The Nucleolus: In Genome Maintenance and Repair

Abstract
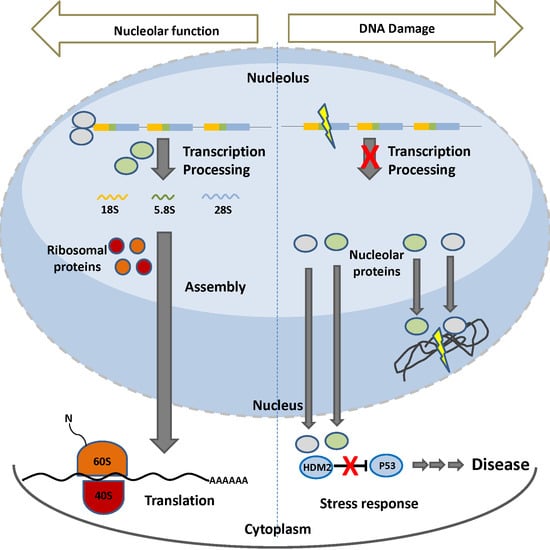
:
{kind=link}
{kind=link}
{kind=link}
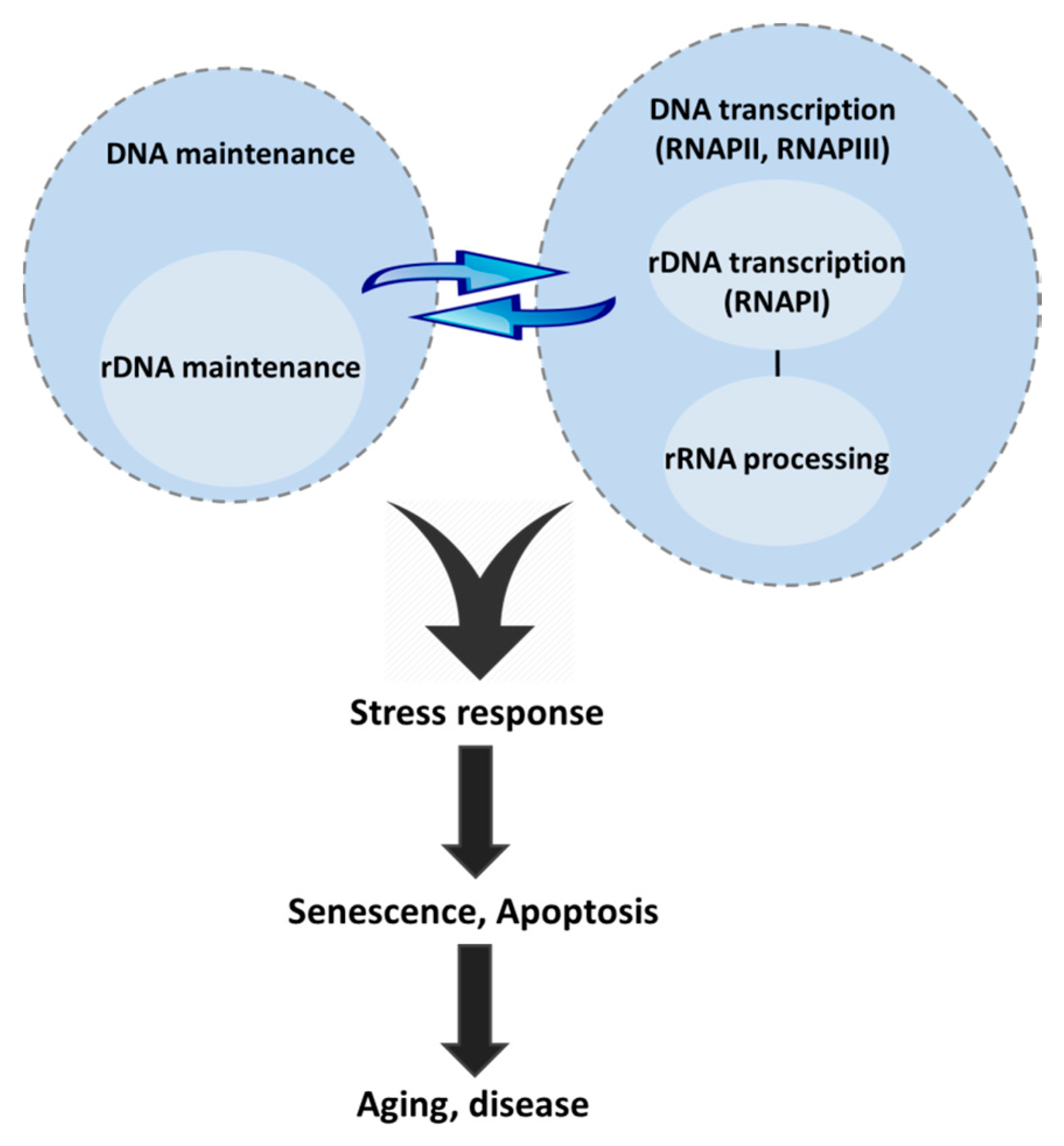
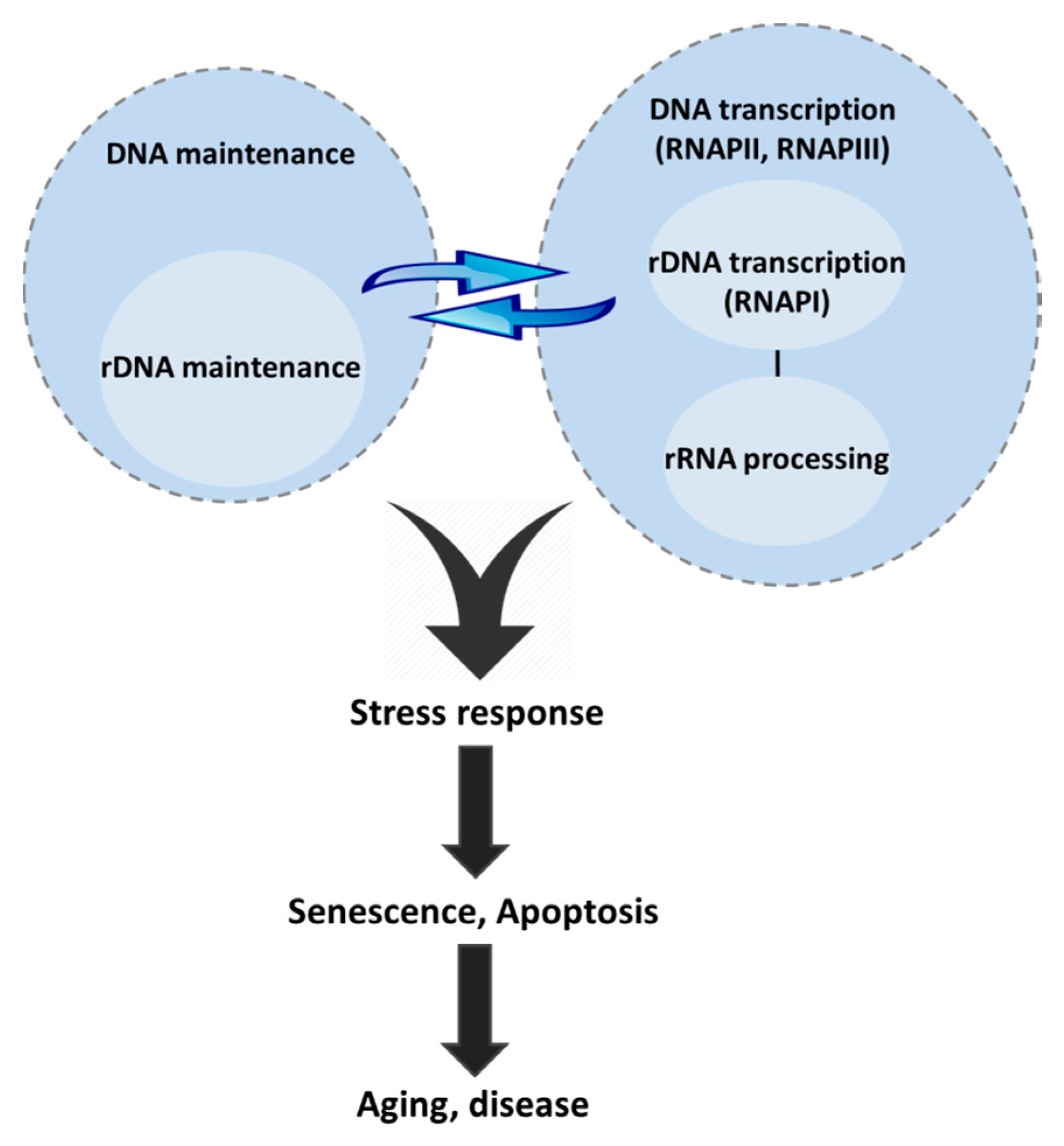{kind=link}
1. Introduction
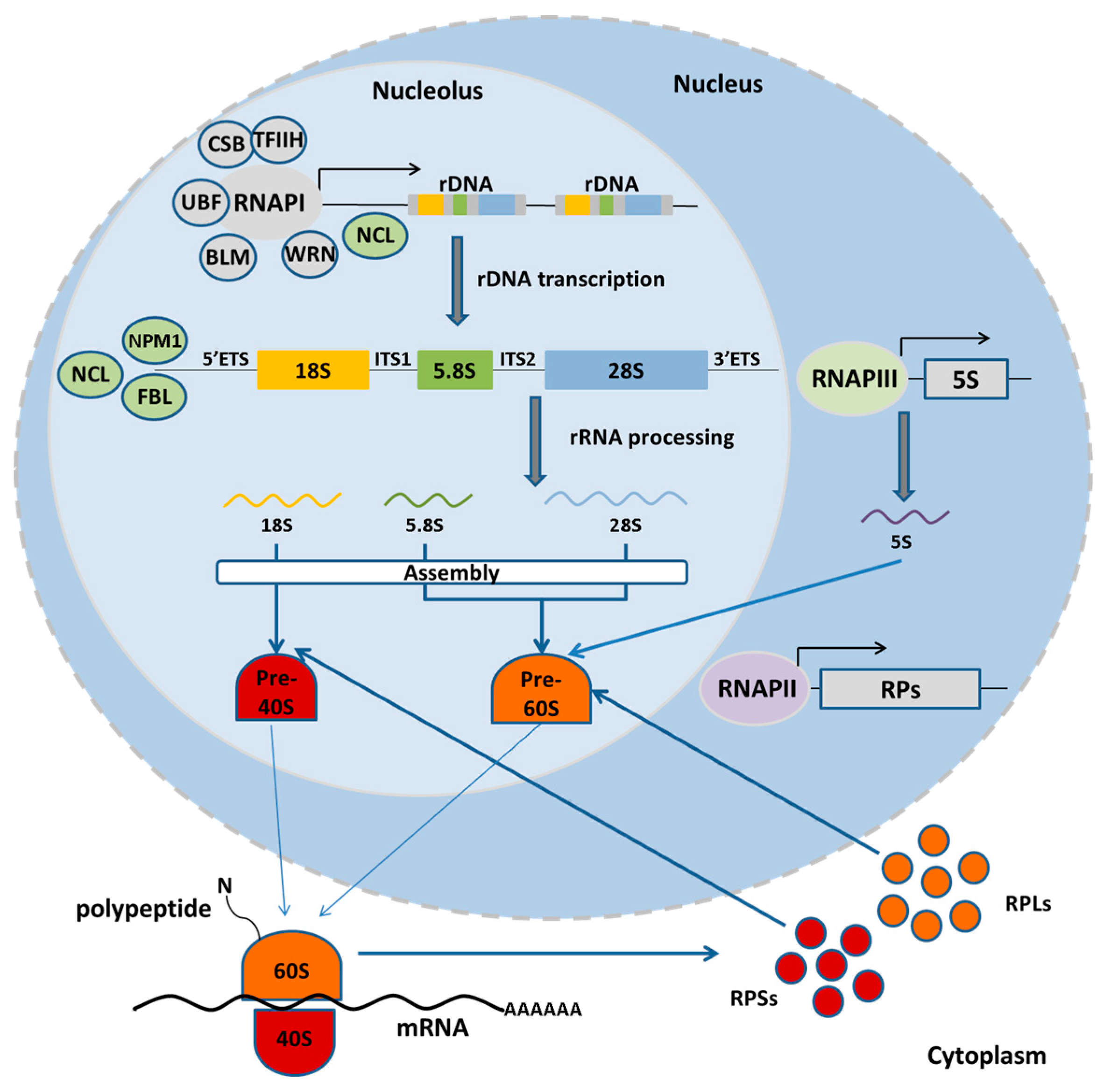
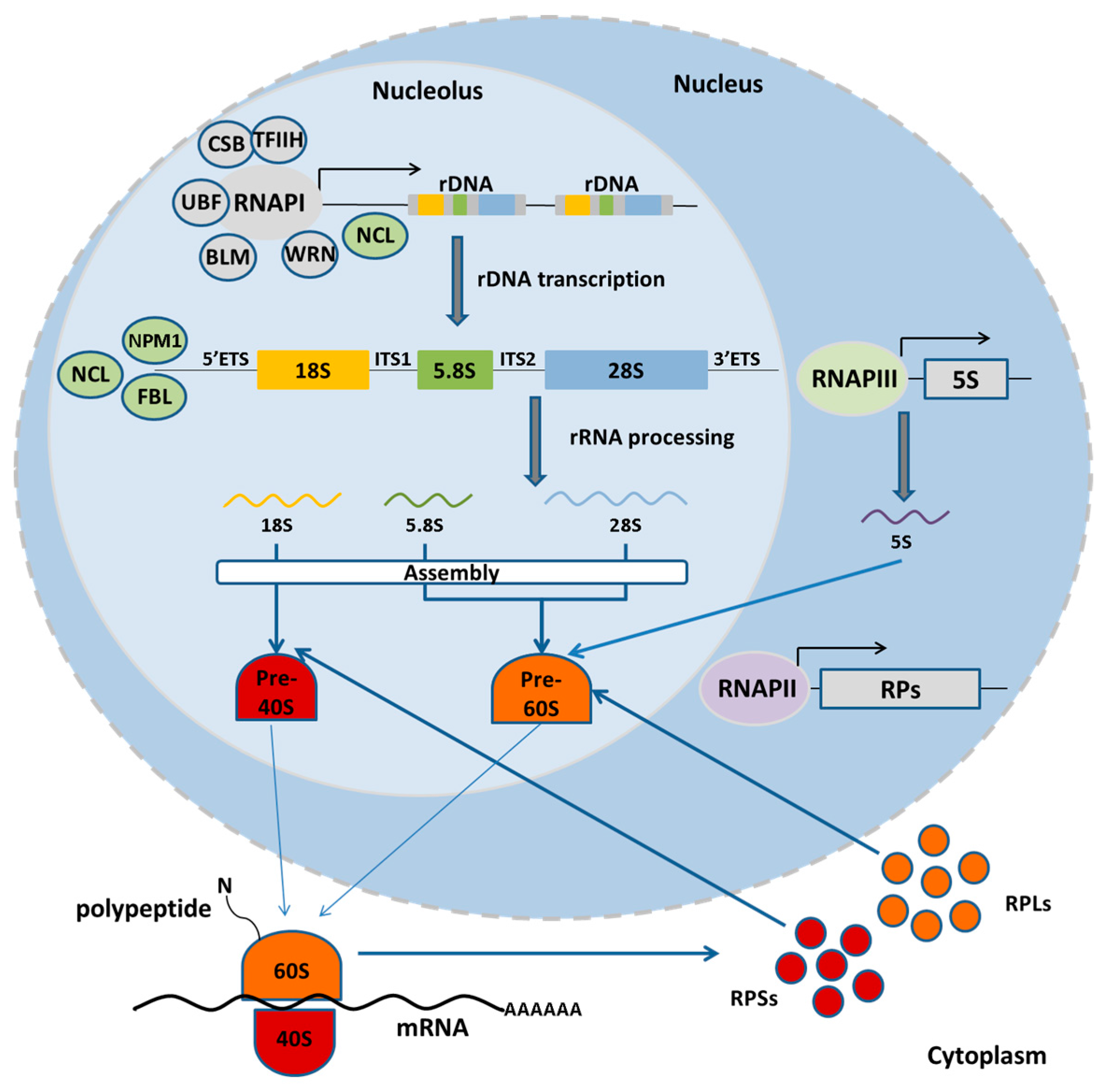
2. Nucleolus and the rDNA: Structure, Function and Organization
3. The Nucleolus Associated Heterochromatin and Its Role in Safeguarding Genome Integrity
4. Genome Maintenance inside the Nucleolus
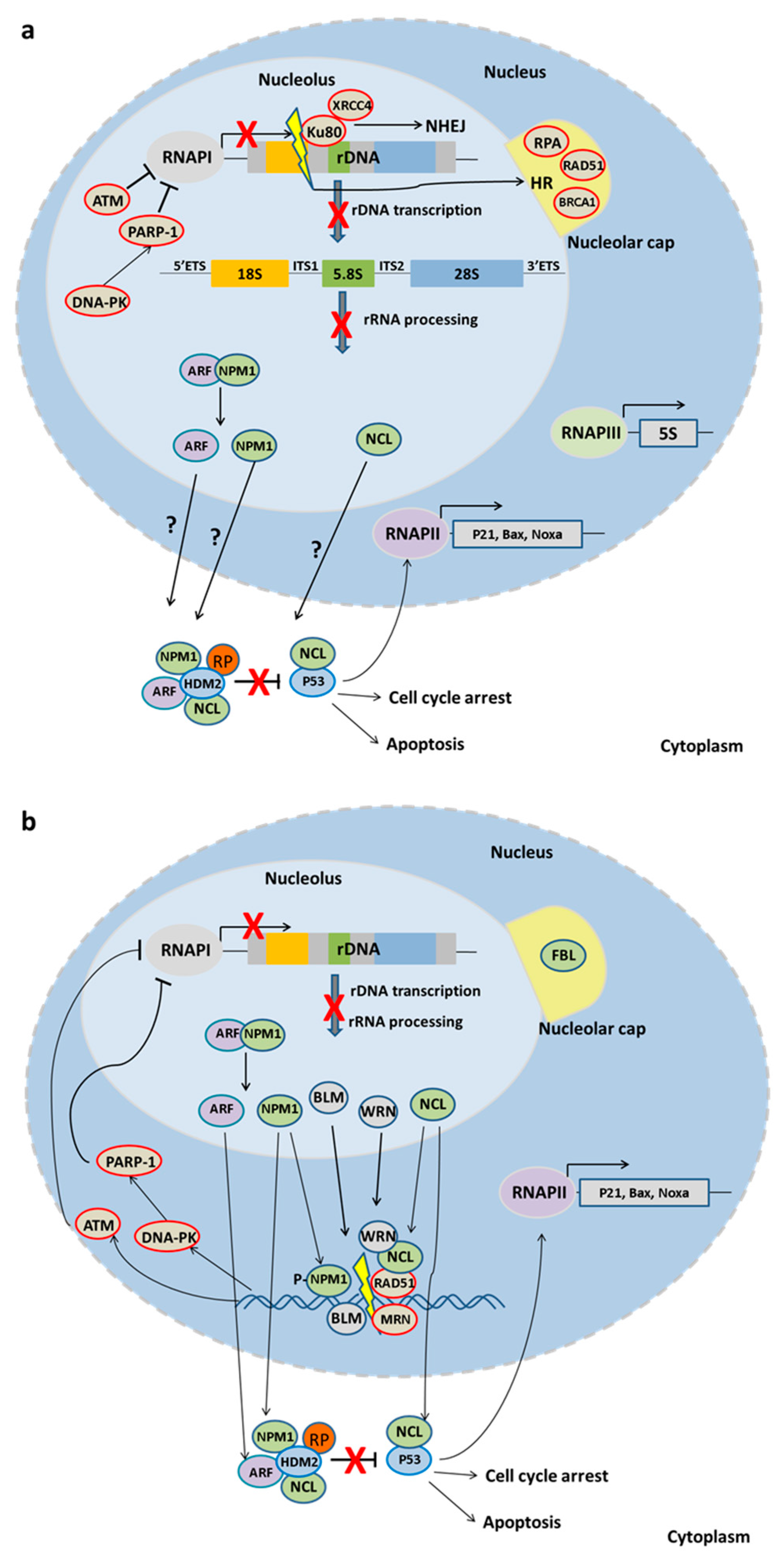
4.1. DSB Repair and the Nucleolus
4.2. Nucleotide Excision Repair and the Nucleolus
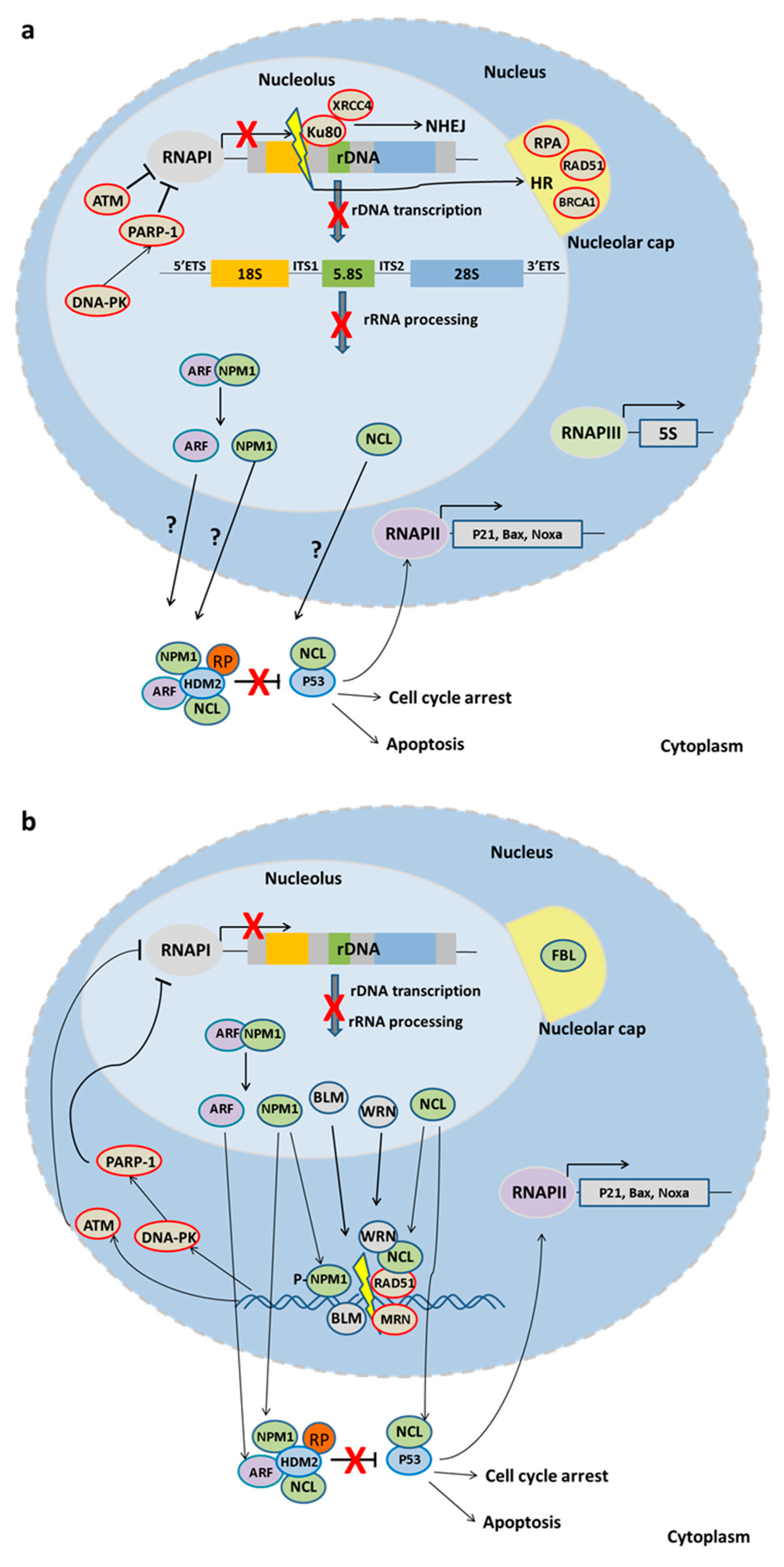
5. The Nucleolus upon Stress
Nucleolar Proteins in DNA Repair
6. rDNA Damage and Disease Onset
DNA Repair Factors Regulating rDNA Transcription and Disease
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hadjiolov, A.A. The Nucleolus and Ribosome Biogenesis Cell Biology Monographs; Springer: New York, NY, USA, 1985. [Google Scholar]
- Fontana, F. Traite sur le Venin de la Viper, sur les Poisons Americains, sur le Laurier-Cerise et sur Quelques Autres Poisons Vegetaux; Gibelin: Florence, Italy, 1781. [Google Scholar]
- Montgomery, T. Comparative cytological studies, with especial regard to the morphology of the nucleolus. J. Morphol. 1898, 15, 265–582. [Google Scholar] [CrossRef]
- Maggi, L.B., Jr.; Weber, J.D. Nucleolar adaptation in human cancer. Cancer Investig. 2005, 23, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Rudra, D.; Warner, J.R. What better measure than ribosome synthesis? Genes Dev. 2004, 18, 2431–2436. [Google Scholar] [CrossRef] [PubMed]
- Heitz, E. Nukleolen und chromosomen in der Gattung Vicia. Planta 1931, 15, 495–505. [Google Scholar] [CrossRef]
- McClintock, B. The relationship of a particular chrmosomal element to the development of the nucleoli in Zea mays. Cell Tissue Res. 1934, 21, 294–326. [Google Scholar]
- Pederson, T. The nucleolus. Cold Spring Harbor Perspect. Biol. 2011, 3, a000638. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.D.; Gurdon, J.B. Absence of Ribosomal Rna Synthesis in the Anucleolate Mutant of Xenopus Laevis. Proc. Natl. Acad. Sci. USA 1964, 51, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F.M.; Spiegelman, S. Localization of DNA Complementary to Ribosomal RNA in the Nucleolus Organizer Region of Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 1965, 53, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Birnstiel, M.L.; Wallace, H.; Sirlin, J.L.; Fischberg, M. Localization of the ribosomal DNA complements in the nucleolar organizer region of Xenopus laevis. Natl. Cancer Inst. Monogr. 1966, 23, 431–447. [Google Scholar] [PubMed]
- Ritossa, F.M.; Atwood, K.C.; Lindsley, D.L.; Spiegelman, S. On the chromosomal distribution of DNA complementary to ribosomal and soluble RNA. Natl. Cancer Inst. Monogr. 1966, 23, 449–472. [Google Scholar] [PubMed]
- Edstrom, J.E.; Grampp, W.; Schor, N. The intracellular distribution and heterogeneity of ribonucleic acid in starfish oocytes. J. Biophys. Biochem. Cytol. 1961, 11, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.P.; Errera, M. The role of the nucleolus in ribonucleic acid-and protein synthesis: I. Incorporation of cytidine into normal and nucleolar inactivated HeLa cells. Biochim. Biophys. Acta 1961, 49, 47–57. [Google Scholar] [CrossRef]
- Pendle, A.F.; Clark, G.P.; Boon, R.; Lewandowska, D.; Lam, Y.W.; Andersen, J.; Mann, M.; Lamond, A.I.; Brown, J.W.; Shaw, P.J. Proteomic analysis of the Arabidopsis nucleolus suggests novel nucleolar functions. Mol. Biol. Cell 2005, 16, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.S.; Lyon, C.E.; Fox, A.H.; Leung, A.K.; Lam, Y.W.; Steen, H.; Mann, M.; Lamond, A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002, 12, 1–11. [Google Scholar] [CrossRef]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.; Ong, S.E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.J.; Eggleton, P.; Young, P.J. Joining the dots: Production, processing and targeting of U snRNP to nuclear bodies. Biochim. Biophys. Acta 2008, 1783, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.R.; Cao, L.G.; Taneja, K.; Singer, R.H.; Wang, Y.L.; Pederson, T. Nuclear domains of the RNA subunit of RNase P. J. Cell Sci. 1997, 110, 829–837. [Google Scholar] [PubMed]
- Bertrand, E.; Houser-Scott, F.; Kendall, A.; Singer, R.H.; Engelke, D.R. Nucleolar localization of early tRNA processing. Genes Dev. 1998, 12, 2463–2468. [Google Scholar] [CrossRef] [PubMed]
- Jarrous, N.; Wolenski, J.S.; Wesolowski, D.; Lee, C.; Altman, S. Localization in the nucleolus and coiled bodies of protein subunits of the ribonucleoprotein ribonuclease P. J. Cell Biol. 1999, 146, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.R.; Pederson, T. Localization of signal recognition particle RNA in the nucleolus of mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 7981–7986. [Google Scholar] [CrossRef] [PubMed]
- Politz, J.C.; Yarovoi, S.; Kilroy, S.M.; Gowda, K.; Zwieb, C.; Pederson, T. Signal recognition particle components in the nucleolus. Proc. Natl. Acad. Sci. USA 2000, 97, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Sommerville, J.; Brumwell, C.L.; Politz, J.C.; Pederson, T. Signal recognition particle assembly in relation to the function of amplified nucleoli of Xenopus oocytes. J. Cell Sci. 2005, 118, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. A new role of the rDNA and nucleolus in the nucleus—rDNA instability maintains genome integrity. Bioessays 2008, 30, 267–272. [Google Scholar] [CrossRef] [PubMed]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Melese, T.; Xue, Z. The nucleolus: An organelle formed by the act of building a ribosome. Curr. Opin. Cell Biol. 1995, 7, 319–324. [Google Scholar] [CrossRef]
- Smetana, K.; Busch, H. The Nucleolus and Nucleolar DNA. In The Cell Nucleus; Busch, H., Ed.; Academic Press: New York, NY, USA, 1974; pp. 73–147. [Google Scholar]
- Stoykova, A.S.; Dabeva, M.D.; Dimova, R.N.; Hadjiolov, A.A. Ribosome biogenesis and nucleolar ultrastructure in neuronal and oligodendroglial rat brain cells. J. Neurochem. 1985, 45, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Casafont, I.; Navascues, J.; Pena, E.; Lafarga, M.; Berciano, M.T. Nuclear organization and dynamics of transcription sites in rat sensory ganglia neurons detected by incorporation of 5-fluorouridine into nascent RNA. Neuroscience 2006, 140, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.O.; Barthelmes, H.U.; Boege, F.; Mielke, C. The N-terminal domain anchors human topoisomerase I at fibrillar centers of nucleoli and nucleolar organizer regions of mitotic chromosomes. J. Biol. Chem. 2002, 277, 35932–35938. [Google Scholar] [CrossRef] [PubMed]
- Roussel, P.; Andre, C.; Masson, C.; Geraud, G.; Hernandez-Verdun, D. Localization of the RNA polymerase I transcription factor hUBF during the cell cycle. J. Cell Sci. 1993, 104, 327–337. [Google Scholar] [PubMed]
- Puvion-Dutilleul, F.; Puvion, E.; Bachellerie, J.P. Early stages of pre-rRNA formation within the nucleolar ultrastructure of mouse cells studied by in situ hybridization with a 5’ETS leader probe. Chromosoma 1997, 105, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Sirri, V.; Urcuqui-Inchima, S.; Roussel, P.; Hernandez-Verdun, D. Nucleolus: The fascinating nuclear body. Histochem. Cell Biol. 2008, 129, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Grummt, I. Cell cycle-dependent regulation of RNA polymerase I transcription: The nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. USA 1999, 96, 6096–6101. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Verdun, D. Assembly and disassembly of the nucleolus during the cell cycle. Nucleus 2011, 2, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Carmo-Fonseca, M.; Mendes-Soares, L.; Campos, I. To be or not to be in the nucleolus. Nat. Cell Biol. 2000, 2, e107–e112. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.L., Jr.; Beatty, B.R. Visualization of nucleolar genes. Science 1969, 164, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O.; Dawid, I.B. Repeated genes in eukaryotes. Annu. Rev. Biochem. 1980, 49, 727–764. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.S.; Warburton, D.; Atwood, K.C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. USA 1972, 69, 3394–3398. [Google Scholar] [CrossRef] [PubMed]
- Dev, V.G.; Tantravahi, R.; Miller, D.A.; Miller, O.J. Nucleolus organizers in Mus musculus subspecies and in the RAG mouse cell line. Genetics 1977, 86, 389–398. [Google Scholar] [PubMed]
- Kurihara, Y.; Suh, D.S.; Suzuki, H.; Moriwaki, K. Chromosomal locations of Ag-NORs and clusters of ribosomal DNA in laboratory strains of mice. Mamm. Genome 1994, 5, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Petes, T.D. Yeast ribosomal DNA genes are located on chromosome XII. Proc. Natl. Acad. Sci. USA 1979, 76, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Horiuchi, T.; Kobayashi, T. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 2003, 17, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.X.; Wang, X.S.; Amemiya, H.M.; Brewer, B.J.; Raghuraman, M.K. rDNA Copy Number Variants Are Frequent Passenger Mutations in Saccharomyces cerevisiae Deletion Collections and de Novo Transformants. G3 (Bethesda) 2016, 6, 2829–2838. [Google Scholar] [CrossRef] [PubMed]
- Paredes, S.; Maggert, K.A. Expression of I-CreI endonuclease generates deletions within the rDNA of Drosophila. Genetics 2009, 181, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Mais, C.; Wright, J.E.; Prieto, J.L.; Raggett, S.L.; McStay, B. UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev. 2005, 19, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Grob, A.; Colleran, C.; McStay, B. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev. 2014, 28, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Blevins, T.; Chandrasekhara, C.; Mozgova, I.; Hassel, C.; Pontes, O.M.; Tucker, S.; Mokros, P.; Muchova, V.; Fajkus, J.; et al. Subnuclear partitioning of rRNA genes between the nucleolus and nucleoplasm reflects alternative epiallelic states. Genes Dev. 2013, 27, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Guibert, S.; Tiwari, V.K.; Ohlsson, R.; Langst, G. Epigenetic regulation of TTF-I-mediated promoter-terminator interactions of rRNA genes. EMBO J. 2008, 27, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Conesa, A.; Santoyo-Lopez, J.; Medina, I.; Montaner, D.; Peterfia, B.; Solovei, I.; Cremer, T.; Dopazo, J.; Langst, G. Initial genomics of the human nucleolus. PLoS Genet. 2010, 6, e1000889. [Google Scholar] [CrossRef] [PubMed]
- Van Koningsbruggen, S.; Gierlinski, M.; Schofield, P.; Martin, D.; Barton, G.J.; Ariyurek, Y.; den Dunnen, J.T.; Lamond, A.I. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell 2010, 21, 3735–3748. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Saiakhova, A.; Manaenkov, P.; Adams, M.D.; Scacheri, P.C. Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 2011, 39, 4949–4960. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Li, J.; Grummt, I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 2002, 32, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Grummt, I. Epigenetic mechanism of rRNA gene silencing: Temporal order of NoRC-mediated histone modification, chromatin remodeling, and DNA methylation. Mol. Cell Biol. 2005, 25, 2539–2546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Santoro, R.; Grummt, I. The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J. 2002, 21, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Strohner, R.; Nemeth, A.; Jansa, P.; Hofmann-Rohrer, U.; Santoro, R.; Langst, G.; Grummt, I. NoRC—A novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 2001, 20, 4892–4900. [Google Scholar] [CrossRef] [PubMed]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Karpen, G.H. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat. Cell Biol. 2007, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Gagnon-Kugler, T.; Langlois, F.; Stefanovsky, V.; Lessard, F.; Moss, T. Loss of human ribosomal gene CpG methylation enhances cryptic RNA polymerase II transcription and disrupts ribosomal RNA processing. Mol. Cell 2009, 35, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Govoni, M.; Farabegoli, F.; Pession, A.; Novello, F. Inhibition of topoisomerase II activity and its effect on nucleolar structure and function. Exp. Cell Res. 1994, 211, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.O.; Krokowski, R.M.; Barthelmes, H.U.; Hock, R.; Boege, F.; Mielke, C. Distinct effects of topoisomerase I and RNA polymerase I inhibitors suggest a dual mechanism of nucleolar/nucleoplasmic partitioning of topoisomerase I. J. Biol. Chem. 2004, 279, 21873–21882. [Google Scholar] [CrossRef] [PubMed]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Stults, D.M.; Killen, M.W.; Pierce, H.H.; Pierce, A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008, 18, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Stults, D.M.; Killen, M.W.; Williamson, E.P.; Hourigan, J.S.; Vargas, H.D.; Arnold, S.M.; Moscow, J.A.; Pierce, A.J. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 2009, 69, 9096–9104. [Google Scholar] [CrossRef] [PubMed]
- Caburet, S.; Conti, C.; Schurra, C.; Lebofsky, R.; Edelstein, S.J.; Bensimon, A. Human ribosomal RNA gene arrays display a broad range of palindromic structures. Genome Res. 2005, 15, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Van Sluis, M.; McStay, B. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015, 29, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Neumaier, T.; Swenson, J.; Pham, C.; Polyzos, A.; Lo, A.T.; Yang, P.; Dyball, J.; Asaithamby, A.; Chen, D.J.; Bissell, M.J.; et al. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proc. Natl. Acad. Sci. USA 2012, 109, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.; Evdokimova, V.N.; Cuenco, K.T.; Nikiforova, M.N.; Kelly, L.M.; Stringer, J.R.; Bakkenist, C.J.; Nikiforov, Y.E. Homologous chromosomes make contact at the sites of double-strand breaks in genes in somatic G0/G1-phase human cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9454–9459. [Google Scholar] [CrossRef] [PubMed]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.O.; Zink, D.; Durante, M.; Lobrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; Sunjevaric, I.; de Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragon, L.; Lisby, M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montano, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Suzuki, K.; Yamauchi, M.; Mitsutake, N.; Yamashita, S. Recruitment of the cohesin loading factor NIPBL to DNA double-strand breaks depends on MDC1, RNF168 and HP1γ in human cells. Biochem. Biophys. Res. Commun. 2011, 411, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Calkins, A.S.; Iglehart, J.D.; Lazaro, J.B. DNA damage-induced inhibition of rRNA synthesis by DNA-PK and PARP-1. Nucleic Acids Res. 2013, 41, 7378–7386. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.; Gottlieb, T.M.; Jackson, S.P.; Grummt, I. DNA-dependent protein kinase: A potent inhibitor of transcription by RNA polymerase I. Genes Dev. 1995, 9, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Michaelidis, T.M.; Grummt, I. Mechanism of inhibition of RNA polymerase I transcription by DNA-dependent protein kinase. Biol. Chem. 2002, 383, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell Proteomics. 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and-independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal 2010, 3, rs3. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.M.; Bai, B.; Boisvert, F.M.; Latonen, L.; Rantanen, V.; Simpson, J.C.; Pepperkok, R.; Lamond, A.I.; Laiho, M. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell Proteomics. 2011, 10, M111009241. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, D.O.; van den Berg, J.; Medema, R.H. Breaks in the 45S rDNA Lead to Recombination-Mediated Loss of Repeats. Cell Rep. 2016, 14, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Kamileri, I.; Karakasilioti, I.; Garinis, G.A. Nucleotide excision repair: New tricks with old bricks. Trends Genet. 2012, 28, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Menoni, H.; Hoeijmakers, J.H.; Vermeulen, W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J. Cell Biol. 2012, 199, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Stevnsner, T.; May, A.; Petersen, L.N.; Larminat, F.; Pirsel, M.; Bohr, V.A. Repair of ribosomal RNA genes in hamster cells after UV irradiation, or treatment with cisplatin or alkylating agents. Carcinogenesis 1993, 14, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Christians, F.C.; Hanawalt, P.C. Lack of transcription-coupled repair in mammalian ribosomal RNA genes. Biochemistry 1993, 32, 10512–10518. [Google Scholar] [CrossRef] [PubMed]
- Fritz, L.K.; Smerdon, M.J. Repair of UV damage in actively transcribed ribosomal genes. Biochemistry 1995, 34, 13117–13124. [Google Scholar] [CrossRef] [PubMed]
- Christians, F.C.; Hanawalt, P.C. Repair in ribosomal RNA genes is deficient in xeroderma pigmentosum group C and in Cockayne’s syndrome cells. Mutat. Res. 1994, 323, 179–187. [Google Scholar] [CrossRef]
- Wei, L.; Levine, A.S.; Lan, L. Transcription-coupled homologous recombination after oxidative damage. DNA Repair 2016, 44, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.O. Sensing cellular stress: Another new function for the nucleolus? Sci. STKE 2004, 224, e10. [Google Scholar] [CrossRef] [PubMed]
- Pederson, T.; Tsai, R.Y. In search of nonribosomal nucleolar protein function and regulation. J. Cell Biol. 2009, 184, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell Biol. 2000, 20, 5930–5938. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.D.; Oeffinger, M. Nucleolin and nucleophosmin: Nucleolar proteins with multiple functions in DNA repair. Biochem. Cell Biol. 2016, 94, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Gjerset, R.A.; Bandyopadhyay, K. Regulation of p14ARF through subnuclear compartmentalization. Cell Cycle 2006, 5, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Daniely, Y.; Dimitrova, D.D.; Borowiec, J.A. Stress-dependent nucleolin mobilization mediated by p53-nucleolin complex formation. Mol. Cell Biol. 2002, 22, 6014–6022. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Rorie, C.J.; Dimitrova, D.; Daniely, Y.; Borowiec, J.A. Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene 2006, 25, 7274–7288. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Park, J.J.; Gu, B.H.; Kim, J.O.; Park, S.G.; Baek, K.H. HAUSP-nucleolin interaction is regulated by p53-Mdm2 complex in response to DNA damage response. Sci. Rep. 2015, 5, 12793. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, K.; Kastan, M.B. Interactions of nucleolin and ribosomal protein L26 (RPL26) in translational control of human p53 mRNA. J. Biol. Chem. 2012, 287, 16467–16476. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Donahue, S.L.; Tabah, A.; Castro, N.E.; Mraz, N.; Cruise, J.L.; Campbell, C. A novel interaction [corrected] of nucleolin with Rad51. Biochem. Biophys. Res. Commun. 2006, 344, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Wabl, M.; Akhmedov, A.T.; Jessberger, R. A B-cell-specific DNA recombination complex. J. Biol. Chem. 1998, 273, 17025–17035. [Google Scholar] [CrossRef] [PubMed]
- Indig, F.E.; Rybanska, I.; Karmakar, P.; Devulapalli, C.; Fu, H.; Carrier, F.; Bohr, V.A. Nucleolin inhibits G4 oligonucleotide unwinding by Werner helicase. PLoS ONE 2012, 7, e35229. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Fujimoto, H.; Sato, J.; Hayashi, I.; Burma, S.; Matsuura, S.; Chen, D.J.; Komatsu, K. Nucleolin participates in DNA double-strand break-induced damage response through MDC1-dependent pathway. PLoS ONE 2012, 7, e49245. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Derheimer, F.A.; Tait-Mulder, J.; Kastan, M.B. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2013, 110, 16874–16879. [Google Scholar] [CrossRef] [PubMed]
- Angelov, D.; Bondarenko, V.A.; Almagro, S.; Menoni, H.; Mongelard, F.; Hans, F.; Mietton, F.; Studitsky, V.M.; Hamiche, A.; Dimitrov, S.; et al. Nucleolin is a histone chaperone with FACT-like activity and assists remodeling of nucleosomes. EMBO J. 2006, 25, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Ui, A.; Nakajima, S.; Hatakeyama, K.; Hoshi, M.; Watanabe, R.; Janicki, S.M.; Ogiwara, H.; Kohno, T.; Kanno, S.; et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol. Cell 2010, 40, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Koike, A.; Nishikawa, H.; Wu, W.; Okada, Y.; Venkitaraman, A.R.; Ohta, T. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res. 2010, 70, 6746–6756. [Google Scholar] [CrossRef] [PubMed]
- Okuwaki, M.; Matsumoto, K.; Tsujimoto, M.; Nagata, K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001, 506, 272–276. [Google Scholar] [CrossRef]
- Holmberg Olausson, K.; Nister, M.; Lindstrom, M.S. Loss of nucleolar histone chaperone NPM1 triggers rearrangement of heterochromatin and synergizes with a deficiency in DNA methyltransferase DNMT3A to drive ribosomal DNA transcription. J. Biol. Chem. 2014, 289, 34601–34619. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, V.; Kishore, A.H.; Febitha, K.K.; Kundu, T.K. Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol. Cell Biol. 2005, 25, 7534–7545. [Google Scholar] [CrossRef] [PubMed]
- Brewer, B.J.; Lockshon, D.; Fangman, W.L. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell 1992, 71, 267–276. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nomura, M.; Horiuchi, T. Identification of DNA cis elements essential for expansion of ribosomal DNA repeats in Saccharomyces cerevisiae. Mol. Cell Biol. 2001, 21, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol. Cell Biol. 2003, 23, 9178–9188. [Google Scholar] [CrossRef] [PubMed]
- Weitao, T.; Budd, M.; Campbell, J.L. Evidence that yeast SGS1, DNA2, SRS2, and FOB1 interact to maintain rDNA stability. Mutat. Res. 2003, 532, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, M.D.; Sogo, J.M. rDNA enhancer affects replication initiation and mitotic recombination: Fob1 mediates nucleolytic processing independently of replication. Mol. Cell 2004, 15, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Horiuchi, T.; Tongaonkar, P.; Vu, L.; Nomura, M. SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 2004, 117, 441–453. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ganley, A.R. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 2005, 309, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Defossez, P.A.; Prusty, R.; Kaeberlein, M.; Lin, S.J.; Ferrigno, P.; Silver, P.A.; Keil, R.L.; Guarente, L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell 1999, 3, 447–455. [Google Scholar] [CrossRef]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef]
- Park, P.U.; Defossez, P.A.; Guarente, L. Effects of mutations in DNA repair genes on formation of ribosomal DNA circles and life span in Saccharomyces cerevisiae. Mol. Cell Biol. 1999, 19, 3848–3856. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. How does genome instability affect lifespan?: Roles of rDNA and telomeres. Genes Cells 2011, 16, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Miyazaki, T.; Maki, H.; Kobayashi, T. Abundance of ribosomal RNA gene copies maintains genome integrity. Science 2010, 327, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Ganley, A.R.; Ide, S.; Saka, K.; Kobayashi, T. The effect of replication initiation on gene amplification in the rDNA and its relationship to aging. Mol. Cell 2009, 35, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, Y.; Kobayashi, T. The Human RNA Polymerase I Transcription terminator complex acts as a replication fork barrier that coordinates the progress of replication with rRNA transcription activity. Mol. Cell Biol. 2015, 35, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Grob, A.; Roussel, P.; Wright, J.E.; McStay, B.; Hernandez-Verdun, D.; Sirri, V. Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J. Cell Sci. 2009, 122, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Killen, M.W.; Stults, D.M.; Adachi, N.; Hanakahi, L.; Pierce, A.J. Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum. Mol. Genet. 2009, 18, 3417–3428. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Pietrzak, M.; Rempala, G.; Nelson, P.T.; Hetman, M. Neurodegeneration-associated instability of ribosomal DNA. Biochim. Biophys. Acta 2014, 1842, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Deng, L.; Xue, Y.; Suzuki, K.; Zhang, W.; Yu, Y.; Wu, J.; Sun, L.; Gong, X.; Luan, H.; et al. Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 2017, 27, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Zafiropoulos, A.; Tsentelierou, E.; Linardakis, M.; Kafatos, A.; Spandidos, D.A. Preferential loss of 5S and 28S rDNA genes in human adipose tissue during ageing. Int. J. Biochem. Cell Biol. 2005, 37, 409–415. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, R.A.; Spitzer, D.; Bar-Am, I.; Sylvester, J.E.; Kaufmann, M.; Wernich, A.; Drexler, H.G. Karyotypic dissection of Hodgkin’s disease cell lines reveals ectopic subtelomeres and ribosomal DNA at sites of multiple jumping translocations and genomic amplification. Leukemia 2000, 14, 1803–1814. [Google Scholar] [CrossRef] [PubMed]
- Rieker, C.; Engblom, D.; Kreiner, G.; Domanskyi, A.; Schober, A.; Stotz, S.; Neumann, M.; Yuan, X.; Grummt, I.; Schutz, G.; et al. Nucleolar disruption in dopaminergic neurons leads to oxidative damage and parkinsonism through repression of mammalian target of rapamycin signaling. J. Neurosci. 2011, 31, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, M.; Rempala, G.; Nelson, P.T.; Zheng, J.J.; Hetman, M. Epigenetic silencing of nucleolar rRNA genes in Alzheimer’s disease. PLoS ONE 2011, 6, e22585. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta 2014, 1842, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA polymerase I transcription machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Sussman, M.A. Stressing on the nucleolus in cardiovascular disease. Biochim. Biophys. Acta 2014, 1842, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, M.; Suzuki, T.; Itoh, C.; Goto, M.; Furuichi, Y.; Matsumoto, T. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I-associated complex. Oncogene 2002, 21, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.D.; Wang, L.; Youssoufian, H.; Martin, G.M.; Oshima, J. Werner helicase is localized to transcriptionally active nucleoli of cycling cells. Exp. Cell Res. 1998, 242, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Lutomska, A.; Lebedev, A.; Scharffetter-Kochanek, K.; Iben, S. The transcriptional response to distinct growth factors is impaired in Werner syndrome cells. Exp. Gerontol. 2008, 43, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Grierson, P.M.; Lillard, K.; Behbehani, G.K.; Combs, K.A.; Bhattacharyya, S.; Acharya, S.; Groden, J. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum. Mol. Genet. 2012, 21, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Grierson, P.M.; Acharya, S.; Groden, J. Collaborating functions of BLM and DNA topoisomerase I in regulating human rDNA transcription. Mutat. Res. 2013, 743–744, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Velez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.W.; Inouye, C.; Yamaguchi, T.; Cattoglio, C.; Grubisic, I.; Tjian, R. A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell 2011, 147, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Kamileri, I.; Karakasilioti, I.; Sideri, A.; Kosteas, T.; Tatarakis, A.; Talianidis, I.; Garinis, G.A. Defective transcription initiation causes postnatal growth failure in a mouse model of nucleotide excision repair (NER) progeria. Proc. Natl. Acad. Sci. USA 2012, 109, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Chatzinikolaou, G.; Apostolou, Z.; Aid-Pavlidis, T.; Ioannidou, A.; Karakasilioti, I.; Papadopoulos, G.L.; Aivaliotis, M.; Tsekrekou, M.; Strouboulis, J.; Kosteas, T.; et al. ERCC1-XPF cooperates with CTCF and cohesin to facilitate the developmental silencing of imprinted genes. Nat. Cell Biol. 2017, 19, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Bradsher, J.; Auriol, J.; Proietti de Santis, L.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829. [Google Scholar] [CrossRef]
- Yuan, X.; Feng, W.; Imhof, A.; Grummt, I.; Zhou, Y. Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol. Cell 2007, 27, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.; Scharffetter-Kochanek, K.; Iben, S. Truncated Cockayne syndrome B protein represses elongation by RNA polymerase I. J. Mol. Biol. 2008, 382, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Assfalg, R.; Lebedev, A.; Gonzalez, O.G.; Schelling, A.; Koch, S.; Iben, S. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012, 40, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Tseng, A.; Borch Jensen, M.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsekrekou, M.; Stratigi, K.; Chatzinikolaou, G. The Nucleolus: In Genome Maintenance and Repair. Int. J. Mol. Sci. 2017, 18, 1411. https://doi.org/10.3390/ijms18071411
Tsekrekou M, Stratigi K, Chatzinikolaou G. The Nucleolus: In Genome Maintenance and Repair. International Journal of Molecular Sciences. 2017; 18(7):1411. https://doi.org/10.3390/ijms18071411
Chicago/Turabian StyleTsekrekou, Maria, Kalliopi Stratigi, and Georgia Chatzinikolaou. 2017. "The Nucleolus: In Genome Maintenance and Repair" International Journal of Molecular Sciences 18, no. 7: 1411. https://doi.org/10.3390/ijms18071411
APA StyleTsekrekou, M., Stratigi, K., & Chatzinikolaou, G. (2017). The Nucleolus: In Genome Maintenance and Repair. International Journal of Molecular Sciences, 18(7), 1411. https://doi.org/10.3390/ijms18071411