Drug Hypersensitivity and Desensitizations: Mechanisms and New Approaches

Abstract
:
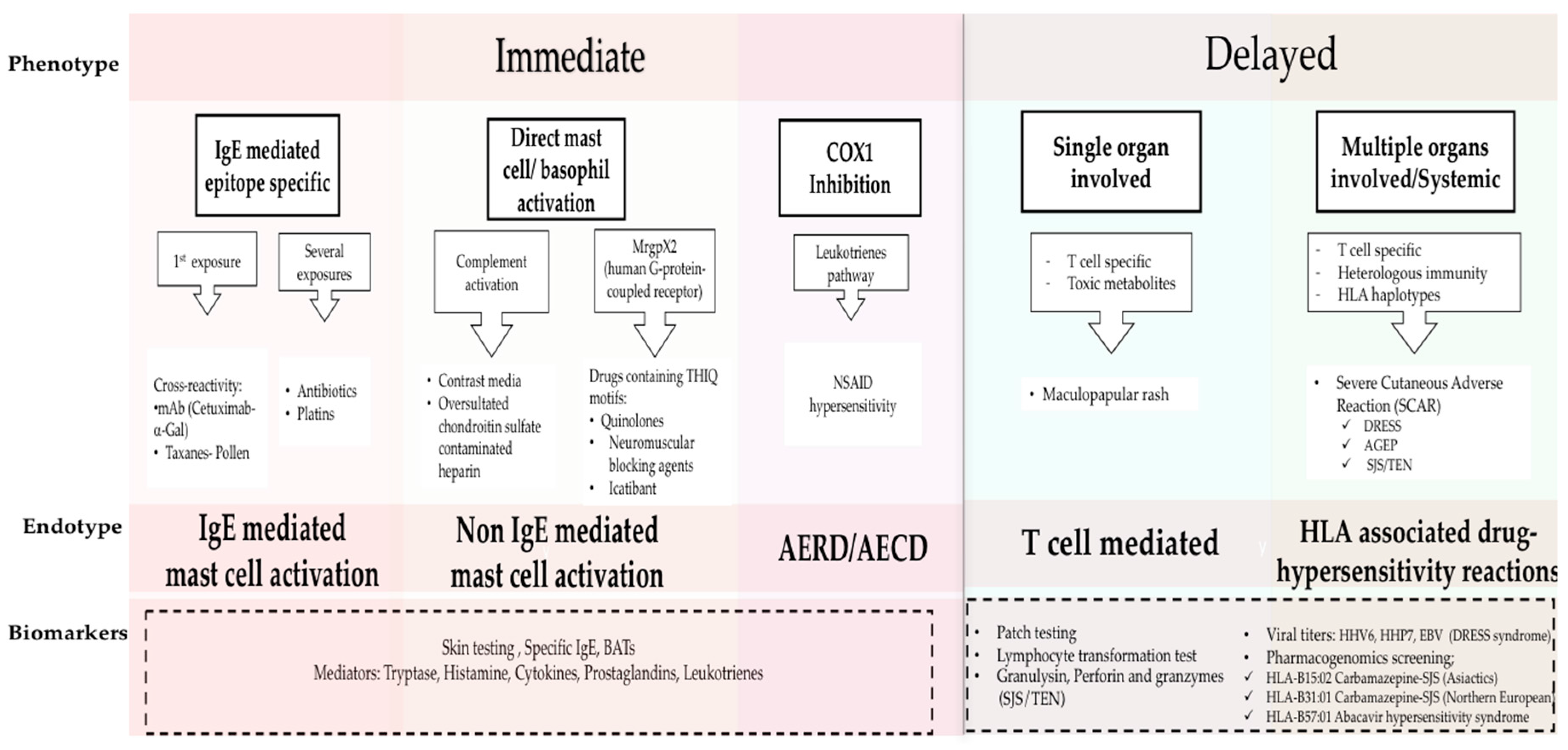
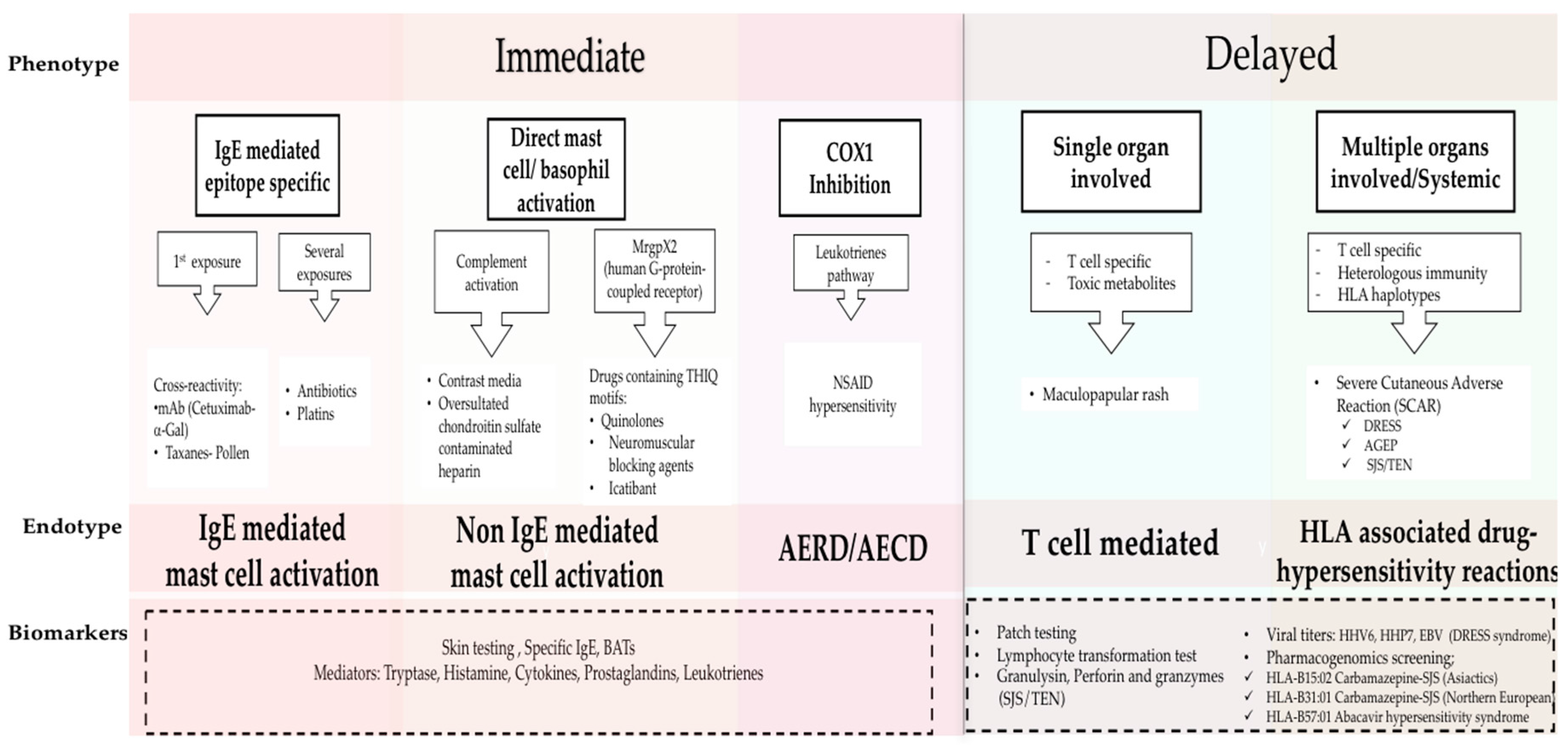
1. Drug Hypersensitivity Reactions: New Clinical Approach Through Phenotypes, Endotypes, and Biomarkers
2. Drug Desensitization: A Revolutionary Approach to the Management of Type I and Type IV Drug Hypersensitivity Reactions
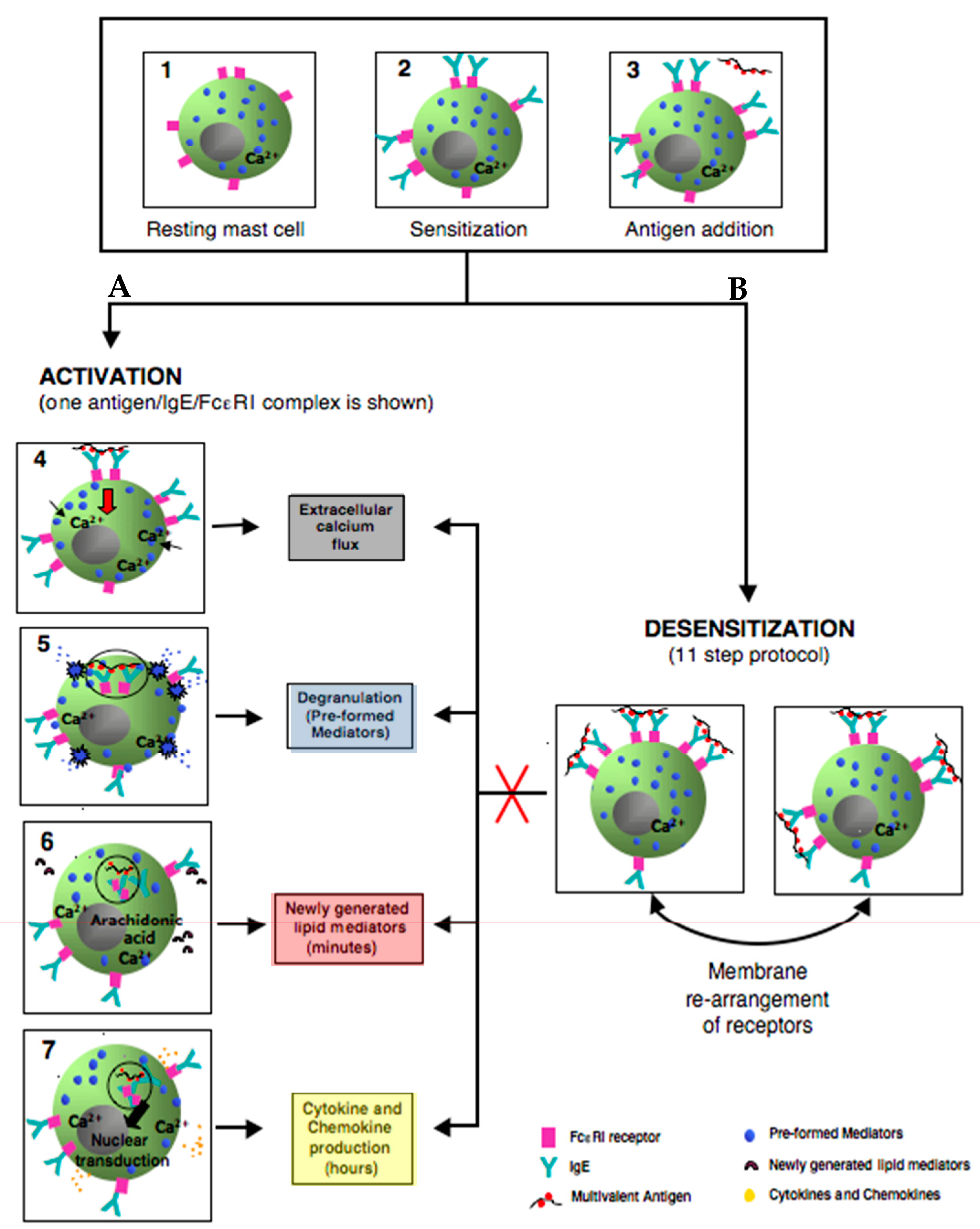
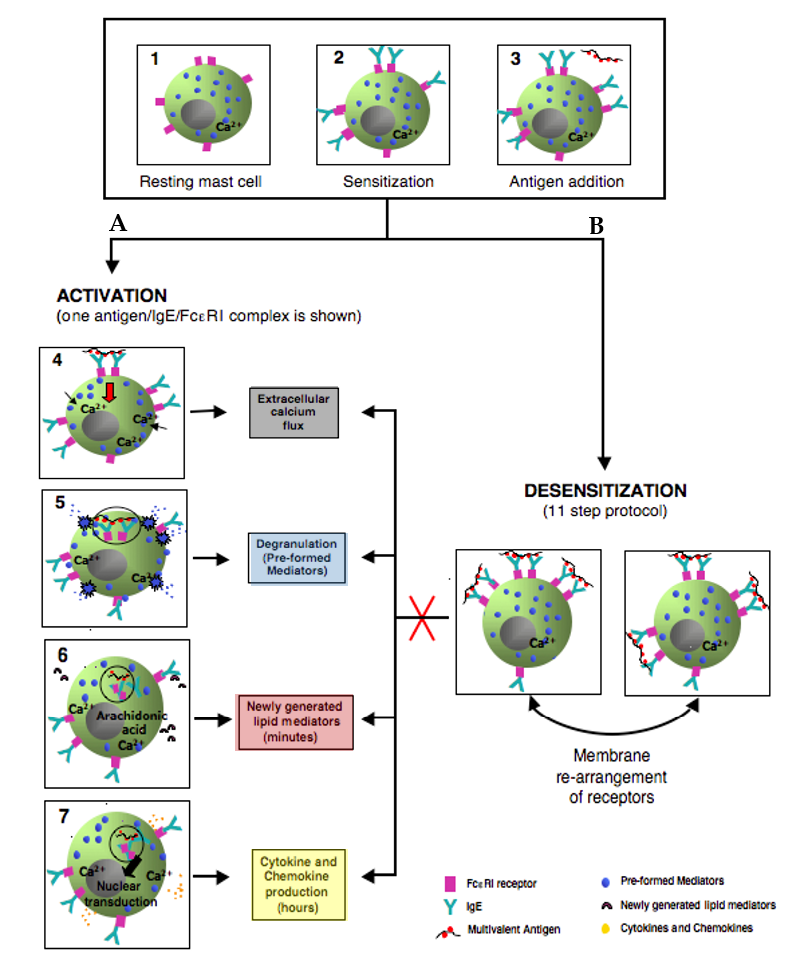
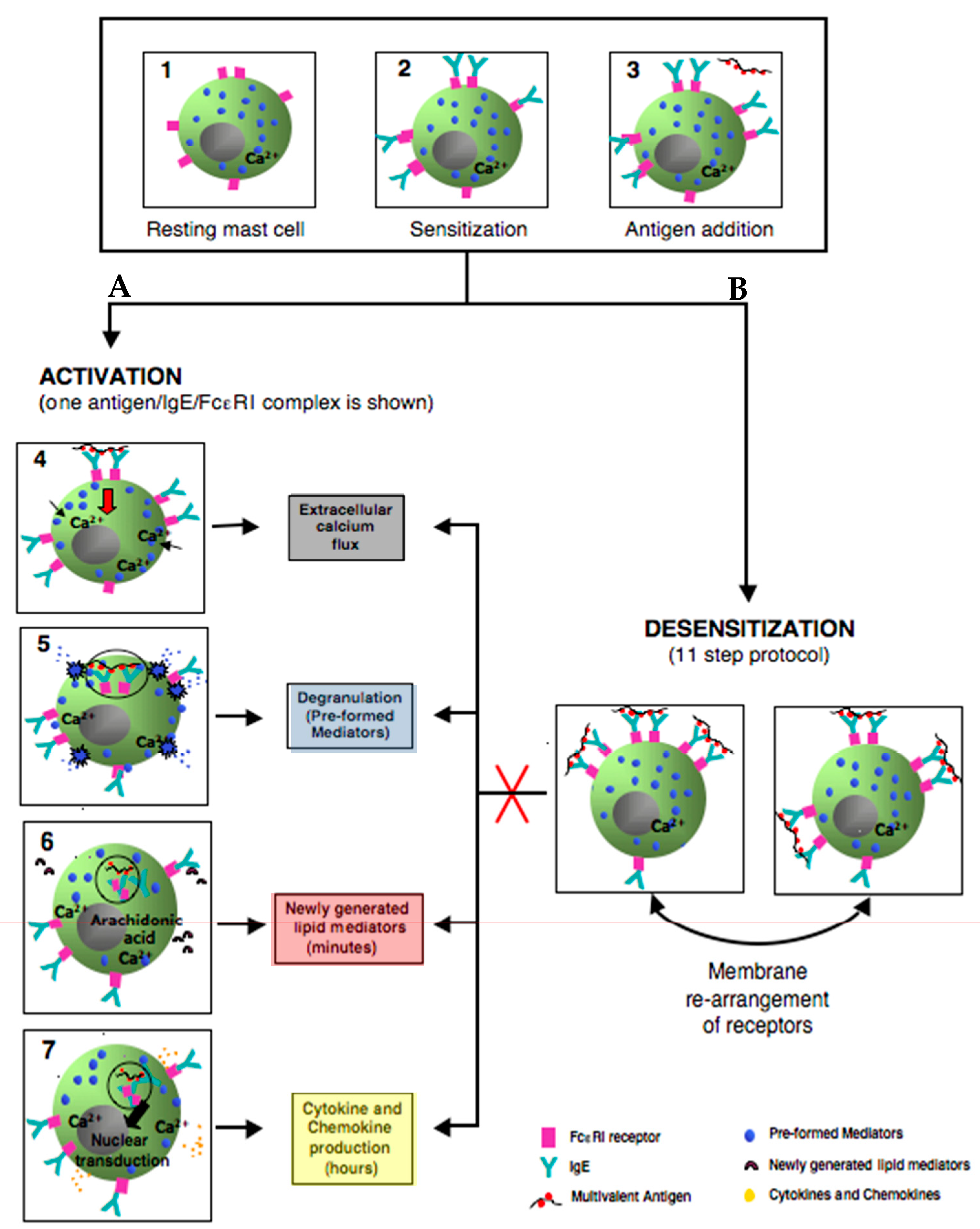
3. Mast Cells: Positive and Negative Regulation is Relevant to Desensitization
3.1. FcεRI Structure
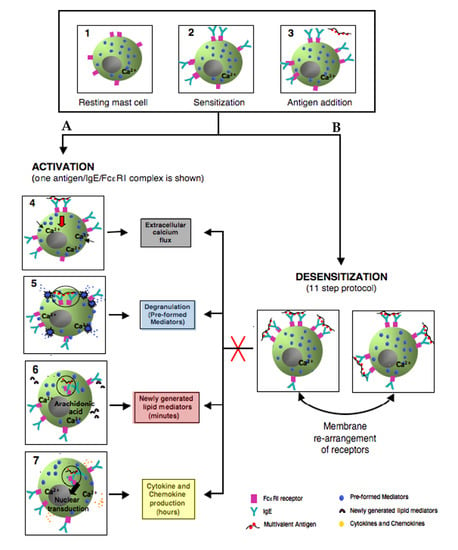
3.2. Mast Cell Activation via FcεRI
3.3. Degranulation, Lipid Mediator, and Cytokine Production
3.4. Negative Regulation of Mast Cell Activation through FcεRI
4. Molecular Mechanisms in IgE Mast Cell Desensitization
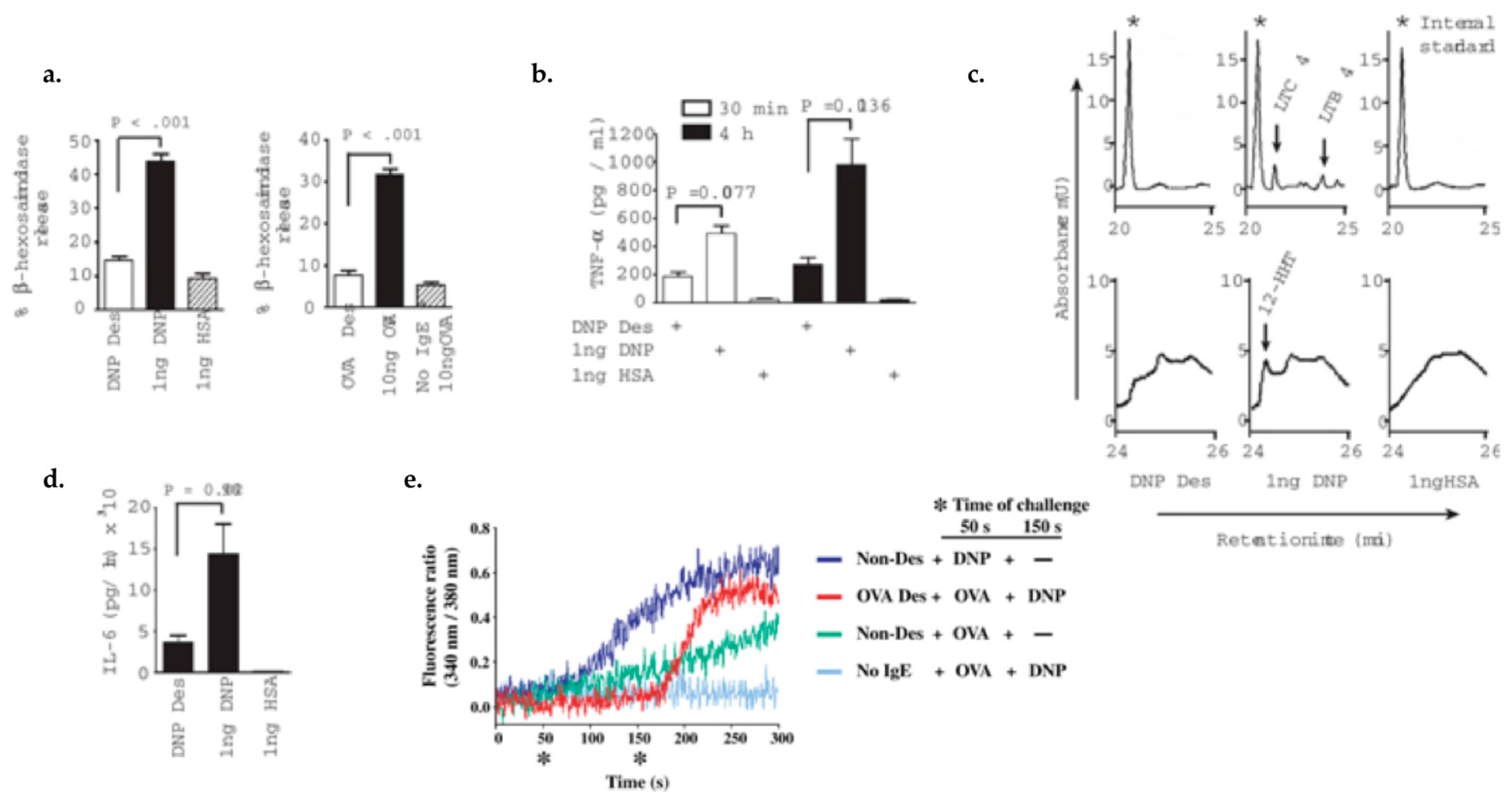
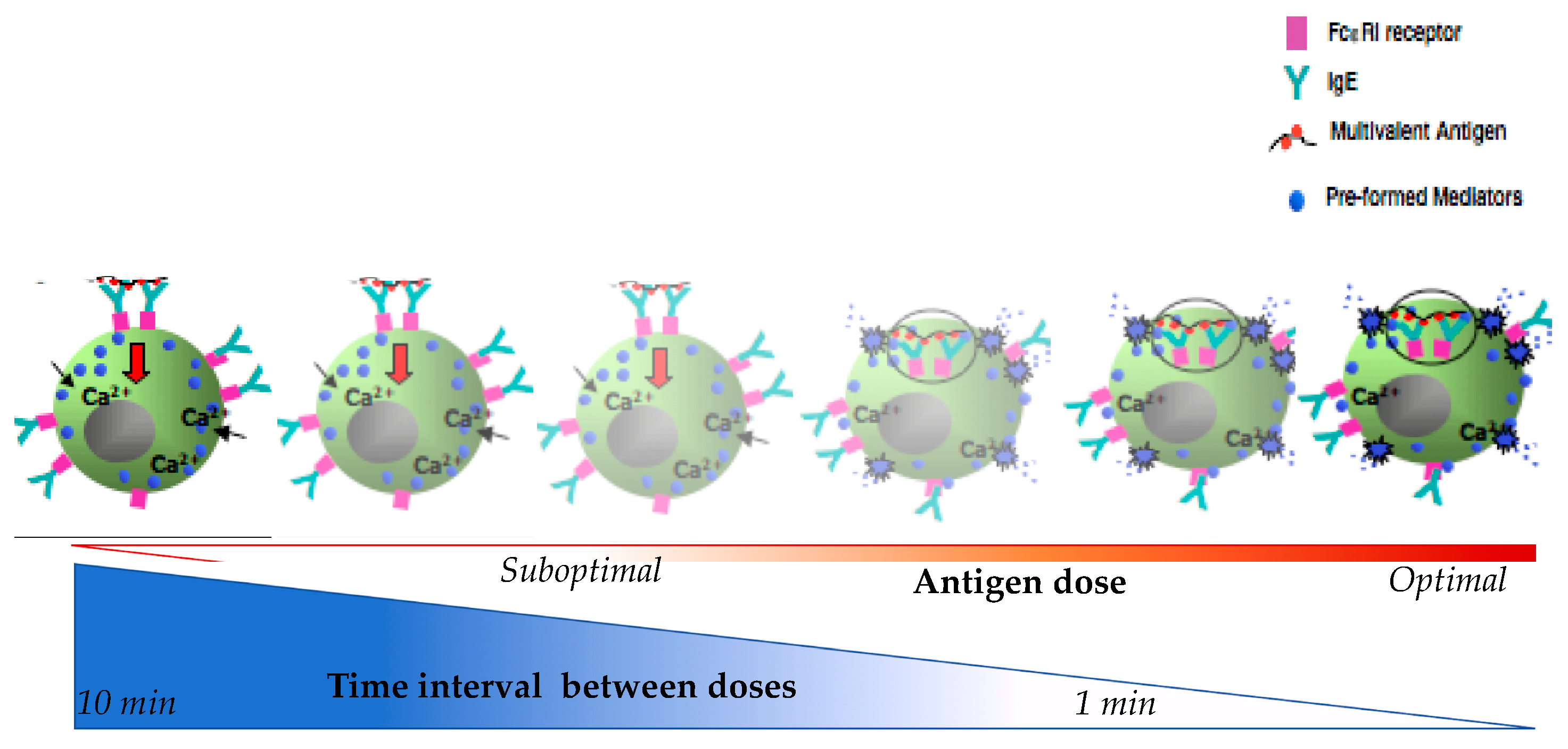
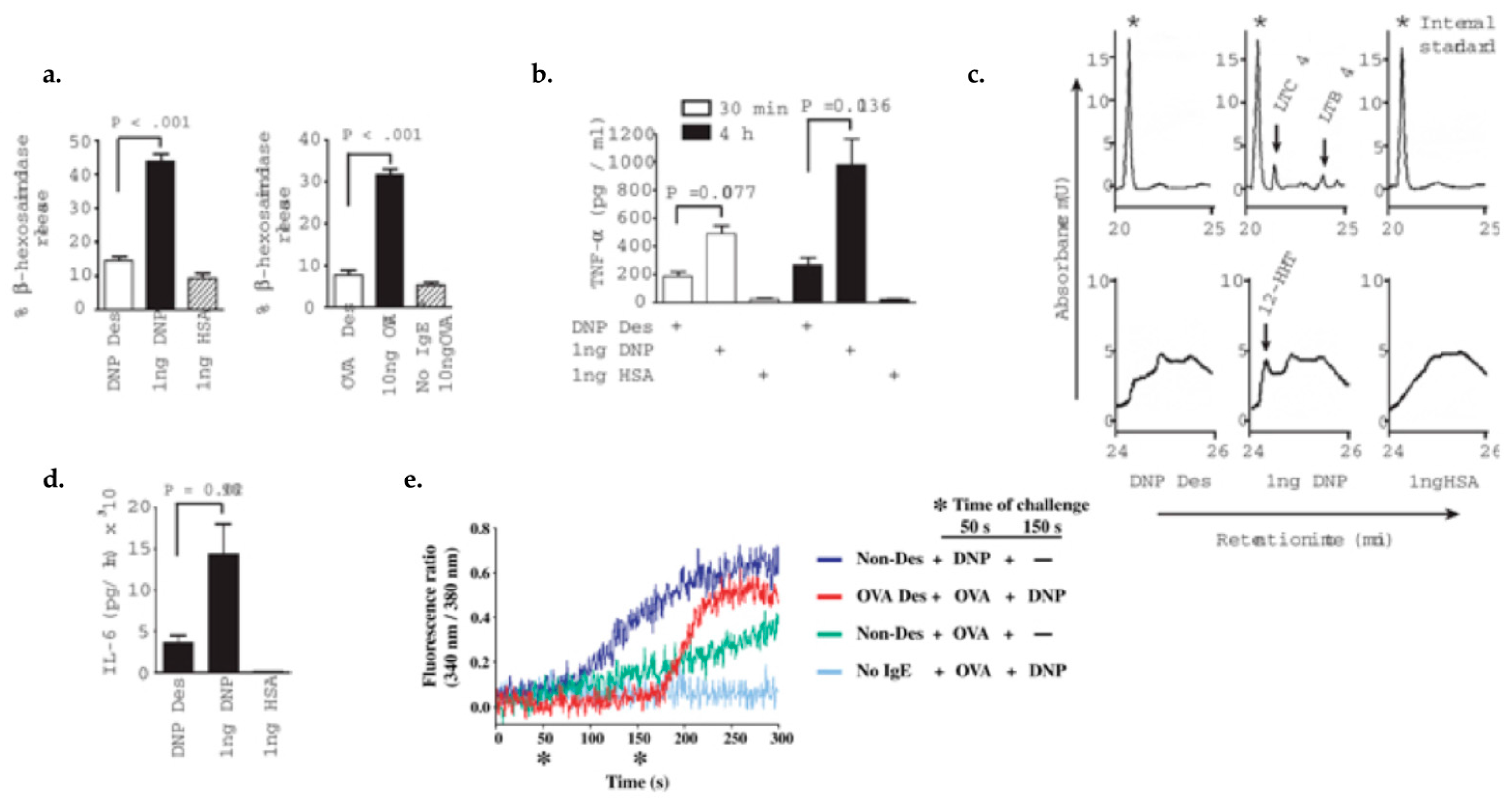
4.1. Characterizing Desensitization Mechanisms through In Vitro and In Vivo Models
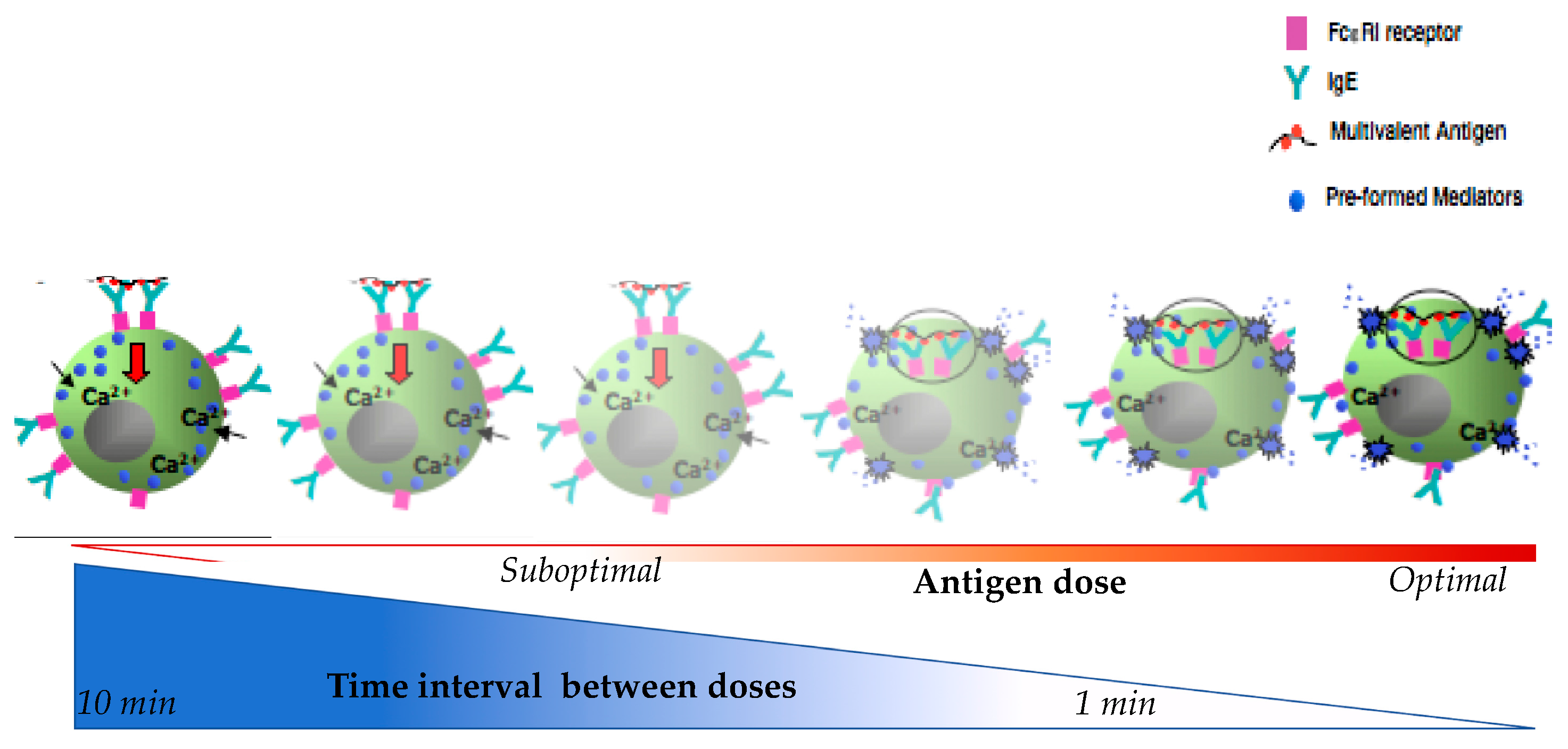
4.2. Proposed Mechanisms of Desensitization
4.2.1. Ag/IgE-FcεRI Complex Mobility
4.2.2. ITAM/ITIM Counter Regulation
4.2.3. Ca Channel Desensitization
4.2.4. Actin Remodeling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CPA | Carboxypeptidase A |
| DHR | Drug hypersensitivity reaction |
| DS | Desensitization |
| FcεRI | Fcε receptor I, high affinity Fc receptor for IgE |
| FcγRIIB | Fc receptors for IgG, type IIb |
| HSR | Hypersensitivity reaction |
| IgE | Immunoglobulin E |
| ITAM | Immune-receptor tyrosine-based activation motif |
| ITIM | Immunoreceptor tyrosine-based inhibitory motif |
| mBMMC | Mouse bone marrow-derived mast cell |
| MC | Mast cell |
| MrgprX2 | Mas-related G-protein coupled receptor member X2 |
| PAF | Platelet activating factor |
| PSA | Passive systemic anaphylaxis |
| SHIP | SH-2 containing inositol 5′ polyphosphatase |
| SHP-1 | SH-2 containing tyrosine phosphatase |
References
- Muraro, A.; Lemanske, R.F.; Castells, M.; Torres, M.J.; Khan, D.; Simon, H.-U.; Bindslev-Jensen, C.; Burks, W.; Poulsen, L.K.; Sampson, H.A.; et al. Precision medicine in allergic disease—Food allergy, drug allergy, and anaphylaxis—PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma and Immunology. Allergy 2017, 72, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Demoly, P.; Adkinson, N.F.; Brockow, K.; Castells, M.; Chiriac, A.M.; Greenberger, P.A.; Khan, D.A.; Lang, D.M.; Park, H.S.; Pichler, W.; et al. International Consensus on drug allergy. Allergy 2014, 69, 420–437. [Google Scholar] [CrossRef] [PubMed]
- Bonamichi-Santos, R.; Castells, M. Diagnoses and Management of Drug Hypersensitivity and Anaphylaxis in Cancer and Chronic Inflammatory Diseases: Reactions to Taxanes and Monoclonal Antibodies. Clin. Rev. Allergy Immunol. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.C.; Tennant, N.M.; Sloane, D.E.; Ida Hsu, F.; Barrett, N.A.; Hong, D.I.; Laidlaw, T.M.; Legere, H.J.; Nallamshetty, S.N.; Palis, R.I.; et al. Hypersensitivity reactions to chemotherapy: Outcomes and safety of rapid desensitization in 413 cases. J. Allergy Clin. Immunol. 2008, 22, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Legere, H.J.; Palis, R.I.; Bouza, T.R.; Uluer, A.Z.; Castells, M.C. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J. Cyst. Fibros. 2010, 8, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Sloane, D.; Govindarajulu, U.; Harrow-Mortelliti, J.; Barry, W.; Hsu, F.I.; Hong, D.; Laidlaw, T.; Palis, R.; Legere, H.; Bunyavanich, S.; et al. Safety, Costs, and Efficacy of Rapid Drug Desensitizations to Chemotherapy and Monoclonal Antibodies. J. Allergy Clin. Immunol. Pract. 2016, 4, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.J.; Romano, A.; Celik, G.; Demoly, P.; Khan, D.A.; Macy, E.; Park, M.; Blumenthal, K. Approach to the diagnosis of drug hypersensitivity reactions: Similarities and differences between Europe and North America. Clin. Transl. Allergy 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Coombs, R.R.A.; Gell, P.G.H. Clinical Aspects of Immunology; Philadelphia: Davis, CA, USA, 1963. [Google Scholar]
- Picard, M.; Pur, L.; Caiado, J.; Giavina-Bianchi, P.; Galvão, V.R.; Berlin, S.T.; Campos, S.M.; Matulonis, U.A.; Castells, M.C. Risk stratification and skin testing to guide re-exposure in taxane-induced hypersensitivity reactions. J. Allergy Clin. Immunol. 2016, 137, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Giavina-Bianchi, P.; Galvão, V.R.; Picard, M.; Caiado, J.; Castells, M.C. Basophil Activation Test Is a Relevant Biomarker of the Outcome of Rapid Desensitization in Platinum Compounds-Allergy. J. Allergy Clin. Immunol. Pract. 2016, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Galvão, V.R.; Phillips, E.; Giavina-Bianchi, P.; Castells, M.C. Carboplatin-allergic patients undergoing desensitization: Prevalence and impact of the BRCA 1/2 mutation. J. Allergy Clin. Immunol. Pract. 2016, 5, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.U.; Long, A.A.; Ling, M.; Wilson, M.T.; Hesterberg, P.; Wong, J.T.; Banerji, A. A Protocol for risk stratification of patients with carboplatin-induced hypersensitivity reactions. J. Allergy Clin. Immunol. 2012, 129, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Leckband, S.G.; Kelsoe, J.R.; Dunnenberger, H.M.; George, A.L., Jr.; Tran, E.; Berger, R.; Müller, D.J.; Whirl-Carrillo, M.; Caudle, K.E.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Carbamazepine Dosing. Clin. Pharmacol. Ther. 2013, 94, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Mallal, S.; Phillips, E.; Carosi, G.; Molina, J.M.; Workman, C.; Tomazic, J.; Jägel-Guedes, E.; Rugina, S.; Kozyrev, O.; Cid, J.F.; et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. N. Engl. J. Med. 2008, 358, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Rajan, T.V. The Gell–Coombs classification of hypersensitivity reactions: A re-interpretation. Trends Immunol. 2003, 24, 376–379. [Google Scholar]
- Brown, S.G.A. Clinical features and severity grading of anaphylaxis. J. Allergy Clin. Immunol. 2004, 114, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Ring, J.; Messmer, K. Incidence and severity of anaphylactoid reactions to colloid volume substitutes. Lancet 1977, 1, 466–469. [Google Scholar] [CrossRef]
- Khan, D.A.; Solensky, R. Drug allergy. J. Allergy Clin. Immunol. 2010, 125, S126–S137. [Google Scholar] [CrossRef] [PubMed]
- Bankova, L.G.; Lai, J.; Yoshimoto, E.; Boyce, J.A.; Austen, K.F.; Kanaoka, Y.; Barrett, N.A. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc. Natl. Acad. Sci. USA 2016, 113, 6242–6247. [Google Scholar] [CrossRef] [PubMed]
- Mastalerz, L.; Setkowicz, M.; Sanak, M.; Szczeklik, A. Hypersensitivity to aspirin: Common eicosanoid alterations in urticaria and asthma. J. Allergy Clin. Immunol. 2004, 113, 771–775. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast cell specific receptor crucial for pseudoallergic drug reaction. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Pichler, W.J.; Hausmann, O. Classification of Drug Hypersensitivity into Allergic, p-i, and Pseudo-Allergic Forms. Int. Arch. Allergy Immunol. 2016, 171, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J.; Bouza, T.R.; Hsu, F.I.; Sloane, D.E.; Castells, M.C. Hypersensitivity reactions to mAbs: 105 desensitizations in 23 patients, from evaluation to treatment. J. Allergy Clin. Immunol. 2009, 124, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Lehloenya, R.J.; Todd, G.; Wallace, J.; Ngwanya, M.R.; Muloiwa, R.; Dheda, K. Diagnostic patch testing following tuberculosis-associated cutaneous adverse drug events induces systemic reactions in HIV-infected persons. Br. J. Dermatol. 2016, 175, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Patil, S.U.; Long, A.A.; Banerji, A. Risk-stratification protocol for carboplatin and oxaliplatin hypersensitivity: Repeat skin testing to identify drug allergy. Ann. Allergy Asthma Immunol. 2015, 115, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.; Ling, M.; Patil, S.; Banerji, A.; Long, A. Oxaliplatin hypersensitivity: Evaluation, implications of skin testing, and desensitization. J. Allergy Clin. Immunol. Pract. 2014, 2, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.; Park, M.A. Penicillin skin testing in the evaluation and management of penicillin allergy. Ann. Allergy Asthma Immunol. 2011, 106, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, K.G.; Wickner, P.G.; Hurwitz, S.; Pricco, N.; Nee, A.E.; Laskowski, K.; Shenoy, E.S.; Walensky, R.P. Tackling inpatient penicillin allergies: Assessing tools for antimicrobial stewardship. J. Allergy Clin. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Caiado, J.; Venemalm, L.; Pereira-Santos, M.C.; Costa, L.; Barbosa, M.P.; Castells, M. Carboplatin-, Oxaliplatin-, and Cisplatin-specific IgE: Cross-reactivity and Value in the Diagnosis of Carboplatin and Oxaliplatin Allergy. J. Allergy Clin. Immunol. Pract. 2013, 1, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, K.M.; Laidlaw, T.M. Update on the Management of Aspirin-Exacerbated Respiratory Disease. Allergy Asthma Immunol. Res. 2016, 8, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Sala-Cunill, A.; Cardona, V. Biomarkers of anaphylaxis, beyond tryptase. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Gueant, J.-L.; Romano, A.; Cornejo-Garcia, J.-A.; Oussalah, A.; Chery, C.; Blanca-Lopez, N.; Gueant-Rodriguez, R.-M.; Gaeta, F.; Rouyer, P.; Josse, T.; et al. HLA-DRA variants predict penicillin allergy in genome-wide fine-mapping genotyping. J. Allergy Clin. Immunol. 2015, 135, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Luque, I.; Leyva, L.; Jose Torres, M.; Rosal, M.; Mayorga, C.; Segura, J.M.; Blanca, M.; Juarez, C. In vitro T-cell responses to β-lactam drugs in immediate and nonimmediate allergic reactions. Allergy 2001, 56, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-H.; Hung, S.-I.; Yang, J.-Y.; Su, S.-C.; Huang, S.-P.; Wei, C.-Y.; Chin, S.-W.; Chiou, C.-C.; Chu, S.-C.; Ho, H.-C.; et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat. Med. 2008, 14, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.J.; Yecies, L.D.; Shatz, G.S.; Parker, C.W.; James Wedner, H. Desensitization of patients allergic to penicillin using orally administered β-lactam antibiotics. J. Allergy Clin. Immunol. 1982, 69, 275–282. [Google Scholar] [CrossRef]
- Wendel, G.D., Jr.; Stark, B.J.; Jamison, R.B.; Molina, R.D.S.T. Penicillin allergy and desensitization in serious infections during pregnancy. N. Engl. J. Med. 1985, 312, 1229–1233. [Google Scholar] [CrossRef] [PubMed]
- Borish, L.; Tamir, R.; Rosenwasser, L.J. Itravenous desensitization to β-lactam antibiotics. J. Allergy Clin. Immunol. 1987, 80, 314–319. [Google Scholar] [CrossRef]
- Castells, M. Desensitization for drug allergy. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Foer, D.; Buchheit, K.M.; Gargiulo, A.R.; Lynch, D.M.; Castells, M.; Wickner, P.G. Progestogen Hypersensitivity in 24 Cases: Diagnosis, Management, and Proposed Renaming and Classification. J. Allergy Clin. Immunol. Pract. 2016, 4, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Daulat, S.; Solensky, R.; Earl, H.S.; Casey, W.; Gruchalla, R.S. Safety of cephalosporin administration to patients with histories of penicillin allergy. J. Allergy Clin. Immunol. 2004, 113, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Castells Guitart, M.C. Rapid Drug Desensitization for Hypersensitivity Reactions to Chemotherapy and Monoclonal Antibodies in the 21st Century. J. Investig. Allergol. Clin. Immunol. 2014, 24, 72–79. [Google Scholar] [PubMed]
- Macy, E.; Romano, A.; Khan, D. Practical Management of Antibiotic Hypersensitivity in 2017. J. Allergy Clin. Immunol. Pract. 2016, 5, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.H.; Lee, J.-M.; Noonan, A.M.; Annunziata, C.M.; Minasian, L.; Houston, N.; Hays, J.L.; Kohn, E.C. Deleterious BRCA1/2 mutation is an independent risk factor for carboplatin hypersensitivity reactions. Br. J. Cancer. 2013, 109, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hesterberg, P.E.; Banerji, A.; Oren, E.; Penson, R.T.; Krasner, C.N.; Seiden, M.V.; Wong, J.T. Risk stratification for desensitization of patients with carboplatin hypersensitivity: Clinical presentation and management. J. Allergy Clin. Immunol. 2009, 123, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Gladys Ang, W.X.; Church, A.M.; Kulis, M.; Choi, H.W.; Wesley Burks, A.; Abraham, S.N. Mast Cell Desensitization Inhibits Calcium Flux and Aberrantly Remodels Actin. J. Clin. Investig. 2016, 126, 4103–4118. [Google Scholar]
- Suzuki, R.; Scheffel, J.; Rivera, J. New Insights on the Signaling and Function of the High-Affinity Receptor for IgE. In IgE Antibodies: Generation and Function; Lafaille, J.J., de Lafaille, M.A., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 63–90. [Google Scholar]
- Kalesnikoff, J.; Galli, S.J. New developments in mast cell biology. Nat. Immunol. 2008, 9, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Kopeć, A.; Panaszek, B.; Fal, A.M. Intracellular signaling pathways in IgE-dependent mast cell activation. Arch. Immunol. Ther. Exp. 2006, 54, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Sibilano, R.; Frossi, B.; Pucillo, C.E. Mast cell activation: A complex interplay of positive and negative signaling pathways. Eur. J. Immunol. 2014, 44, 2558–2566. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.C. Analysis of Mitogen-Activated Protein Kinase Activation. In Mast Cells: Methods and Protocols; Krishnaswamy, G., Chi, D.S., Eds.; Humana Press: Totowa, NJ, USA, 2005; pp. 151–163. [Google Scholar]
- Katz, H.R. Inhibitory receptors and allergy. Curr. Opin. Immunol. 2002, 14, 698–704. [Google Scholar] [CrossRef]
- Del Carmen Sancho-Serra, M.; Simarro, M.; Castells, M. Rapid IgE desensitization is antigen specific and impairs early and late mast cell responses targeting FccεRI internalization. Eur. J. Immunol. 2011, 41, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Rios, E.J.; Tsai, M.; Kalesnikoff, J.; Galli, S.J. Rapid desensitization induces internalization of antigen-specific IgE on mouse mast cells. J. Allergy Clin. Immunol. 2017, 132, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.R.; Shah, N.; Castells, M. Antigen-IgE desensitization in signal transducer and activator of transcription 6-deficient mast cells by suboptimal doses of antigen. Ann. Allergy Asthma Immunol. 2005, 94, 575–580. [Google Scholar] [CrossRef]
- MacGlashan, J. Subthreshold desensitization of human basophils re-capitulates the loss of SYK and FccεRI expression characterized by other methods of desensitization. Clin. Exp. Allergy 2012, 42, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- MacGlashan, D.; Miura, K. Loss of syk kinase during IgE-mediated stimulation of human basophils. J. Allergy Clin. Immunol. 2004, 114, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Hishinuma, S.; Nozawa, H.; Akatsu, C.; Shoji, M. C-terminal of human histamine H1 receptors regulates their agonist-induced clathrin-mediated internalization and G-protein signaling. J. Neurochem. 2016, 139, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.; Monczor, F.; Baldi, A.; Davio, C.; Shayo, C. Histamine H2 receptor trafficking: Role of arrestin, dynamin, and clathrin in histamine H2 receptor internalization. Mol. Pharmacol. 2008, 74, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Agier, J.; Rozalska, S.; Wodz, K.; Brzezinska-Blaszczyk, E. Leukotriene receptor expression in mast cells is affected by their agonists. Cell Immunol. 2017, 317, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-W.; Matulonis, U.A.; Castells, M.C. Carboplatin hypersensitivity: A 6-h 12-step protocol effective in 35 desensitizations in patients with gynecological malignancies and mast cell/IgE-mediated reactions. Gynecol. Oncol. 2017, 95, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Shalit, M.; Levi-Schaffer, F. Challenge of mast cells with increasing amounts of antigen induces desensitization. Clin. Exp. Allergy 1995, 25, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Barua, D.; Cutler, P.; Lidke, D.S.; Espinoza, F.A.; Pehlke, C.; Grattan, R.; Kawakami, Y.; Tung, C.S.; Bradbury, A.R.M.; et al. Optimal aggregation of FccεRI with a structurally defined trivalent ligand overrides negative regulation driven by phosphatases. ACS Chem. Biol. 2014, 9, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.L.; Pfeiffer, J.R.; Martinez, A.M.; Davis, R.W.; Kawakami, T.; Oliver, J.M.; Wilson, B.S.; Lidke, D.S. Small, Mobile FcεRI Receptor Aggregates Are Signaling Competent. Immunity 2009. [Google Scholar] [CrossRef] [PubMed]
- Katz, H.R. Inhibition of anaphylactic inflammation by the gp49B1 receptor on mast cells. Mol. Immunol. 2002, 38, 1301–1305. [Google Scholar] [CrossRef]
- Lu-Kuo, J.M.; Joyal, D.M.; Austen, K.F.; Katz, H.R. gp49b1 Inhibits IgE-initiated Mast Cell Activation through Both Immunoreceptor Tyrosine-based Inhibitory Motifs, Recruitment ofsrc Homology 2 Domain-containing Phosphatase-1, and Suppression of Early and Late Calcium Mobilization. J. Biol. Chem. 1999, 274, 5791–5796. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Medication: Penicillin | |||
|---|---|---|---|
| Stock Solution | Voulum Per Stock Solution (mL) | Concentration (units/mL) | Total Dose Per Stock Solution (units) |
| 1 | 30 | 1000 | 30,000 |
| 2 | 30 | 10,000 | 300,000 |
| 3 | 30 | 80,000 | 2,400,000 |
| Target dose (units) = 1,296,700 | |||
| Step | Stock Solution | Time (min) | Cumulative Time (min) | Voulum Given Per Step (mL) | Dose Given Per Step (units) | Cumulative Dose (units) | Fold Increase Per Step |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 15 | 15 | 0.1 | 100 | 100 | 0 |
| 2 | 1 | 15 | 30 | 0.2 | 200 | 300 | 2 |
| 3 | 1 | 15 | 45 | 0.4 | 400 | 700 | 2 |
| 4 | 1 | 15 | 60 | 0.8 | 800 | 1500 | 2 |
| 5 | 1 | 15 | 75 | 1.6 | 1600 | 3100 | 2 |
| 6 | 1 | 15 | 90 | 3.2 | 3200 | 6300 | 2 |
| 7 | 1 | 15 | 105 | 6.4 | 6400 | 12,700 | 2 |
| 8 | 2 | 15 | 120 | 1.2 | 12,000 | 24,700 | 1.875 |
| 9 | 2 | 15 | 135 | 2.4 | 24,000 | 48,700 | 2 |
| 10 | 2 | 15 | 150 | 4.8 | 48,000 | 96,700 | 2 |
| 11 | 3 | 15 | 165 | 1 | 80,000 | 176,700 | 1.67 |
| 12 | 3 | 15 | 180 | 2 | 160,000 | 336,700 | 2 |
| 13 | 3 | 15 | 195 | 4 | 320,000 | 656,700 | 2 |
| 14 | 3 | 15 | 210 | 8 | 640,000 | 1,296,700 | 2 |
| Total time (h) = 3.5 | |||||||
| Name of Medication: Obinutuzumab | |
|---|---|
| Target dose (mg) | 750 |
| Standard volume per bag (mL) | 250 |
| Final rate of infusion (mL/h) | 80 |
| Calculated target concentration (mg/mL) | 3 |
| Standard time of infusion (min) | 187.5 |
| Bag | Volumen Per Bag (mL) | Concentration (mg/mL) | Total Dose Per Bag (mg) | Amount of Bag Infused (mL) |
|---|---|---|---|---|
| 1 | 250 | 0.003 | 0.75 | 4.69 |
| 2 | 250 | 0.03 | 7.5 | 9.38 |
| 3 | 250 | 0.3 | 75 | 18.75 |
| 4 | 250 | 2.97632 | 744.08 | 250 |
| Step | Bag | Rate (mL/h) | Time (min) | Cumulative Time (min) | Volume Infused Per step (mL) | Dose Administrated with This Step (mg) | Cumulative Dose (mg) | Fold Increase Per Step |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1.3 | 15 | 15 | 0.31 | 0.0009 | 0.0009 | 0 |
| 2 | 1 | 2.5 | 15 | 30 | 0.63 | 0.0019 | 0.0028 | 2 |
| 3 | 1 | 5 | 15 | 45 | 1.25 | 0.0038 | 0.0066 | 2 |
| 4 | 1 | 10 | 15 | 60 | 2.5 | 0.0075 | 0.0141 | 2 |
| 5 | 2 | 2.5 | 15 | 75 | 0.63 | 0.0188 | 0.0328 | 2.5 |
| 6 | 2 | 5 | 15 | 90 | 1.25 | 0.0375 | 0.0703 | 2 |
| 7 | 2 | 10 | 15 | 105 | 2.5 | 0.075 | 0.1453 | 2 |
| 8 | 2 | 20 | 15 | 120 | 5 | 0.15 | 0.2953 | 2 |
| 9 | 3 | 5 | 15 | 135 | 1.25 | 0.375 | 0.6703 | 2.5 |
| 10 | 3 | 10 | 15 | 150 | 2.5 | 0.75 | 1.4203 | 2 |
| 11 | 3 | 20 | 15 | 165 | 5 | 1.5 | 2.9203 | 2 |
| 12 | 3 | 40 | 15 | 180 | 10 | 3 | 5.9203 | 2 |
| 13 | 4 | 10 | 15 | 195 | 2.5 | 7.4408 | 13.3611 | 2.48 |
| 14 | 4 | 20 | 15 | 210 | 5 | 14.8816 | 28.2427 | 2 |
| 15 | 4 | 40 | 15 | 225 | 10 | 29.7632 | 58.0059 | 2 |
| 16 | 4 | 80 | 174.375 | 399.38 | 232.5 | 691.9941 | 750 | 2 |
| Total time (h) = 6.66 | ||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De las Vecillas Sánchez, L.; Alenazy, L.A.; Garcia-Neuer, M.; Castells, M.C. Drug Hypersensitivity and Desensitizations: Mechanisms and New Approaches. Int. J. Mol. Sci. 2017, 18, 1316. https://doi.org/10.3390/ijms18061316
De las Vecillas Sánchez L, Alenazy LA, Garcia-Neuer M, Castells MC. Drug Hypersensitivity and Desensitizations: Mechanisms and New Approaches. International Journal of Molecular Sciences. 2017; 18(6):1316. https://doi.org/10.3390/ijms18061316
Chicago/Turabian StyleDe las Vecillas Sánchez, Leticia, Leila A. Alenazy, Marlene Garcia-Neuer, and Mariana C. Castells. 2017. "Drug Hypersensitivity and Desensitizations: Mechanisms and New Approaches" International Journal of Molecular Sciences 18, no. 6: 1316. https://doi.org/10.3390/ijms18061316
APA StyleDe las Vecillas Sánchez, L., Alenazy, L. A., Garcia-Neuer, M., & Castells, M. C. (2017). Drug Hypersensitivity and Desensitizations: Mechanisms and New Approaches. International Journal of Molecular Sciences, 18(6), 1316. https://doi.org/10.3390/ijms18061316