] | |
| CDKN1A/p21 | in vitro normal breast MCF-10A, prostate cancer PC-3, DU145, colon cancer SW620, ovarian cancer IGROV, breast cancer MCF-7, lung cancer A549 | FR901228 | [44] | |
| p53 | in vitro colon cancer HCT116 + knock out DNMT1−/−, DNMT3B−/−, DNMT1−/− and DNMT3B−/− HCT116 | TSA | [46] | |
| p53 | in vitro lung cancer A549 | depsipeptide | [47] | |
| p53 | in vitro lung cancer H1299, osteosarcoma U2OS, human embryonal kidney HEK293 | TSA, nicotinamide | [48] | |
| p21 | in vitro colon cancer HCT116 + knock out p53−/−, p21−/− | sodium butyrate | [49] | |
| p21 | in vitro gastric cancer TMK-1, MKN-1, MKN-7, MKN-28, MKN-74, MKN-45, KATO III, HSC-39, oral squamous carcinoma HSC-4, Ho-1-N-1, Ho-1-U-1 | TSA | [50] | |
| RUNX3 | in vitro pancreatic endocrine tumors- insulinoma CM, carcinoid BON, somatostatinoma QCP-1 | TSA | [52] | |
| RUNX3 | in vitro prostate cancer DU-145, LNCaP, PC-3 | TSA | [55] | |
| Apoptosis | death receptor | in vitro leukemic cells HL60 | apicidin | [60] |
| death receptor | in vitro AML cells from patients, normal CD34+ progenitors | MS275 | [61] | |
| intrinsic pathway | in vitro leukemic cells U937 | SAHA | [42] | |
| intrinsic pathway | in vitro T cell leukemia CEM-CCRF and doxorubicin derived P-gp+(CEM-P-gp) | SAHA | [63] | |
| intrinsic pathway | in vitro fetal lung fibroblasts IMR90, osteosarcoma U2OS and Saos-2, colon cancer HCT116 p53−/− | SAHA, TSA | [64] | |
| intrinsic pathway | in vitro leukemic cells Jurkat, ML-1 | MS275 | [66] | |
| intrinsic pathway | in vitro leukemic cells Jurkat, U937, HL-60, K562, U937 expressing Bcl-2, Bcl-XL, CrmA, C8DN, p21 antisense | MS275 | [67] | |
| intrinsic pathway | in vitro prostate cancer LNCaP, bladder cancer T24, breast cancer MCF7 | SAHA | [68] | |
| intrinsic pathway | in vitro neuroblastoma UKF-NB-4, normoxic and hypoxic condition | VPA | [71] | |
| Autophagy | FOXO3 | in vitro 293T, mice embryonal fibroblasts wild t. and SIRT−/−, Rat1 fibroblasts inducible expressing FOXO3 | Nicotinamide, BML-210, splitomicin, TSA | [74] |
| Atg5 & Beclin1 | in vitro rat cardiomyocytes, in vivo model- a-MHC Beclin transgenic mice | TSA | [75] | |
| unknown | in vitro cervical cancer HeLa | siRNA HDAC1, FK228, SAHA | [76] | |
| Atg4D | in vitro neuroblastoma BE(2)-C, Kelly, IMR32 | siRNA HDAC10 | [77] | |
| Atg5, 7 & 8 | in vitro cervical cancer HeLa, colon cancer HCT116- SIRT1 transfected, starvation | - | [78] | |
| FOXO1 | in vitro colon cancer HCT116 Tp53−/− and +/+, mouse embryonic fibroblasts, liver cancer HepG2 | TSA | [79] | |
| Akt/mTOR, ULK1 | in vitro liver cancer Hep3B, HepG2, Huh7, mouse embryonic fibroblasts | SAHA, TSA, MS275 | [80] | |
| mTOR | in vitro endometrial stromal sarcoma ESS-1, endometrial stromal cells HESCs | SAHA | [81] | |
| p53 | in vitro uterine sarcoma MES-SA, ESS-1, cervical cancer HeLa, PANC-1, leukemic cells, pancreatic cancer Jurkat, HL-60, U937 | SAHA | [82] | |
| ULK1 | in vitro glioblastoma T98G, MEF cells, ULK1/2 knockout MEF, ATG3 knockout MEFs | SAHA | [83] | |
| ULK1 | in vitro leukemic cells Jurkat | SAHA | [84] | |
| NF- κB | in vitro PC3, DU145, and HCT116, mouse embryonic fibroblasts and MEF Atg5−/− | SAHA, MS275 | [85] | |
| Akt/mTOR | short term culture glioblastoma cells xenografts in nude mice | SAHA | [87] | |
| Effect on non-coding RNA | miR-129-5p → GALNT1 & SOX4 | in vitro thyroid carcinoma BCPAP, TPC-1, 8505C, CAL62 | TSA, SAHA | [88] |
| miR-31→BMI1 | in vitro breast cancer MDA-MB-231, MCF7, 293T, embryonal lung fibroblasts | sodium butyrate, panobiostat | [89] | |
| miR-15 & let-7 → Myc | in vitro and mice xenografts leukemic cells/lymphoma Daudi, Ramos, Raji, Su-DHL-6, NIH3T3, P493-6 | RGFP966, depsipetide | [90] | |
| Drosha/DGCR8 | in vitro 293FT embryonal kidney cells HDAC1 transfected | / | [91] | |
| miR-449a → HDAC1 | in vitro prostate cancer PC-3 | siRNA HDAC1 | [92] | |
| uc002mbe.2 lncRNA | in vitro liver cancer Huh7, Bel7402, Bel7721, and HepG2, ShRNA uc002mbe.2 lncRNA | TSA | [95] | |
| lncRNA Xist | in vitro stem cells from 16 breast cancer cell lines | SAHA, VPA, abexinostat | [97] | |
| Effect on signal pathways | c-Jun | in vitro neuroblastoma SH-SY5Y | VPA | [98] |
| ERK | in vitro neuroblastoma SH-SY5Y | VPA | [65] | |
| APC/β-catenin/c-Myc | in vitro colon cancer HT-29 HDAC2 transfected, mice C57BL/6J-APC-/- | VPA | [10] | |
| GSK3 β | in vitro hematopoetic stem cells | VPA | [104] | |
| ubiquitin–proteasome | in vitro embryonal kidney cells HEK293T | VPA, TSA | [105] | |
| Anti-angiogenic effect | Gja1, Irf1, Gbp2 | in vitro embryonic stem cells HDAC1 mutated and wild type | / | [106] |
| ACNA2D2 | HDAC1 or HDAC2 mutated C57BL/6 mice | / | [107] | |
| PPAR, ERR | HDAC3 conditional C57BL/6 mice | / | [108] | |
| eNOS, Akt | in vitro endothelial cells, Akt transfected, mutatnteNOS | / | [110] | |
| eNOS | in vitro HUVEC | TSA, MS275 | [112] | |
| MMP-2/VEGF | in vitro melanoma A375, toxicity tests mice | Compound 8 | [119] | |
| semaphoring III | in vitro HUVEC | TSA, SAHA | [113] | |
| thrombospondin-1, activin | in vitro, neuroblastoma BE(2)-C | VPA | [114] | |
| Modulation of immune response | TAP1 & 2, LMP-2, tapasin | in vitro+mice xenografts mouse lung TC-1, rat pancreas D11, mouse fibroblasts A9, endothelial cells PA, mouse colinic cells LMD, mouse melanoma B16F10, B16F10/TAP-1 | TSA | [118] |
| MHC class I | mice C57BL and B6.CB17-Prkdc, human melanoma samples, mouse melanoma B16-F10-luc | MGCD0103, LBH589, TSA | [120] | |
| STAT3/IL-10 | BALB/c and C57BL/6 mice, TCR transgenic mice, HDAC6 recombinant mutants | / | [121] | |
| Tumor associated antigens | in vitro prostate cancer LNCaP, breast cancer MDA-MB-231 | SAHA, entinostat | [123] | |
| MHC genes | in vitro prostate cancer PCa and DU-145 | VPA | [126] |
| HDAC Inhibitor | Other Anticancer Therapy | Model | Effect | Ref. |
|---|---|---|---|---|
| SAHA, tubacin | etoposide, doxorubicin | in vitro prostate cancer LNCaP, breast cancer MCF-7, normal human foreskin fibroblast cells | Potentiation in ca cells not in fibroblasts | [140] |
| TSA | 5-aza-2′-deoxycytidine | in vitro pancreatic endocrine cancer cells | Potentiation | [52] |
| TSA | 5-aza-2′-deoxycytidine | in vitro LNCaP, DU-145, and PC-3 prostate cancer | Potentiation | [55] |
| SAHA, TSA | 5-aza-2′-deoxycytidine | in vitro ovarian cancer Hey, SKOv3 | Synergism | [142] |
| VPA | 5-aza-2′-deoxycytidine | in vitro leukemic cells HL-60, MOLT4 | Synergism | [143] |
| VPA | 5-aza-2′-deoxycytidine | Ptch knockout mice | Potentiation | [144] |
| Depsipeptide | 5-aza-2′-deoxycytidine | in vitro Kasumi-1 cells and blasts from patient with t(8;21) AML | Potentiation | [145] |
| TSA | 5-aza-2′-deoxycytidine | in vitro lung cancer A549, H719 | Synergism | [146] |
| SAHA | β-phenylethyl isothiocyanate | in vitro leukemia cells HL60, HL60/LR, HL60/C6F, U937, ML1, samples from patients with acute myeloid leukemia | Potentiation | [147] |
| Panobinostat | erlotinib | in vitro lung cancer HCC827, A549, NCI-H460 (EGFR wild type and mutant) | Synergism/Potentiation | [148] |
| SAHA | 3-deazaneplanocin A (EZH2 inhibitor) | in vitro and BALB/cAJcl-nu/nu mice xenografts lung cancer NCI-H1299, NCI-H1975, A549, PC-3 | Synergism | [149] |
| sodium butyrate, SAHA | bortezomib | in vitro multiple myeloma U266, RPMI8226 and patient samples | Synergism | [150] |
| SAHA | bortezomib | in vitro and nude mice xenografts, pancreatic cancer L3.6pl, pancreatic epitelium HPDE6-E6E7 | Potentiation | [151] |
| SAHA | carfilzomib | in vitro and nude mice xenografts, lymphoma cell SUDHL16, SUDHL4 , SUDHL6, OCI-LY10, OCI-LY3 bortezomib-resistant SUDHL16-10BR, OCI-LY10-40BR and lymphoma cells frompatients | Potentiation | [152] |
| SNDX-275, SAHA | carfilzomib, bortezomib | in vitro and nude mice xenografts, lymphoma cell Granta 519, Rec-1, HF-4B, JVM-2, MINO, JVM-13 | Potentiation | [153] |
| VPA | cisplatin, etoposide | in vitro neuroblastoma cell UKF-NB-4 | Potentiation-schedule dependent | [154] |
| belinostat, romidepsin | cisplatin, etoposide | in vitro lung cancer H82, H146, H526 and H446 | Synergism-schedule dependent | [155] |
| TSA, SAHA | VP-16, ellipticine, 5fluorouracil, doxorubicin, cisplatin, cyclophosphamide, campthotecin | in vitro glioblastoma U118, brest cancer MCF-7, normal breast MCF-12F, normal intestinal epithelia FHs74Int | Potentiation-schedule dependent, except of 5fluorouracil, cyclophosphamide, campthotecin | [156] |
| VPA | paclitaxel | in vitro anaplastic thyroid carcinoma CAL-62, ARO | Potentiation | [157] |
| VPA | Gene therapy (MSC + HSV-TK) | glioblastoma U87 xenografts in nude mice | Potentiation | [158] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. https://doi.org/10.3390/ijms18071414
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences. 2017; 18(7):1414. https://doi.org/10.3390/ijms18071414
Chicago/Turabian StyleEckschlager, Tomas, Johana Plch, Marie Stiborova, and Jan Hrabeta. 2017. "Histone Deacetylase Inhibitors as Anticancer Drugs" International Journal of Molecular Sciences 18, no. 7: 1414. https://doi.org/10.3390/ijms18071414
APA StyleEckschlager, T., Plch, J., Stiborova, M., & Hrabeta, J. (2017). Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences, 18(7), 1414. https://doi.org/10.3390/ijms18071414