Montelukast Induces Apoptosis-Inducing Factor-Mediated Cell Death of Lung Cancer Cells

 ,
,  , and
, and
Abstract
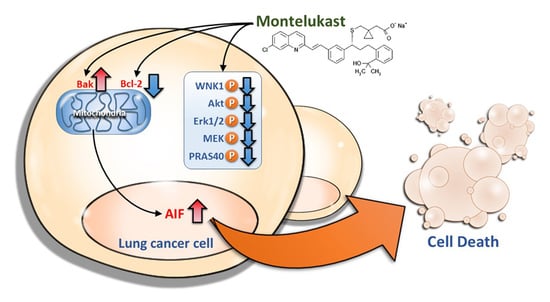
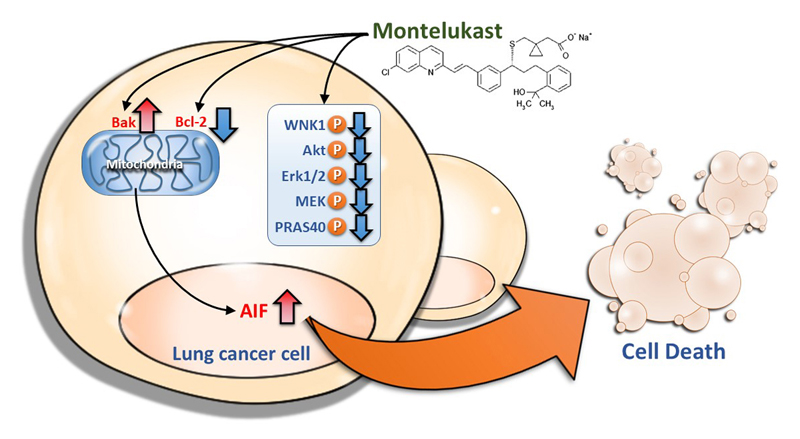
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
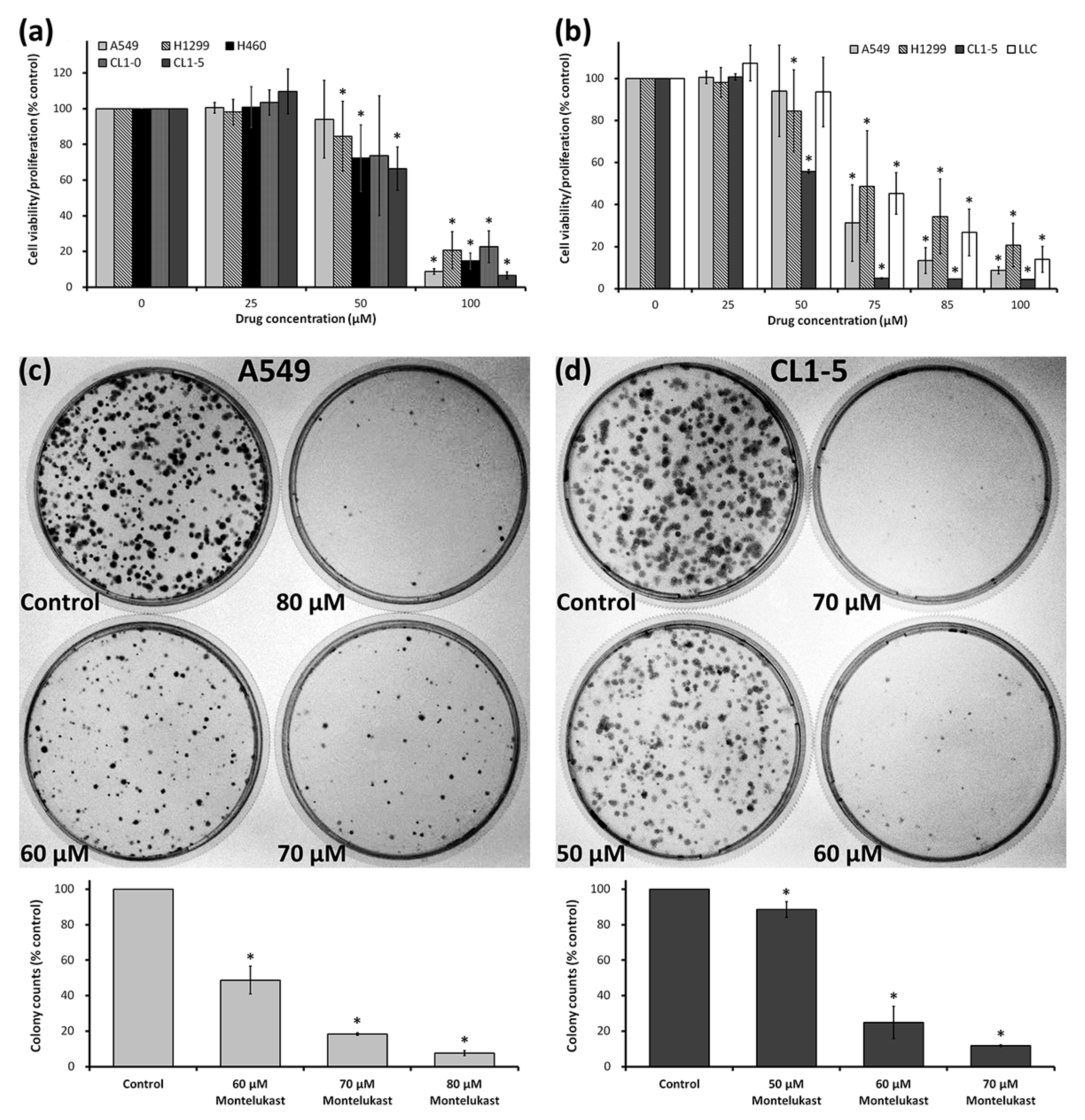
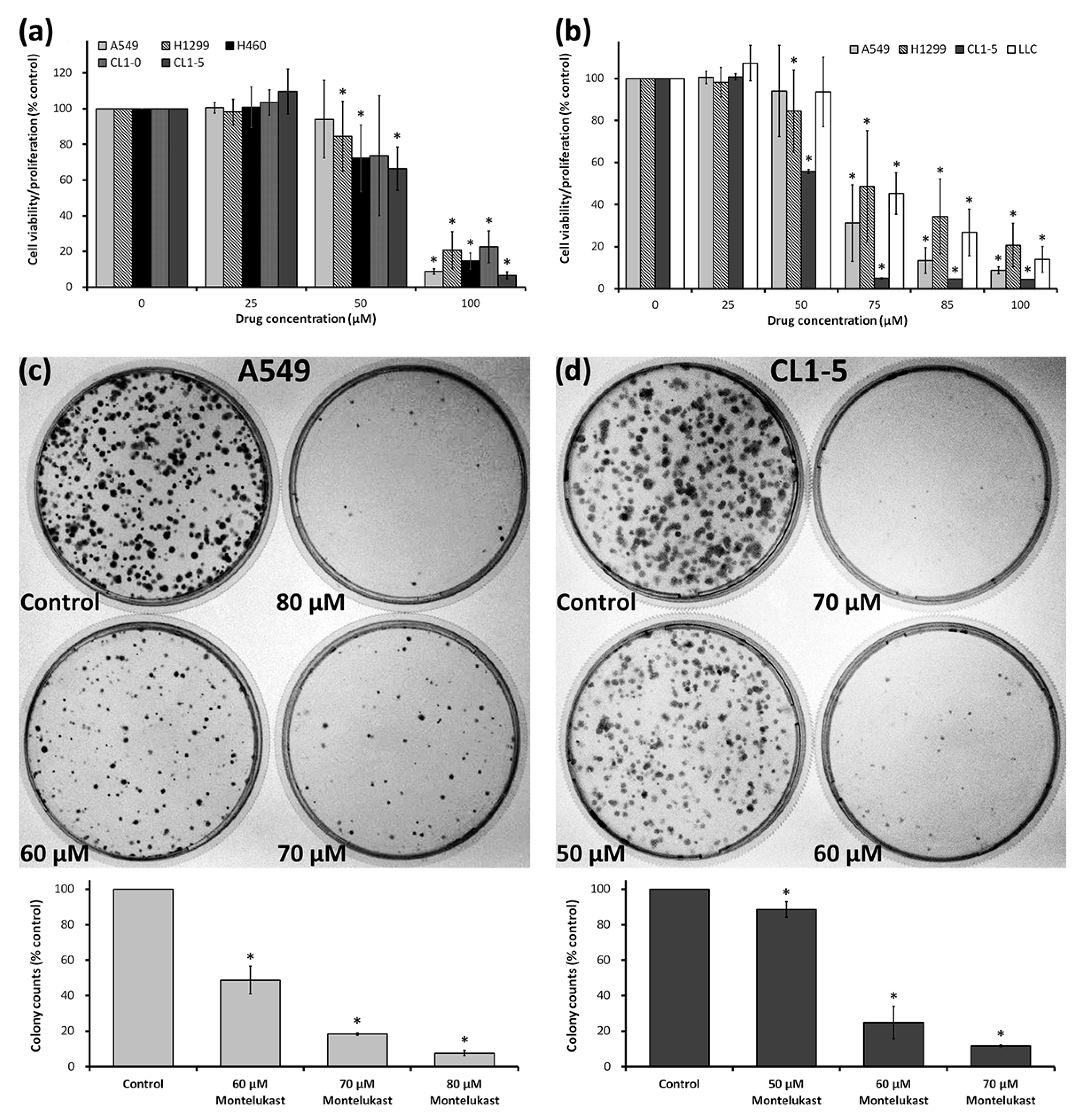
2.1. Montelukast Induced Cell Death of Lung Cancer Cells
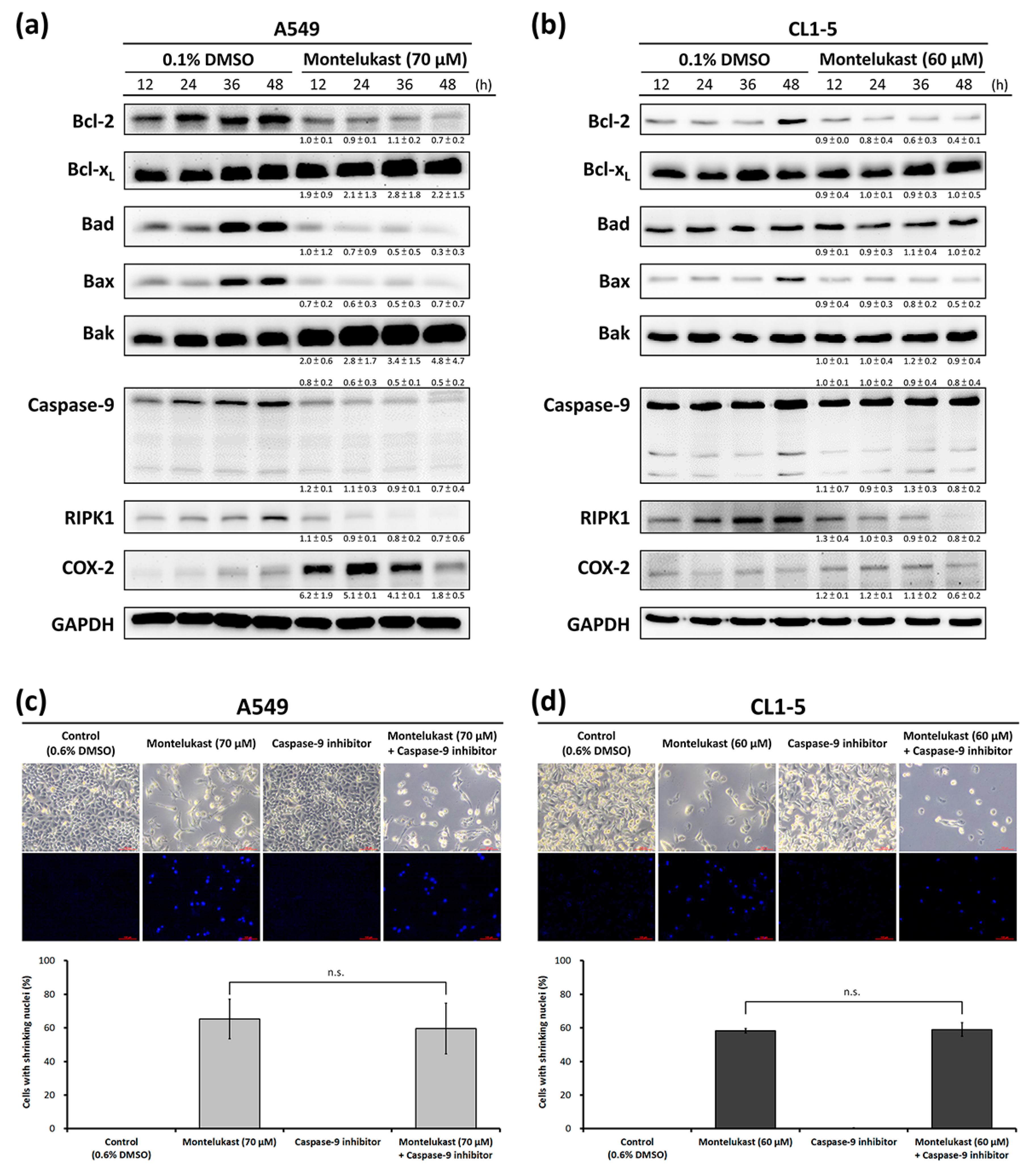
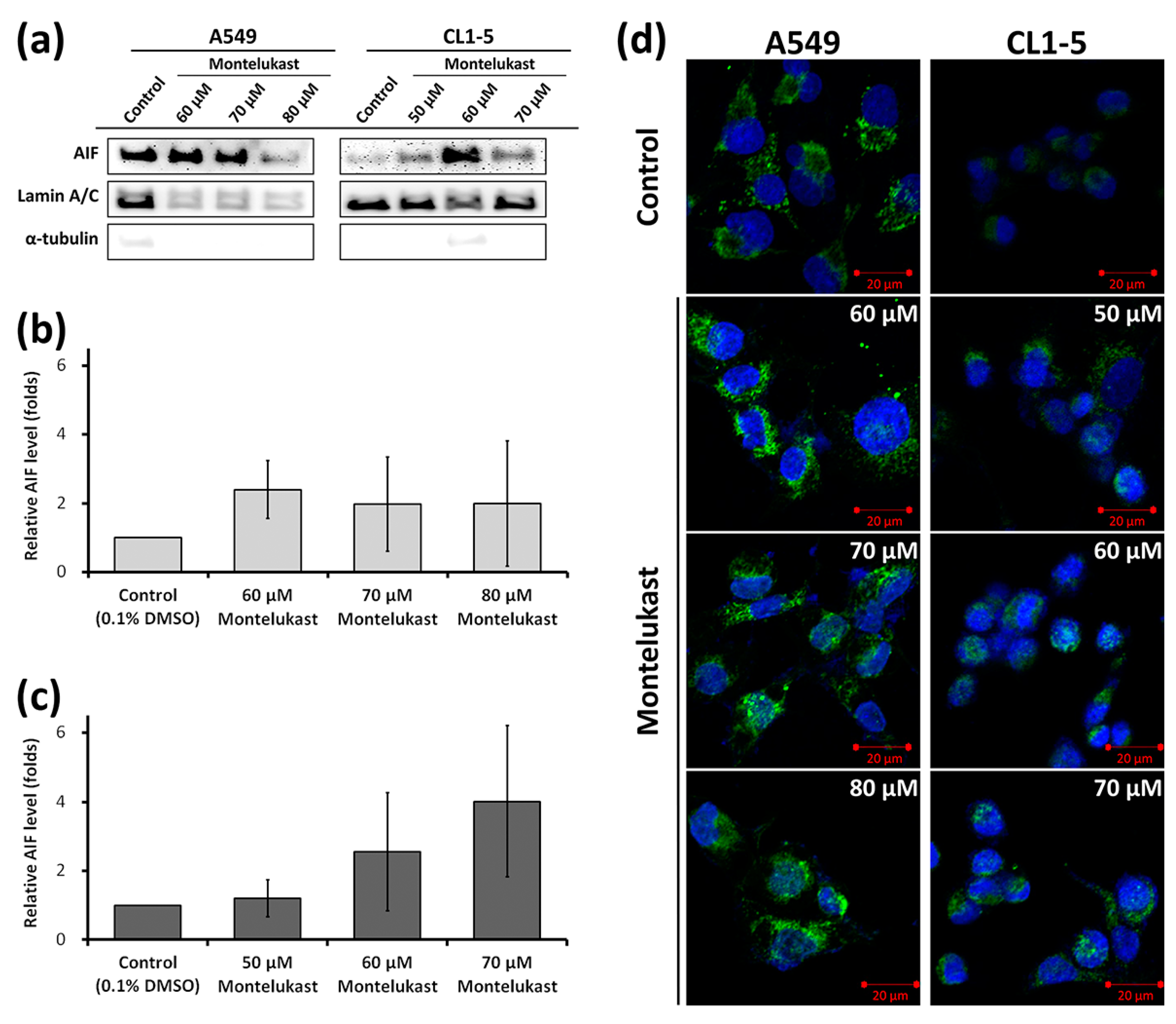
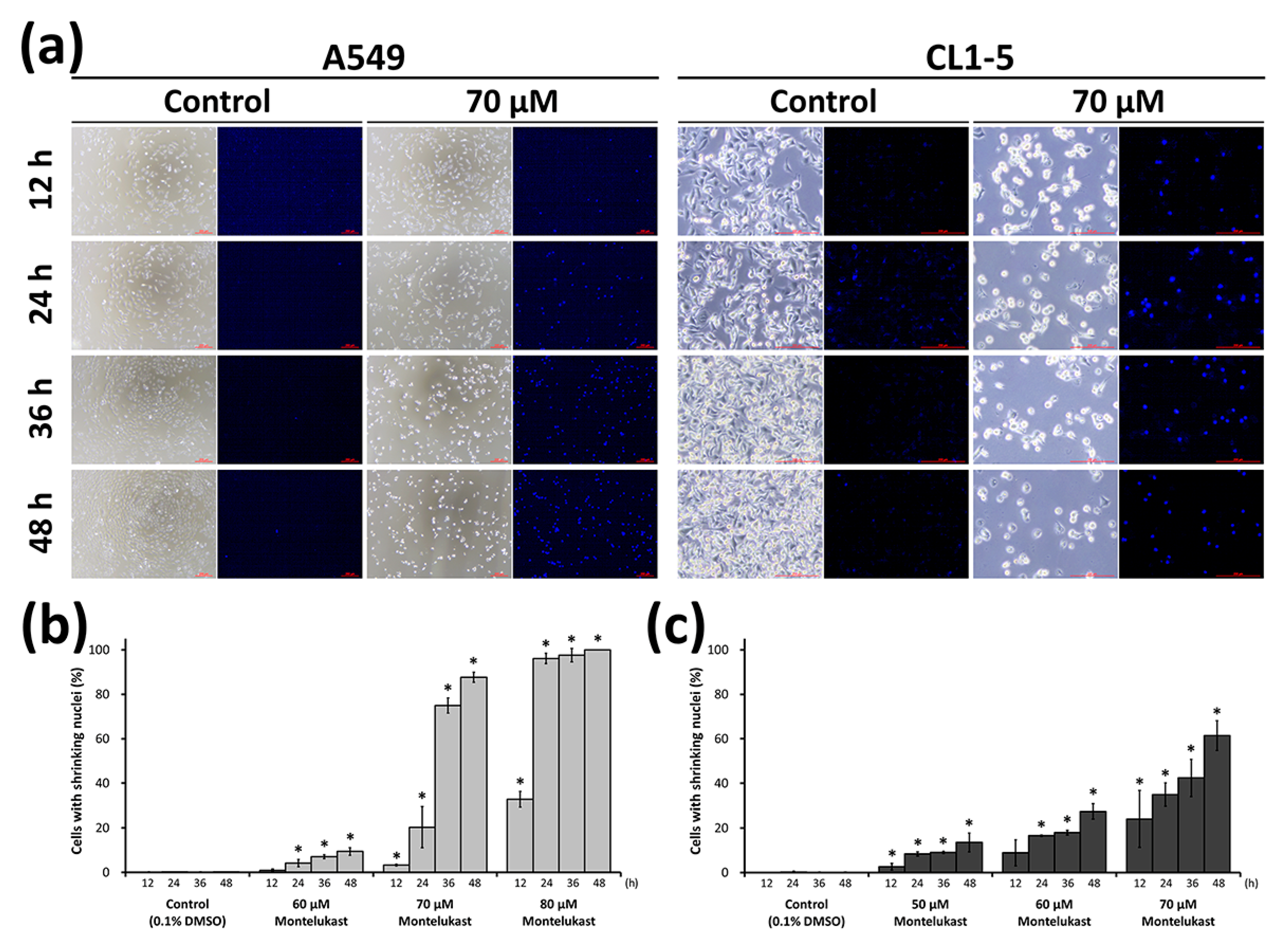
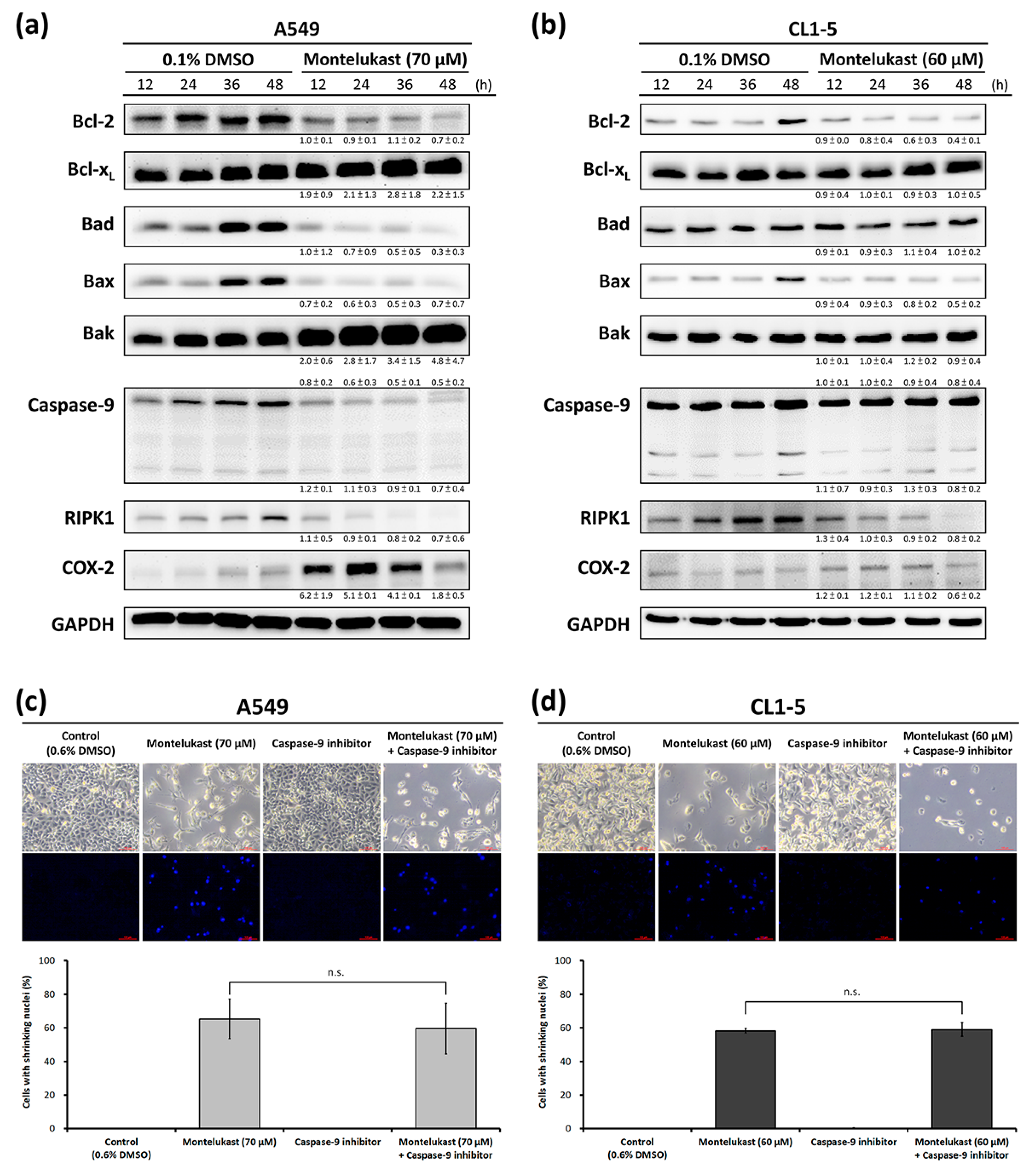
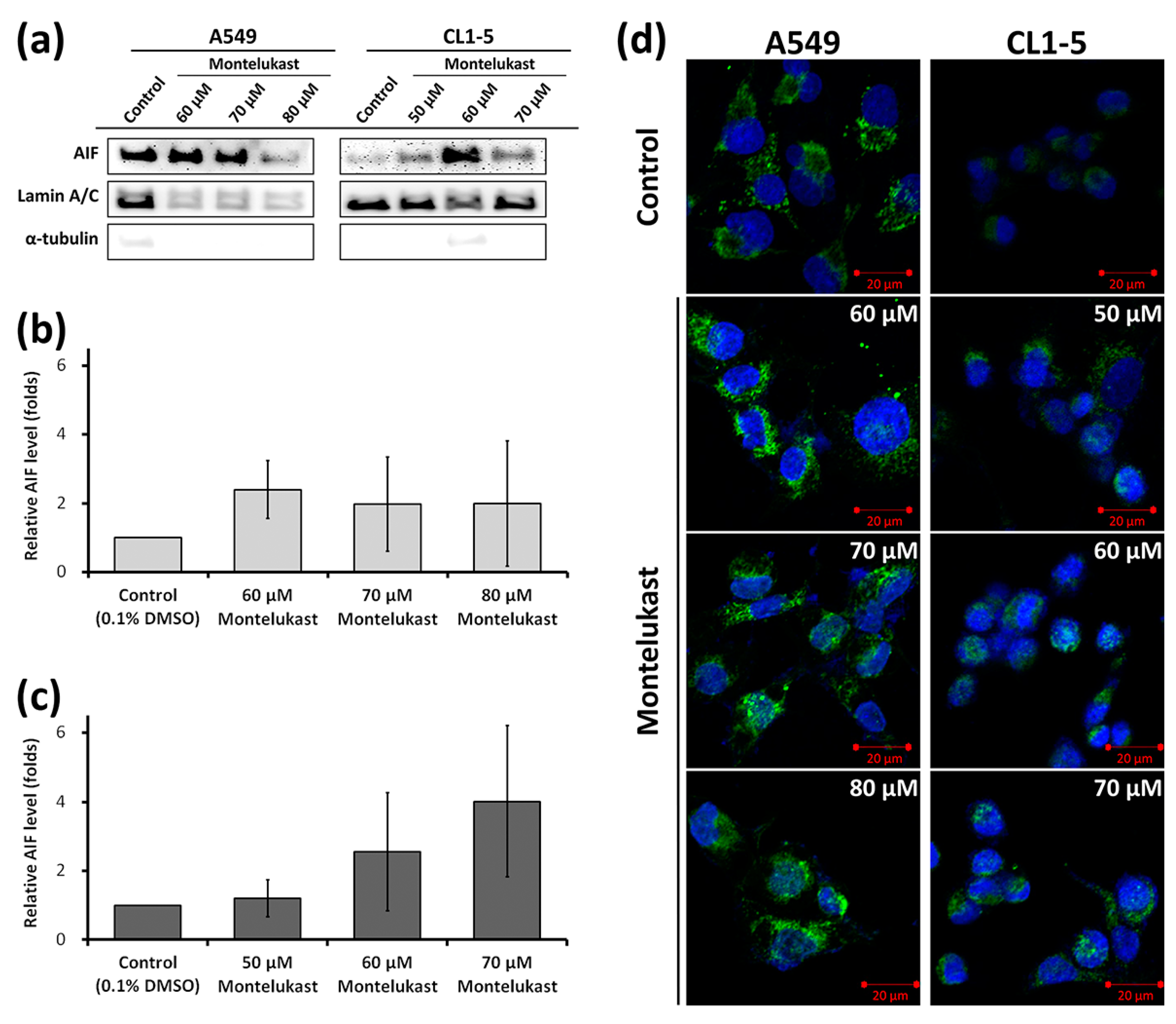
2.2. Montelukast Induced Cell Death of Lung Cancer Cells via Nuclear Translocation of Apoptosis-Inducing Factor
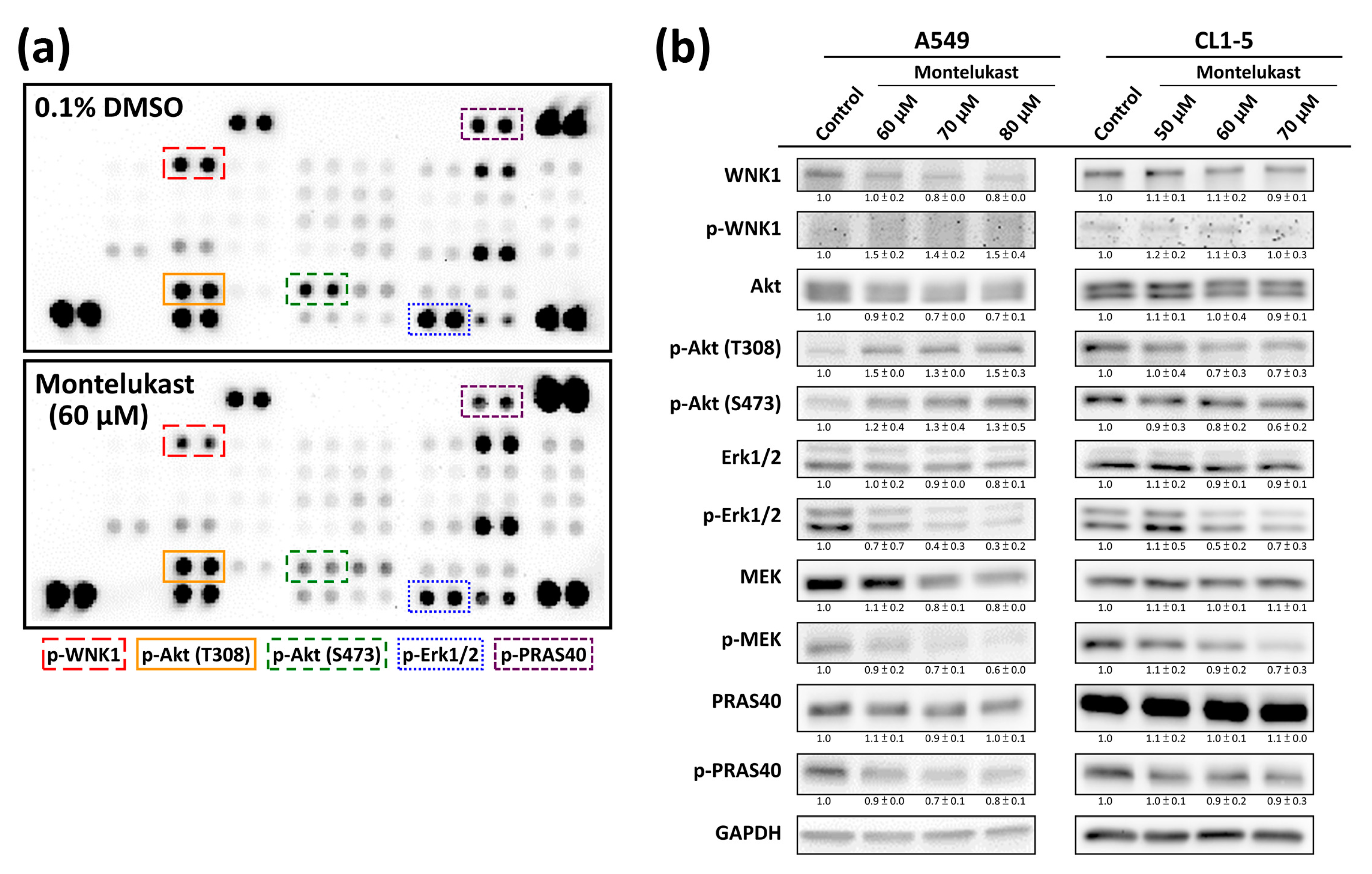
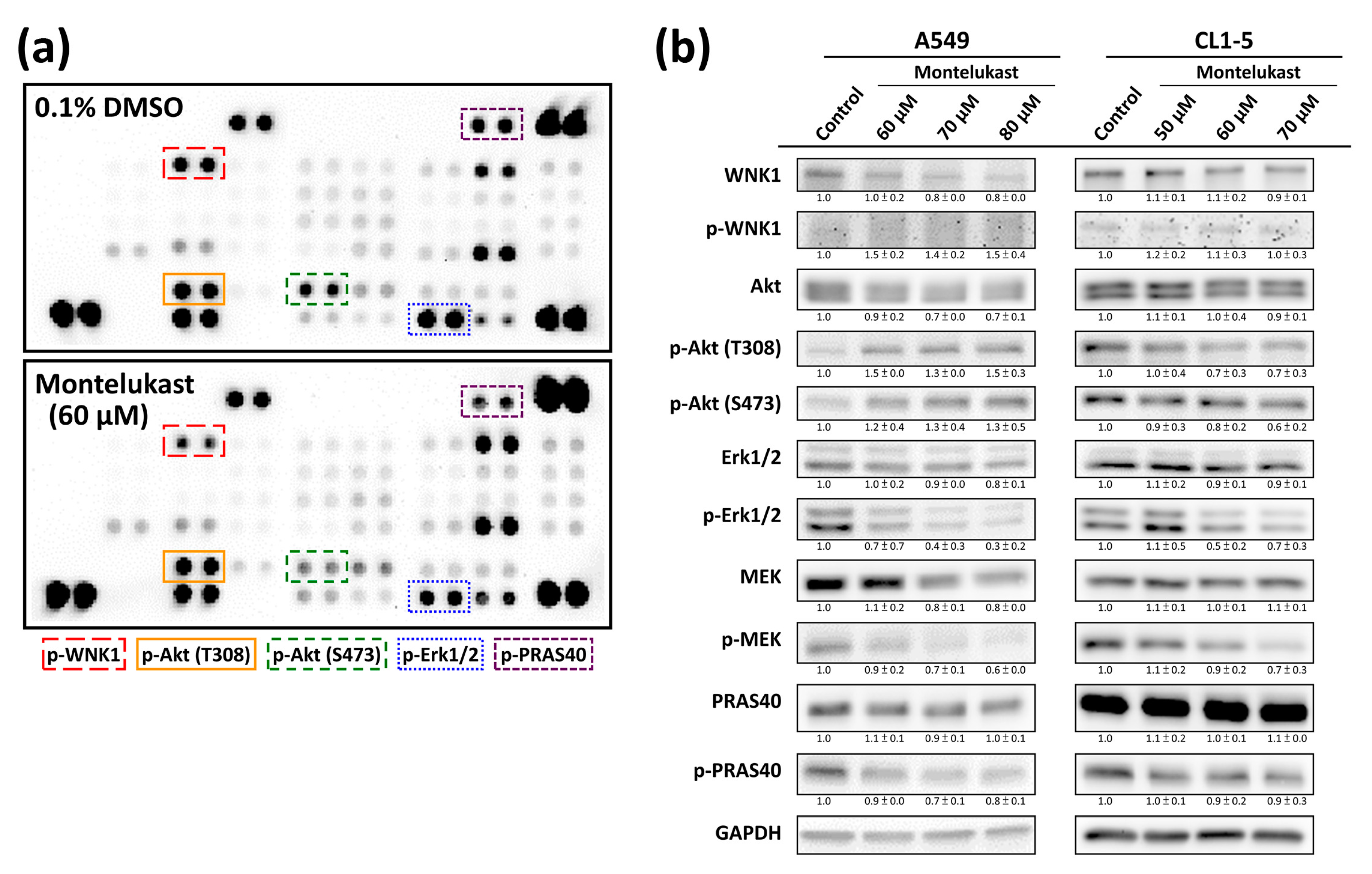
2.3. Montelukast Decreased Phosphorylation of With No Lysine 1 (WNK1), Protein Kinase B (Akt), Extracellular Signal-Regulated Kinase 1/2 (Erk1/2), MAPK/Erk kinase (MEK), and Proline-Rich Akt Substrate of 40-kDa (PRAS40) in Lung Cancer Cells
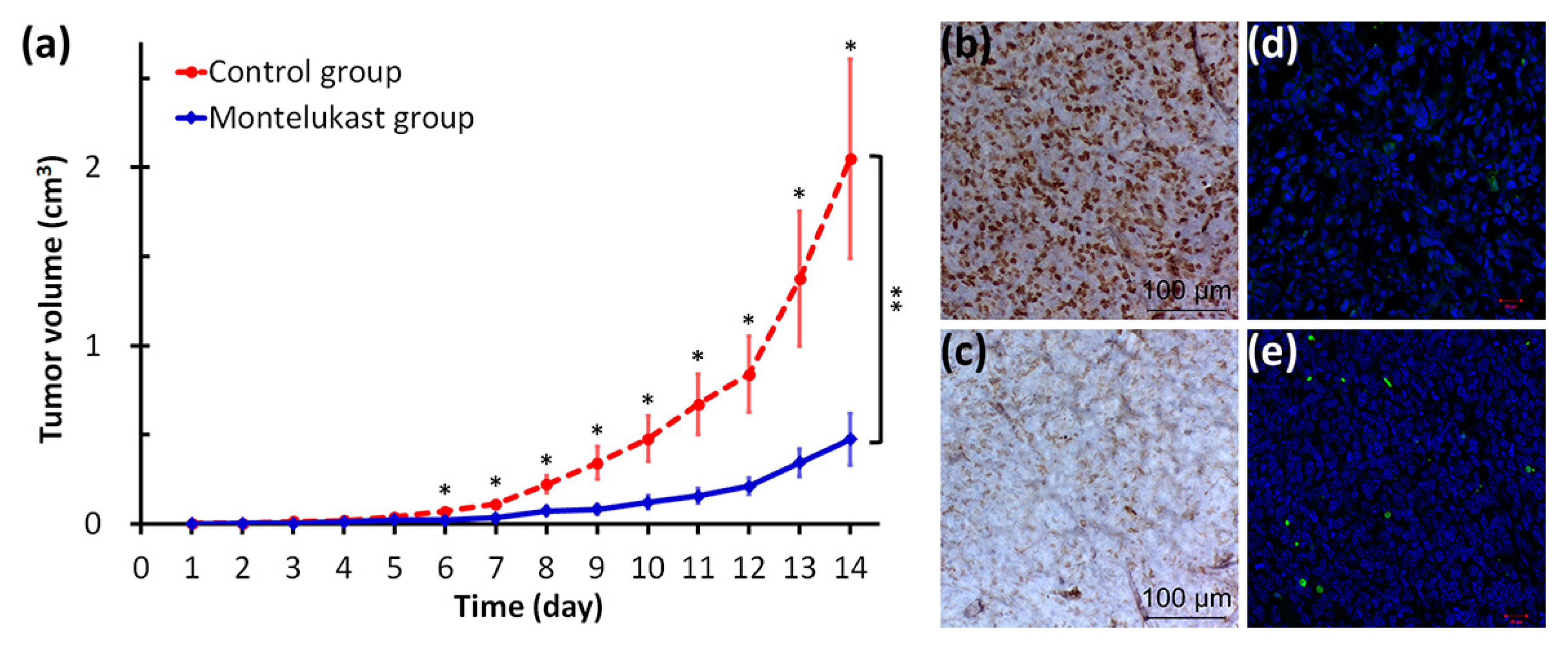
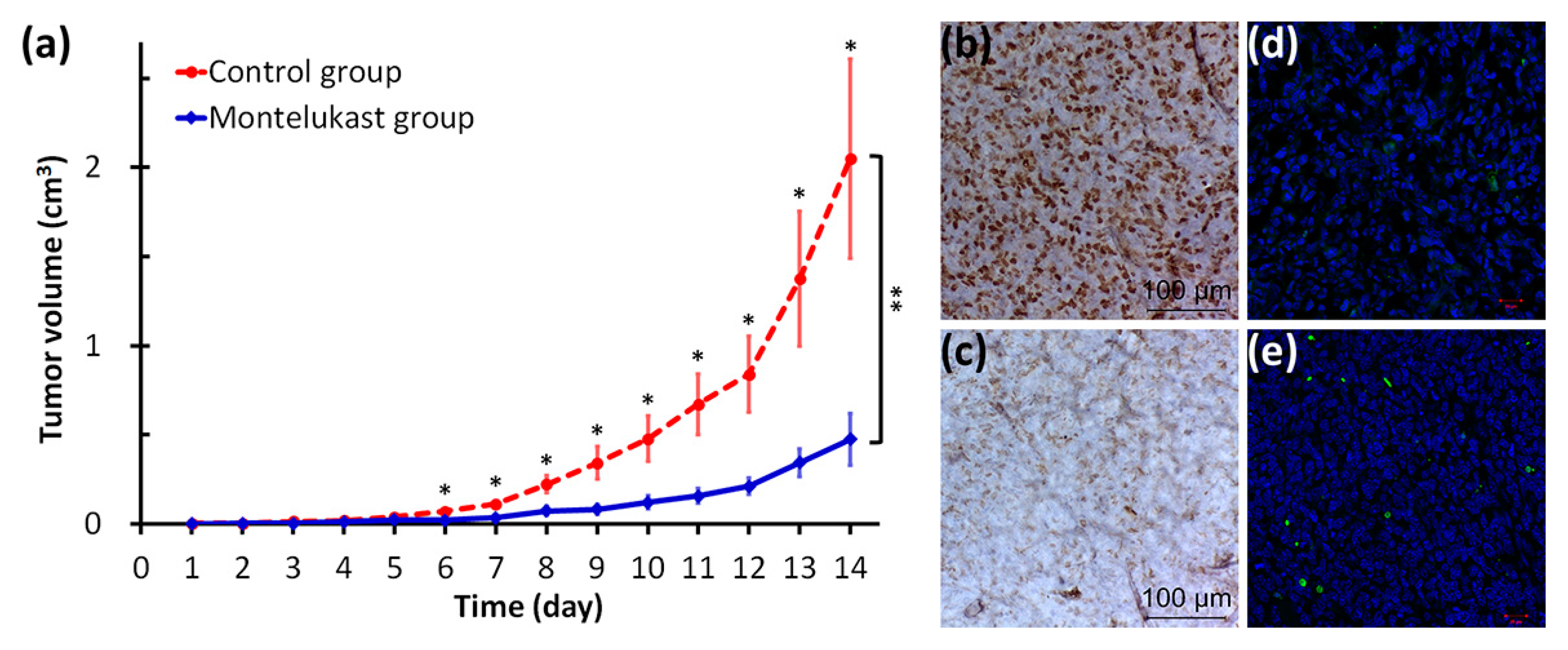
2.4. Montelukast Inhibited Tumor Growth in Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals
4.3. Cell Proliferation Assay
4.4. Clonogenic Assay
4.5. Immunofluorescence Analysis
4.6. Immunoblot Assay
4.7. Subcellular Fractionation
4.8. Immunofluorescence Confocal Microscopy
4.9. Human Phosphor-Kinase Array
4.10. Animal Study
4.11. Statistical Analysis
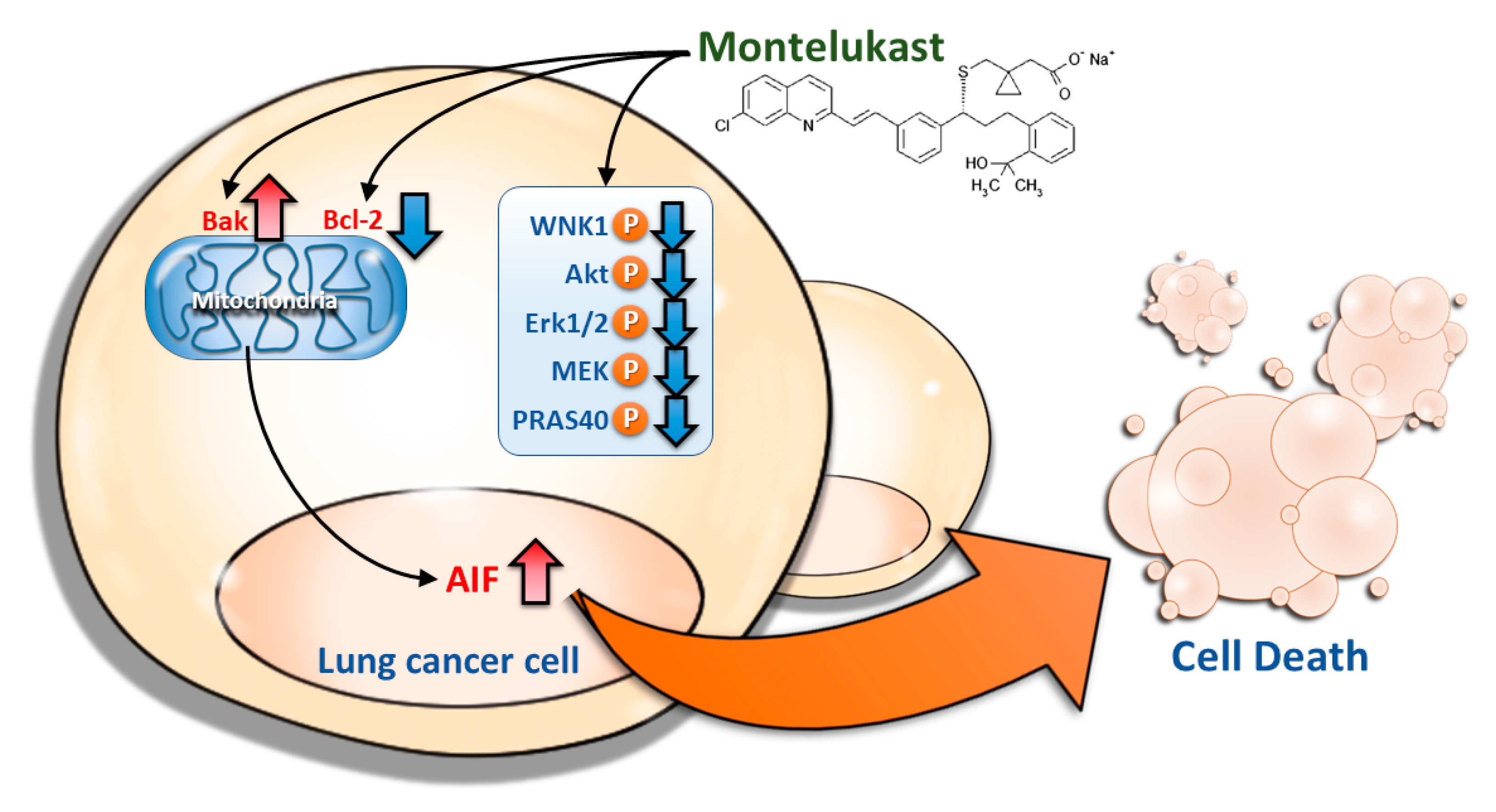
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| AIF | apoptosis-inducing factor |
| Bak | Bcl-2 homologous antagonist/killer |
| Bcl-2 | B-cell lymphoma 2 |
| BSA | bovine serum albumin |
| COX | cyclooxygenase |
| CXCL5 | C-X-C motif chemokine 5 |
| CysLT1R | cysteinyl leukotriene receptor 1 |
| DAPI | 4’,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| EndoG | endonuclease G |
| Erk | extracellular signal-regulated kinase |
| FBS | fetal bovine serum |
| LLC | Lewis lung carcinoma |
| LOX | lipoxygenase |
| LT | cysteinyl leukotriene |
| LTRA | cysteinyl leukotriene receptor antagonist |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/Erk kinase |
| MEM | modified Eagle’s medium |
| mTOR | mammalian target of rapamycin |
| PBS | phosphate-buffered saline |
| PRAS40 | proline-rich Akt substrate of 40-kDa |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| WNK1 | with no lysine 1 |
| WST-1 | water-soluble tetrazolium salt-1 |
References
- Tsai, M.J.; Wu, P.H.; Sheu, C.C.; Hsu, Y.L.; Chang, W.A.; Hung, J.Y.; Yang, C.J.; Yang, Y.H.; Kuo, P.L.; Huang, M.S. Cysteinyl leukotriene receptor antagonists decrease cancer risk in asthma patients. Sci. Rep. 2016, 6, 23979. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Yang, C.J.; Kung, Y.T.; Sheu, C.C.; Shen, Y.T.; Chang, P.Y.; Huang, M.S.; Chiu, H.C. Metformin decreases lung cancer risk in diabetic patients in a dose-dependent manner. Lung Cancer 2014, 86, 137–143. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cancer. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 29 April 2017).
- Scott, J.P.; Peters-Golden, M. Antileukotriene agents for the treatment of lung disease. Am. J. Respir. Crit. Care Med. 2013, 188, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, S.E.; Dahlen, B.; Drazen, J.M. Asthma treatment guidelines meet the real world. N. Engl. J. Med. 2011, 364, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; Yoshimura, R. Cysteinyl-leukotriene1 receptor is a potent target for the prevention and treatment of human urological cancer. Mol. Med. Rep. 2010, 3, 245–251. [Google Scholar] [PubMed]
- Funao, K.; Matsuyama, M.; Naganuma, T.; Kawahito, Y.; Sano, H.; Nakatani, T.; Yoshimura, R. The cysteinyllt1 receptor in human renal cell carcinoma. Mol. Med. Rep. 2008, 1, 185–189. [Google Scholar] [PubMed]
- Matsuyama, M.; Funao, K.; Hayama, T.; Tanaka, T.; Kawahito, Y.; Sano, H.; Takemoto, Y.; Nakatani, T.; Yoshimura, R. Relationship between cysteinyl-leukotriene-1 receptor and human transitional cell carcinoma in bladder. Urology 2009, 73, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; Hayama, T.; Funao, K.; Kawahito, Y.; Sano, H.; Takemoto, Y.; Nakatani, T.; Yoshimura, R. Overexpression of cysteinyl LT1 receptor in prostate cancer and CysLT1R antagonist inhibits prostate cancer cell growth through apoptosis. Oncol. Rep. 2007, 18, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; Funao, K.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.L.; Nakatani, T.; Yoshimura, R. Expression of cysteinylLT1 receptor in human testicular cancer and growth reduction by its antagonist through apoptosis. Mol. Med. Rep. 2009, 2, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Savari, S.; Liu, M.; Zhang, Y.; Sime, W.; Sjolander, A. CysLT1R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS ONE 2013, 8, e73466. [Google Scholar] [CrossRef] [PubMed]
- Gunning, W.T.; Kramer, P.M.; Steele, V.E.; Pereira, M.A. Chemoprevention by lipoxygenase and leukotriene pathway inhibitors of vinyl carbamate-induced lung tumors in mice. Cancer Res. 2002, 62, 4199–4201. [Google Scholar] [PubMed]
- Nozaki, M.; Yoshikawa, M.; Ishitani, K.; Kobayashi, H.; Houkin, K.; Imai, K.; Ito, Y.; Muraki, T. Cysteinyl leukotriene receptor antagonists inhibit tumor metastasis by inhibiting capillary permeability. Keio J. Med. 2010, 59, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Jose, M.A.; Amathi, R.; Sathyamurthy, D.; Kumar, B.N. Chemopreventive effect of montelukast in n-nitroso n-methyl urea induced mammary carcinogenesis in female sprague-dawley rats. Indian J. Pharmacol. 2013, 45, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Boujrad, H.; Gubkina, O.; Robert, N.; Krantic, S.; Susin, S.A. AIF-mediated programmed necrosis: A highly regulated way to die. Cell Cycle 2007, 6, 2612–2619. [Google Scholar] [CrossRef] [PubMed]
- Baritaud, M.; Boujrad, H.; Lorenzo, H.K.; Krantic, S.; Susin, S.A. Histone h2ax: The missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle 2010, 9, 3166–3173. [Google Scholar] [CrossRef] [PubMed]
- Artus, C.; Boujrad, H.; Bouharrour, A.; Brunelle, M.N.; Hoos, S.; Yuste, V.J.; Lenormand, P.; Rousselle, J.C.; Namane, A.; England, P.; et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010, 29, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- D’Anneo, A.; Carlisi, D.; Lauricella, M.; Emanuele, S.; Di Fiore, R.; Vento, R.; Tesoriere, G. Parthenolide induces caspase-independent and AIF-mediated cell death in human osteosarcoma and melanoma cells. J. Cell Physiol. 2013, 228, 952–967. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.Y.; Pidgeon, G.P. Cross-talk between cancer cells and the tumour microenvironment: The role of the 5-lipoxygenase pathway. Int. J. Mol. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.L.; Huang, M.S.; Hung, J.Y.; Chou, S.H.; Chiang, S.Y.; Huang, Y.F.; Yang, C.J.; Tsai, M.J.; Chang, W.A.; Hsu, Y.L. Synergistic effect of lung tumor-associated dendritic cell-derived HB-EGF and CXCL5 on cancer progression. Int. J. Cancer 2014, 135, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Poczobutt, J.M.; Gijon, M.; Amin, J.; Hanson, D.; Li, H.; Walker, D.; Weiser-Evans, M.; Lu, X.; Murphy, R.C.; Nemenoff, R.A. Eicosanoid profiling in an orthotopic model of lung cancer progression by mass spectrometry demonstrates selective production of leukotrienes by inflammatory cells of the microenvironment. PLoS ONE 2013, 8, e79633. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Yang, Y.H.; Cheng, C.L.; Ho, P.S.; Ko, Y.C. The role of chemoprevention by selective cyclooxygenase-2 inhibitors in colorectal cancer patients—A population-based study. BMC Cancer 2012, 12, 582. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Janakiram, N.B.; Mohammed, A. Lipoxygenase and cyclooxygenase pathways and colorectal cancer prevention. Curr. Colorectal Cancer Rep. 2012, 8, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R.; Sjolander, A. The role of leukotriene receptor signaling in inflammation and cancer. Sci. World J. 2007, 7, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Wu, N.; Sood, S.; Wang, P.; Jin, Z.; Beer, D.G.; Giordano, T.J.; Lin, Y.; Shih, W.C.; et al. Overexpression of 5-lipoxygenase in rat and human esophageal adenocarcinoma and inhibitory effects of zileuton and celecoxib on carcinogenesis. Clin. Cancer Res. 2004, 10, 6703–6709. [Google Scholar] [CrossRef] [PubMed]
- Larre, S.; Tran, N.; Fan, C.; Hamadeh, H.; Champigneulles, J.; Azzouzi, R.; Cussenot, O.; Mangin, P.; Olivier, J.L. PGE2 and LTB4 tissue levels in benign and cancerous prostates. Prostaglandins Other Lipid Mediat 2008, 87, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Venerito, M.; Helmke, C.; Jechorek, D.; Wex, T.; Rosania, R.; Antweiler, K.; Weigt, J.; Malfertheiner, P. Leukotriene receptor expression in esophageal squamous cell cancer and non-transformed esophageal epithelium: A matched case control study. BMC Gastroenterol. 2016, 16, 85. [Google Scholar] [CrossRef] [PubMed]
- El-Hakim, I.; Zakrzewski, J.; Langdon, J.; Piper, P.; Costello, J. A possible role for leukotriene B4 in head and neck cancer. Br. J. Cancer 1989, 59, 833. [Google Scholar] [CrossRef] [PubMed]
- Venerito, M.; Kuester, D.; Harms, C.; Schubert, D.; Wex, T.; Malfertheiner, P. Upregulation of leukotriene receptors in gastric cancer. Cancers 2011, 3, 3156–3168. [Google Scholar] [CrossRef] [PubMed]
- Bortuzzo, C.; Hanif, R.; Kashfi, K.; Staiano-Coico, L.; Shiff, S.J.; Rigas, B. The effect of leukotrienes b and selected hetes on the proliferation of colon cancer cells. Biochim. Biophys. Acta 1996, 1300, 240–246. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, D.; Li, H.; Jie, S. Circulating LTD4 in patients with hepatocellular carcinoma. Tumour. Biol. 2011, 32, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Bellamkonda, K.; Chandrashekar, N.K.; Osman, J.; Selvanesan, B.C.; Savari, S.; Sjolander, A. The eicosanoids leukotriene D4 and prostaglandin E2 promote the tumorigenicity of colon cancer-initiating cells in a xenograft mouse model. BMC Cancer 2016, 16, 425. [Google Scholar] [CrossRef] [PubMed]
- Osman, J.; Savari, S.; Chandrashekar, N.K.; Bellamkonda, K.; Douglas, D.; Sjolander, A. Cysteinyl leukotriene receptor 1 facilitates tumorigenesis in a mouse model of colitis-associated colon cancer. Oncotarget 2017, 8, 34773–34786. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, C.; Liu, J.; Ehrnstrom, R.; Manjer, J.; Jirstrom, K.; Andersson, T.; Sjolander, A. Cysteinyl leukotriene receptor expression pattern affects migration of breast cancer cells and survival of breast cancer patients. Int. J. Cancer 2011, 129, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, C.; Mezhybovska, M.; Lorinc, E.; Fernebro, E.; Nilbert, M.; Sjolander, A. Low expression of CysLT1R and high expression of CysLT2R mediate good prognosis in colorectal cancer. Eur. J. Cancer 2010, 46, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.K.; Ohd, J.F.; Wikstrom, K.; Massoumi, R.; Paruchuri, S.; Juhas, M.; Sjolander, A. The leukotriene receptor cysLT1 and 5-lipoxygenase are upregulated in colon cancer. Adv. Exp. Med. Biol. 2003, 525, 201–204. [Google Scholar] [PubMed]
- Sveinbjornsson, B.; Rasmuson, A.; Baryawno, N.; Wan, M.; Pettersen, I.; Ponthan, F.; Orrego, A.; Haeggstrom, J.Z.; Johnsen, J.I.; Kogner, P. Expression of enzymes and receptors of the leukotriene pathway in human neuroblastoma promotes tumor survival and provides a target for therapy. FASEB J. 2008, 22, 3525–3536. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.J.; Kuo, P.L.; Hsu, Y.C.; Huang, Y.F.; Tsai, E.M.; Hsu, Y.L. Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic. Biol. Med. 2014, 67, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prevost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [PubMed]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease g is an apoptotic dnase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Paruchuri, S.; Hallberg, B.; Juhas, M.; Larsson, C.; Sjolander, A. Leukotriene D4 activates MAPK through a Ras-independent but pkcepsilon-dependent pathway in intestinal epithelial cells. J. Cell Sci. 2002, 115, 1883–1893. [Google Scholar] [PubMed]
- Savari, S.; Vinnakota, K.; Zhang, Y.; Sjolander, A. Cysteinyl leukotrienes and their receptors: Bridging inflammation and colorectal cancer. World J. Gastroenterol. 2014, 20, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yang, L.F.; Song, Z.Q.; Shah, S.Z.A.; Cui, Y.Y.; Li, C.S.; Zhao, H.F.; Gao, H.L.; Zhou, X.M.; Zhao, D.M. Pras40 alleviates neurotoxic prion peptide-induced apoptosis via mTOR-Akt signaling. CNS Neurosci. Ther. 2017, 23, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, J.Y.; Chiang, S.Y.; Jian, S.F.; Wu, C.Y.; Lin, Y.S.; Tsai, Y.M.; Chou, S.H.; Tsai, M.J.; Kuo, P.L. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 2016, 7, 27584–27598. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.M.; Chong, I.W.; Hung, J.Y.; Chang, W.A.; Kuo, P.L.; Tsai, M.J.; Hsu, Y.L. Syringetin suppresses osteoclastogenesis mediated by osteoblasts in human lung adenocarcinoma. Oncol. Rep. 2015, 34, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Skeen, J.E.; Bhaskar, P.T.; Chen, C.C.; Chen, W.S.; Peng, X.D.; Nogueira, V.; Hahn-Windgassen, A.; Kiyokawa, H.; Hay, N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell 2006, 10, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.N.; Wu, W.K.; Shin, V.Y.; Bruce, I.C.; Wong, B.C.; Cho, C.H. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 2005, 26, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Valero, A.; Picado, C. Usefulness of montelukast to prevent adverse reactions to COX-2 selective inhibitors: A case report. J. Investig. Allergol. Clin. Immunol. 2005, 15, 156–157. [Google Scholar] [PubMed]
- Tsai, M.J.; Hsu, Y.L.; Wang, T.N.; Wu, L.Y.; Lien, C.T.; Hung, C.H.; Kuo, P.L.; Huang, M.S. Aryl hydrocarbon receptor (AhR) agonists increase airway epithelial matrix metalloproteinase activity. J. Mol. Med. 2014, 92, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, M.-J.; Chang, W.-A.; Tsai, P.-H.; Wu, C.-Y.; Ho, Y.-W.; Yen, M.-C.; Lin, Y.-S.; Kuo, P.-L.; Hsu, Y.-L. Montelukast Induces Apoptosis-Inducing Factor-Mediated Cell Death of Lung Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1353. https://doi.org/10.3390/ijms18071353
Tsai M-J, Chang W-A, Tsai P-H, Wu C-Y, Ho Y-W, Yen M-C, Lin Y-S, Kuo P-L, Hsu Y-L. Montelukast Induces Apoptosis-Inducing Factor-Mediated Cell Death of Lung Cancer Cells. International Journal of Molecular Sciences. 2017; 18(7):1353. https://doi.org/10.3390/ijms18071353
Chicago/Turabian StyleTsai, Ming-Ju, Wei-An Chang, Pei-Hsun Tsai, Cheng-Ying Wu, Ya-Wen Ho, Meng-Chi Yen, Yi-Shiuan Lin, Po-Lin Kuo, and Ya-Ling Hsu. 2017. "Montelukast Induces Apoptosis-Inducing Factor-Mediated Cell Death of Lung Cancer Cells" International Journal of Molecular Sciences 18, no. 7: 1353. https://doi.org/10.3390/ijms18071353
APA StyleTsai, M.-J., Chang, W.-A., Tsai, P.-H., Wu, C.-Y., Ho, Y.-W., Yen, M.-C., Lin, Y.-S., Kuo, P.-L., & Hsu, Y.-L. (2017). Montelukast Induces Apoptosis-Inducing Factor-Mediated Cell Death of Lung Cancer Cells. International Journal of Molecular Sciences, 18(7), 1353. https://doi.org/10.3390/ijms18071353