Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes

 ,
,
Abstract
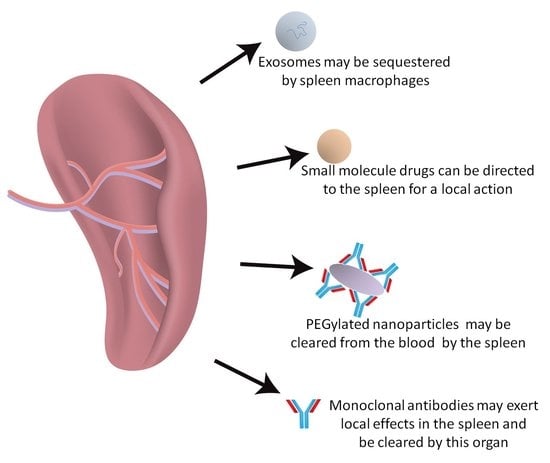
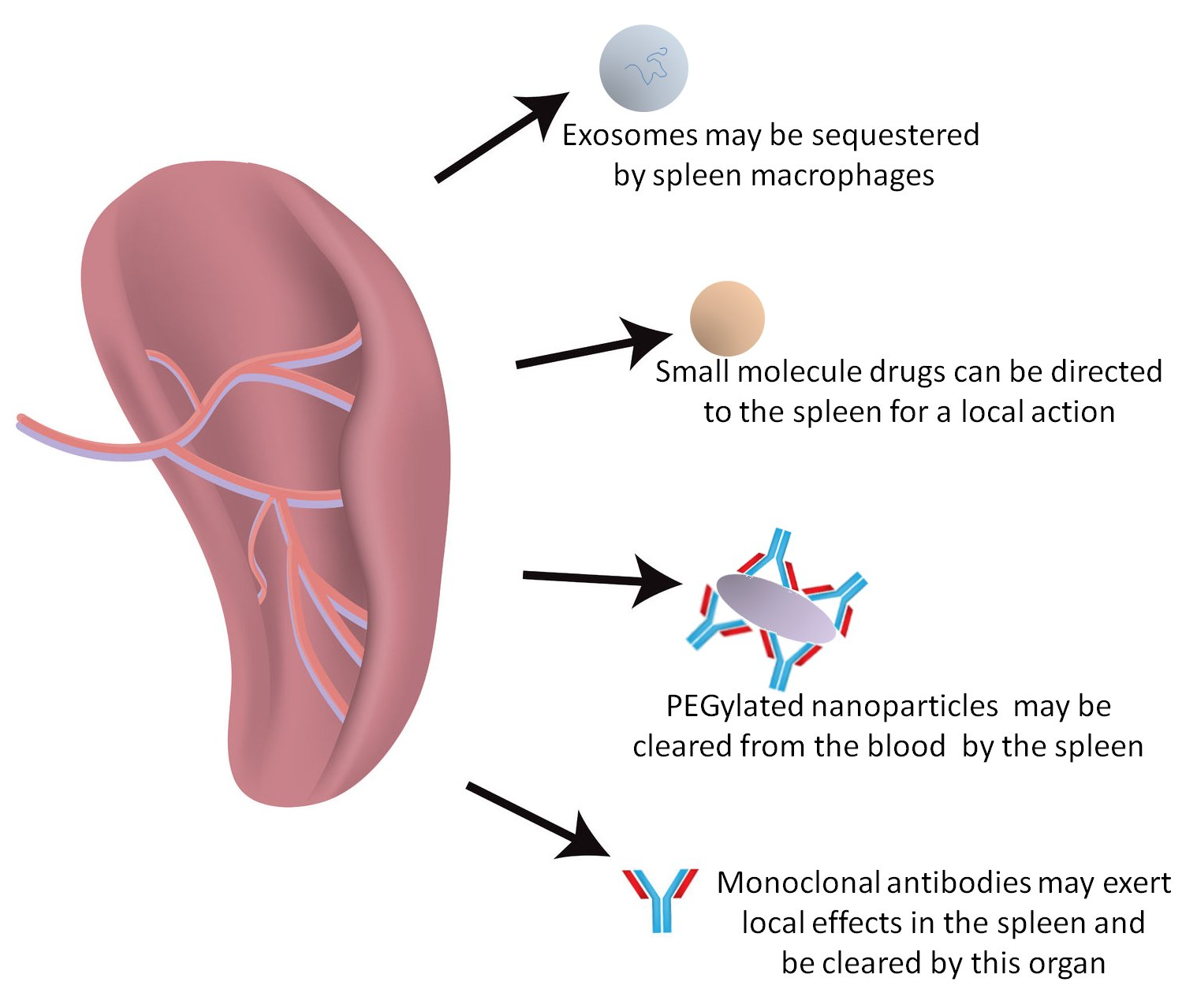
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
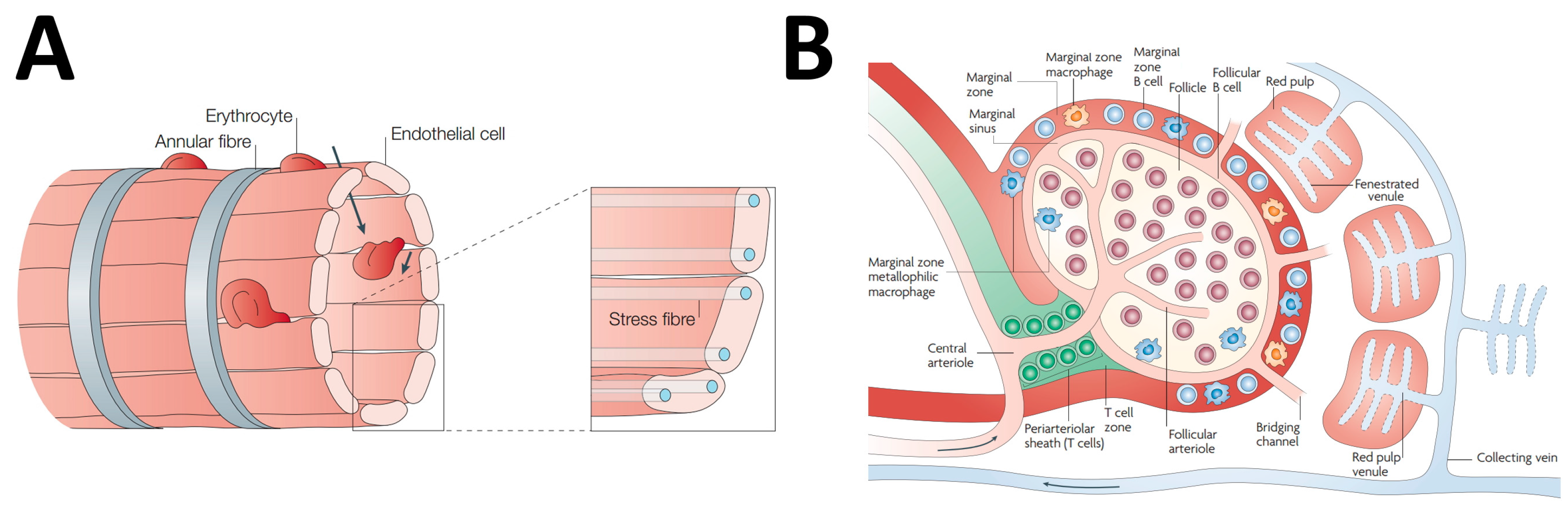
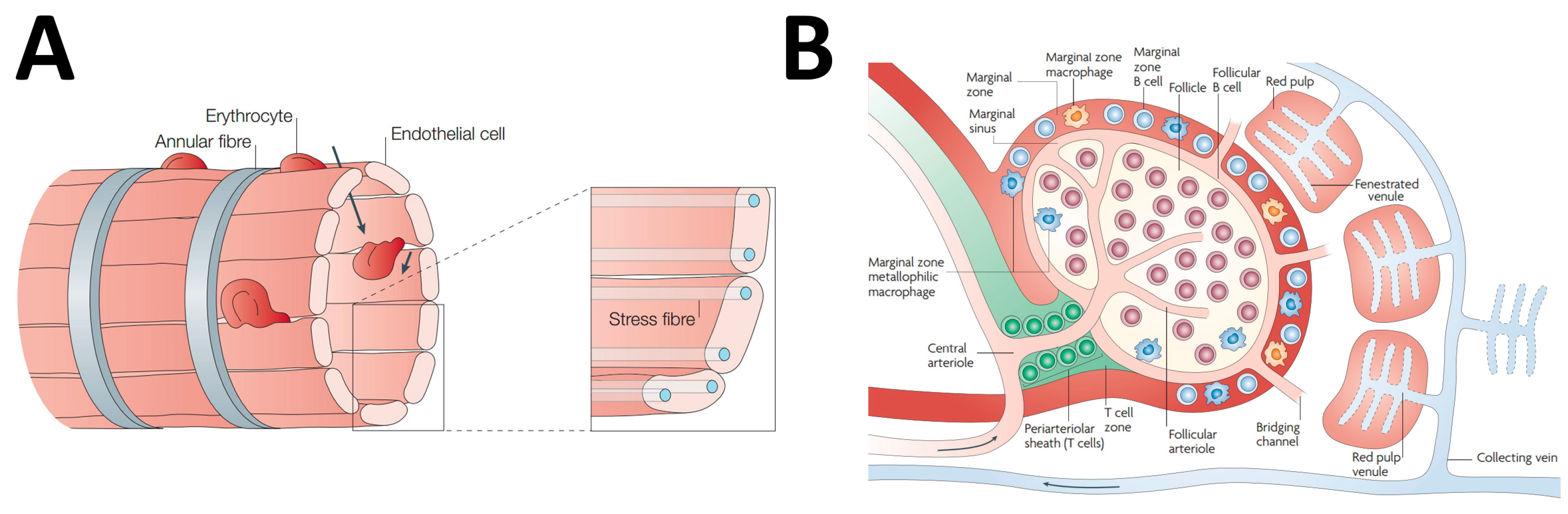
2. Spleen Microanatomy: The Pharmacologist Point of View
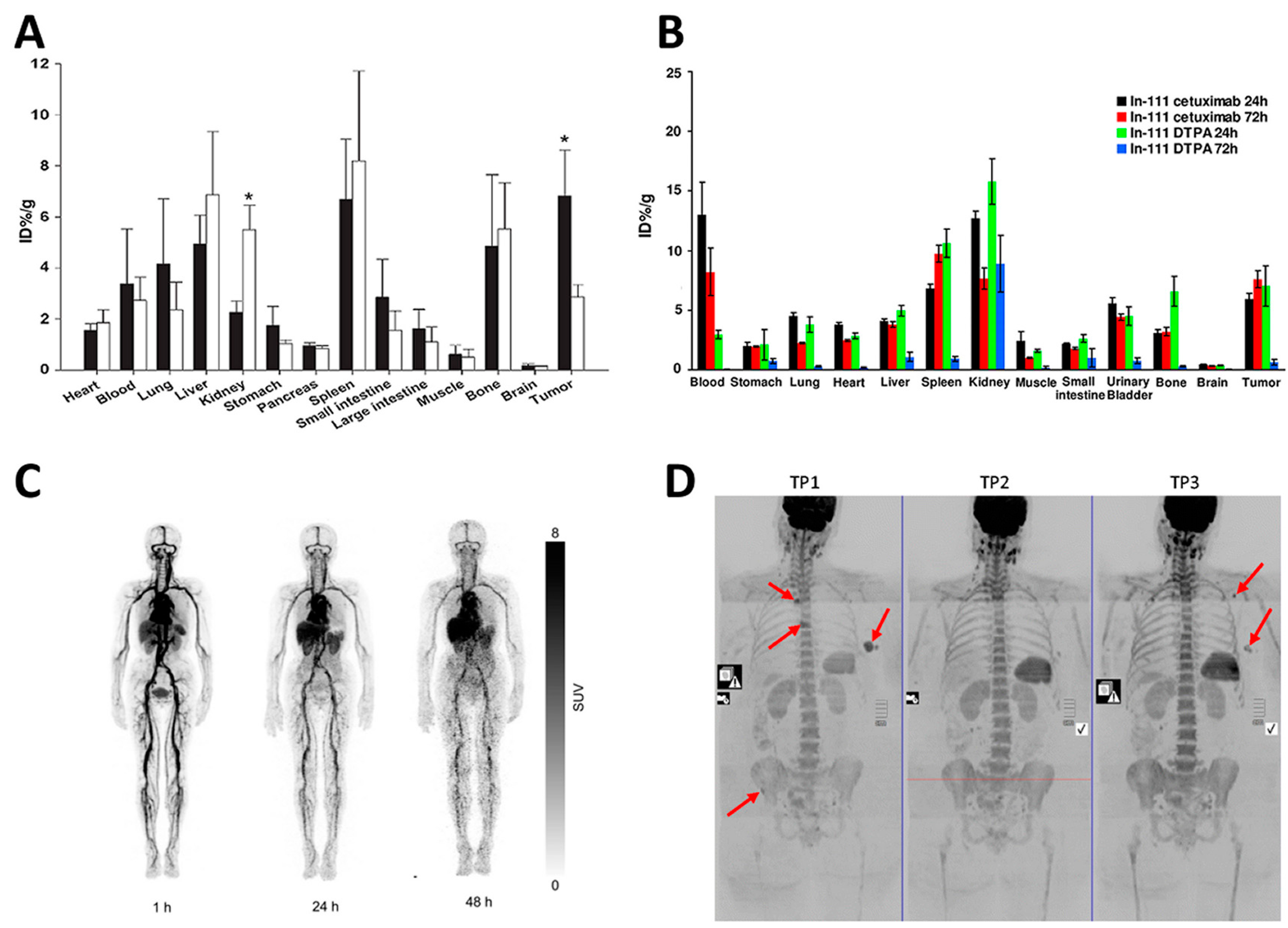
3. Role of the Spleen in the Disposition of Monoclonal Antibodies
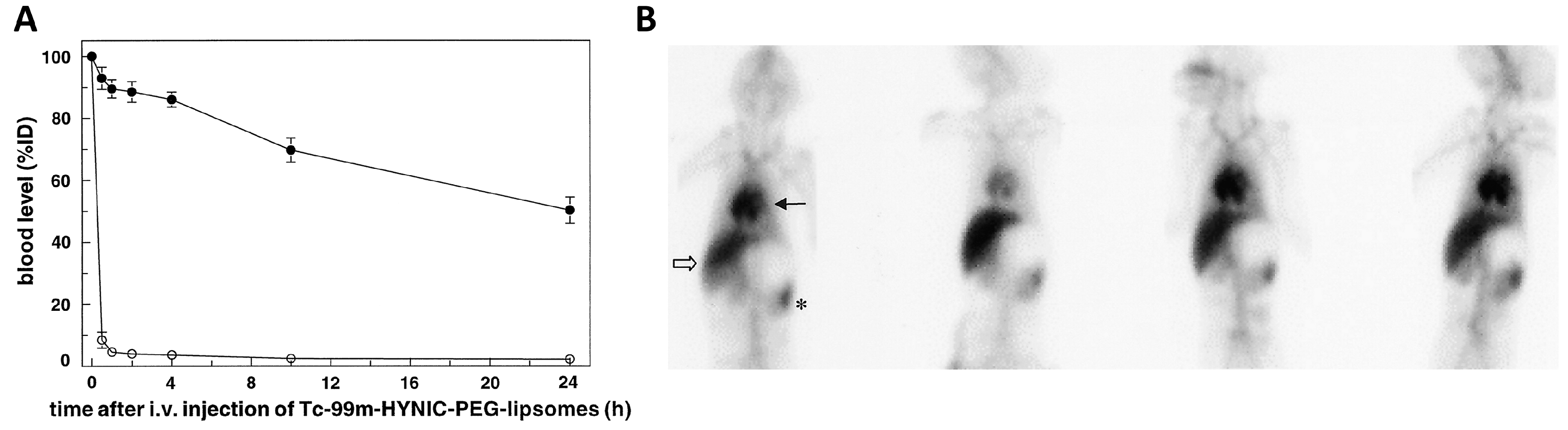
4. Role of the Spleen in the Pharmacokinetics of Nanoparticle Drugs
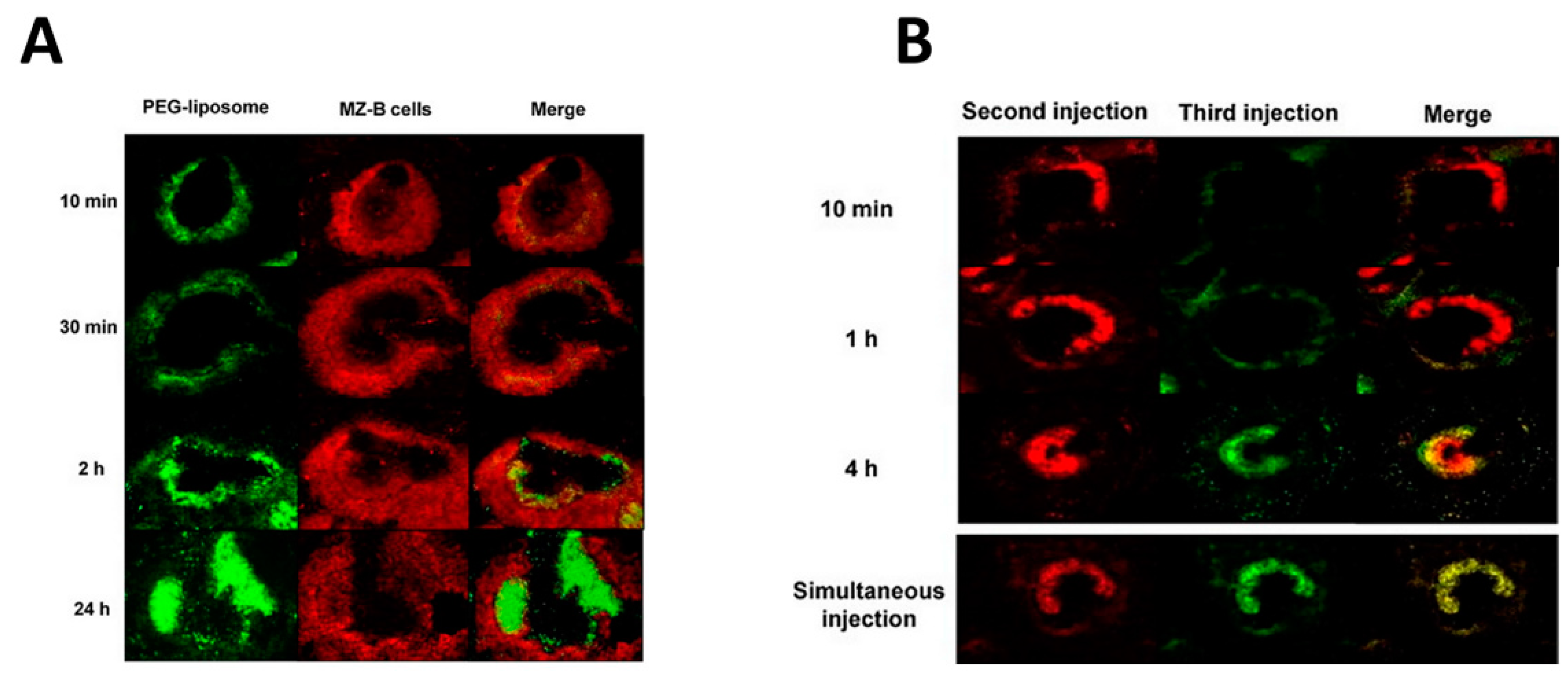
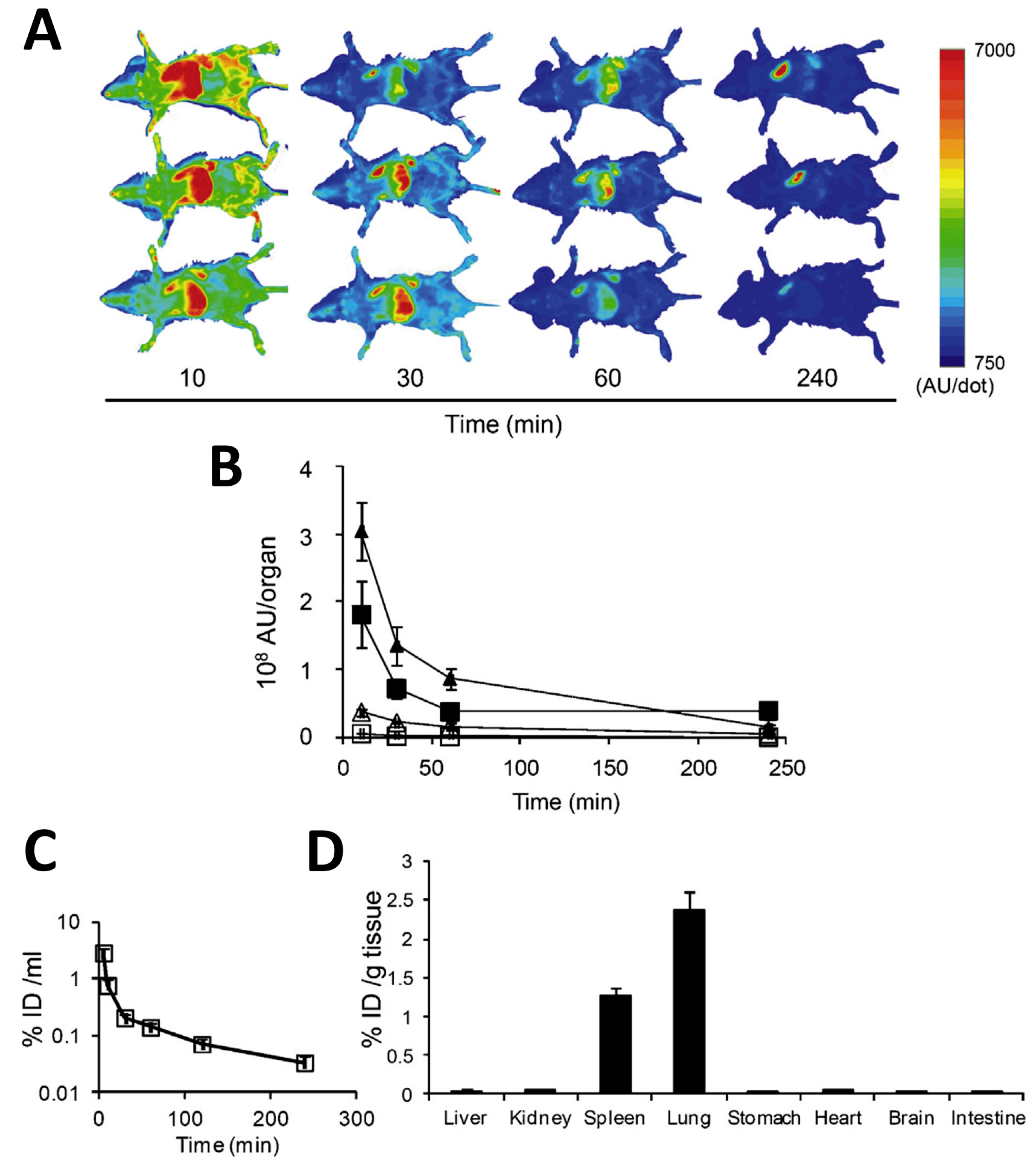
5. Role of the Spleen in the Pharmacokinetics of Exosomes
6. General Conclusions
Conflicts of Interest
References
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Bergman, R.A.; Heidger, P.M., Jr.; Scott-Conner, C.E.H. The Anatomy of the Spleen. In The Complete Spleen, 2nd ed.; Springer Science + Business Media: New York, NY, USA, 2002; pp. 3–10. [Google Scholar]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Groom, A.C.; MacDonald, I.C.; Schmidt, E.E. Splenic Microcirculatory Blood Flow and Function with Respect to Red Blood Cells. In The Complete Spleen, 2nd ed.; Springer Science + Business Media: New York, NY, USA, 2002; pp. 23–50. [Google Scholar]
- Steiniger, B.S. Human spleen microanatomy: Why mice do not suffice. Immunology 2015, 145, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Tablin, F.; Chamberlain, J.K.; Weiss, L. The Microanatomy of the Mammalian Spleen: Mechanisms of Splenic Clearance. In The Complete Spleen, 2nd ed.; Springer Science + Business Media: New York, NY, USA, 2002; pp. 11–22. [Google Scholar]
- Steiniger, B.; Bette, M.; Schwarzbach, H. The open microcirculation in human spleens: A three-dimensional approach. J. Histochem. Cytochem. 2011, 59, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Pivkin, I.V.; Peng, Z.; Karniadakis, G.E.; Buffet, P.A.; Dao, M.; Suresh, S. Biomechanics of red blood cells in human spleen and consequences for physiology and disease. Proc. Natl. Acad. Sci. USA 2016, 113, 7804–7809. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T. A scanning electron microscope study of the human spleen. Arch. Histol. Jpn. 1974, 3, 187–216. [Google Scholar] [CrossRef]
- Fujita, T.; Kashimura, M.; Adachi, K. Scanning electron microscopy and terminal circulation. Experientia 1985, 41, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, S.; Koga, D.; Kanda, T.; Ushiki, T. Three-dimensional reconstruction of serial sections for analysis of the microvasculature of the white pulp and the marginal zone in the human spleen. Biomed. Res. 2015, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, T.K.; Brevé, J.J.; Damoiseaux, J.G.; Döpp, E.A.; Kelm, S.; Crocker, P.R.; Dijkstra, C.D.; Kraal, G. Sialoadhesin on macrophages: Its identification as a lymphocyte adhesion molecule. J. Exp. Med. 1992, 176, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Van Krieken, J.H.; te Velde, J. Immunohistology of the human spleen: An inventory of the localization of lymphocyte subpopulations. Histopathology 1986, 10, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Steiniger, B.; Barth, P.; Hellinger, A. The perifollicular and marginal zones of the human splenic white pulp: Do fibroblasts guide lymphocyte immigration? Am. J. Pathol. 2001, 159, 501–512. [Google Scholar] [CrossRef]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009, 9, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Huchzermeyer, H.; Schmitz-Feuerhake, I.; Reblin, T. Determination of splenic blood flow by inhalation of radioactive rare gases. Eur. J. Clin. Investig. 1977, 7, 345–349. [Google Scholar] [CrossRef]
- Manoharan, A.; Gill, R.W.; Griffiths, K.A. Splenic blood flow measurements by Doppler ultrasound. Cardiovasc. Res. 1987, 21, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Oguro, A.; Taniguchi, H.; Koyama, H.; Tanaka, H.; Miyata, K.; Takeuchi, K.; Inaba, T.; Nakahashi, H.; Takahashi, T. Quantification of human splenic blood flow (quantitative measurement of splenic blood flow with H2(15)O and a dynamic state method: 1). Ann. Nucl. Med. 1993, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.M.; Klonizakis, I.; Lavender, J.P. Use of 111Indium-labelled platelets to measure spleen function. Br. J. Haematol. 1980, 46, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Taniguch, H.; Kunishima, S.; Koh, T.; Oguro, A.; Yamagishi, H. Reproducibility of repeated human regional splenic blood flow measurements using [15O] water and positron emission tomography. Nucl. Med. Commun. 2001, 22, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Crean, P.A.; Pratt, T.; Davies, G.J.; Myers, M.; Lavender, P.; Maseri, A. The fractional distribution of the cardiac output in man using microspheres labelled with technetium 99m. Br. J. Radiol. 1986, 59, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.E.; MacDonald, I.C.; Groom, A.C. Comparative aspects of splenic microcirculatory pathways in mammals: The region bordering the white pulp. Scanning Microsc. 1993, 7, 613–628. [Google Scholar] [PubMed]
- Harms, G.; Hardonk, M.J.; Timens, W. In vitro complement-dependent binding and in vivo kinetics of pneumococcal polysaccharide TI-2 antigens in the rat spleen marginal zone and follicle. Infect. Immun. 1996, 64, 4220–4225. [Google Scholar] [PubMed]
- Semaeva, E.; Tenstad, O.; Skavland, J.; Enger, M.; Iversen, P.O.; Gjertsen, B.T.; Wiig, H. Access to the spleen microenvironment through lymph shows local cytokine production, increased cell flux, and altered signaling of immune cells during lipopolysaccharide-induced acute inflammation. J. Immunol. 2010, 184, 4547–4556. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.; Rosania, G.R. Macrophages sequester clofazimine in an intracellular liquid crystal-like supramolecular organization. PLoS ONE 2012, 7, e47494. [Google Scholar] [CrossRef] [PubMed]
- Conklin, L.S.; Cuffari, C.; Okazaki, T.; Miao, Y.; Saatian, B.; Chen, T.E.; Tse, M.; Brant, S.R.; Li, X. 6-Mercaptopurine transport in human lymphocytes: Correlation with drug-induced cytotoxicity. J. Dig. Dis. 2012, 13, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Dorian, M.; Grellet, J.; Saux, M.C. Uptake of quinolones by in vitro human monocyte derived macrophages. J. Pharm. Pharmacol. 2001, 53, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.; Boffito, M.; Wildfire, A.; Hill, A.; Back, D.; Khoo, S.; Nelson, M.; Moyle, G.; Gazzard, B.; Pozniak, A. Intracellular and plasma pharmacokinetics of saquinavir-ritonavir, administered at 1600/100 milligrams once daily in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 2004, 48, 2388–2393. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Heremans, M.F.; Caceres, N.E.; Mingeot-Leclercq, M.P.; Tulkens, P.M.; Van Bambeke, F. Cellular accumulation and activity of quinolones in ciprofloxacin-resistant J774 macrophages. Antimicrob. Agents Chemother. 2006, 50, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Le Vee, M.; Jouan, E.; Parmentier, Y.; Fardel, O. Drug transporter expression in human macrophages. Fundam. Clin. Pharmacol. 2011, 25, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Hodges, V.M.; Molloy, G.Y.; Wickramasinghe, S.N. Demonstration of mRNA for five species of cytochrome P450 in human bone marrow, bone marrow-derived macrophages and human haemopoietic cell lines. Br. J. Haematol. 2000, 108, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.N. Evidence of drug metabolism by macrophages: Possible role of macrophages in the pathogenesis of drug-induced tissue damage and in the activation of environmental procarcinogens. Clin. Lab. Haematol. 1987, 9, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.N.; Barden, G.; Gardner, B. Ability of unstimulated and phorbol-ester-stimulated human blood-monocyte-derived macrophages to metabolize drugs and its implications. Clin. Lab. Haematol. 1991, 13, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.I.; Raguz, S.; Higgins, C.F. Multidrug transporter activity in lymphocytes. Br. J. Pharmacol. 2004, 143, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Siest, G.; Jeannesson, E.; Marteau, J.B.; Samara, A.; Marie, B.; Pfister, M.; Visvikis-Siest, S. Transcription factor and drug-metabolizing enzyme gene expression in lymphocytes from healthy human subjects. Drug Metab. Dispos. 2008, 36, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Reff, M.E.; Hariharan, K.; Braslawsky, G. Future of monoclonal antibodies in the treatment of hematologic malignancies. Cancer Control 2002, 9, 152–166. [Google Scholar] [PubMed]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Levick, J.R. Revision of the Starling principle: New views of tissue fluid balance. J. Physiol. 2004, 557, 704. [Google Scholar] [CrossRef] [PubMed]
- Levick, J.R.; Michel, C.C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 2012, 39, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.; Deng, Y. Splenic control of intravascular volume in the rat. J. Physiol. 1993, 468, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, M.; Bornstein, G.G.; Suria, H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 2010, 12, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Ghetie, V.; Hubbard, J.G.; Kim, J.K.; Tsen, M.F.; Lee, Y.; Ward, E.S. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur. J. Immunol. 1996, 26, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Junghans, R.P.; Anderson, C.L. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 5512–5516. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Devanaboyina, S.C.; Ober, R.J. Targeting FcRn for the modulation of antibody dynamics. Mol. Immunol. 2015, 67, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Christianson, G.J.; Roopenian, D.C.; Shaw, A.S. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J. Immunol. 2007, 179, 4580–4588. [Google Scholar] [CrossRef] [PubMed]
- Yip, V.; Palma, E.; Tesar, D.B.; Mundo, E.E.; Bumbaca, D.; Torres, E.K.; Reyes, N.A.; Shen, B.Q.; Fielder, P.J.; Prabhu, S.; et al. Quantitative cumulative biodistribution of antibodies in mice: Effect of modulating binding affinity to the neonatal Fc receptor. MAbs 2014, 6, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Beers, S.A.; Chan, C.H.; French, R.R.; Cragg, M.S.; Glennie, M.J. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin. Hematol. 2010, 47, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Benoist, C.; Mathis, D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc. Natl. Acad. Sci. USA 2010, 107, 4658–4663. [Google Scholar] [CrossRef] [PubMed]
- Malviya, G.; Anzola, K.L.; Podestà, E.; Laganà, B.; Del Mastro, C.; Dierckx, R.A.; Scopinaro, F.; Signore, A. (99m)Tc-labeled rituximab for imaging B lymphocyte infiltration in inflammatory autoimmune disease patients. Mol. Imaging Biol. 2012, 14, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, J.G.; Fisher, D.R.; Gopal, A.K.; Durack, L.D.; Press, O.W.; Eary, J.F. High-dose (131)I-tositumomab (anti-CD20) radioimmunotherapy for non-Hodgkin’s lymphoma: Adjusting radiation absorbed dose to actual organ volumes. J. Nucl. Med. 2004, 45, 1059–1064. [Google Scholar] [PubMed]
- Cioc, A.M.; Vanderwerf, S.M.; Peterson, B.A.; Robu, V.G.; Forster, C.L.; Pambuccian, S.E. Rituximab-induced changes in hematolymphoid tissues found at autopsy. Am. J. Clin. Pathol. 2008, 130, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Schechter, G.P. Treatment of splenic marginal zone lymphoma: Splenectomy versus rituximab. Semin. Hematol. 2010, 47, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Kalpadakis, C.; Pangalis, G.A.; Dimopoulou, M.N.; Vassilakopoulos, T.P.; Kyrtsonis, M.C.; Korkolopoulou, P.; Kontopidou, F.N.; Siakantaris, M.P.; Dimitriadou, E.M.; Kokoris, S.I.; et al. Rituximab monotherapy is highly effective in splenic marginal zone lymphoma. Hematol. Oncol. 2007, 25, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Sonnenday, C.J.; Warren, D.S.; Cooper, M.; Samaniego, M.; Haas, M.; King, K.E.; Shirey, R.S.; Simpkins, C.E.; Montgomery, R.A. Plasmapheresis, CMV hyperimmune globulin, and anti-CD20 allow ABO-incompatible renal transplantation without splenectomy. Am. J. Transplant. 2004, 4, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Toki, D.; Ishida, H.; Horita, S.; Setoguchi, K.; Yamaguchi, Y.; Tanabe, K. Impact of low-dose rituximab on splenic B cells in ABO-incompatible renal transplant recipients. Transpl. Int. 2009, 22, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Godeau, B.; Porcher, R.; Fain, O.; Lefrère, F.; Fenaux, P.; Cheze, S.; Vekhoff, A.; Chauveheid, M.P.; Stirnemann, J.; Galicier, L.; et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: Results of a prospective multicenter phase 2 study. Blood 2008, 112, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Kneitz, C.; Wilhelm, M.; Tony, H.P. Effective B cell depletion with rituximab in the treatment of autoimmune diseases. Immunobiology 2002, 206, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Leandro, M.J.; de la Torre, I. Translational Mini-Review Series on B Cell-Directed Therapies: The pathogenic role of B cells in autoantibody-associated autoimmune diseases--lessons from B cell-depletion therapy. Clin. Exp. Immunol. 2009, 157, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.L.; Mahévas, M.; Lee, S.Y.; Stasi, R.; Cunningham-Rundles, S.; Godeau, B.; Kanter, J.; Neufeld, E.; Taube, T.; Ramenghi, U.; et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012, 119, 5989–5995. [Google Scholar] [CrossRef] [PubMed]
- Thatayatikom, A.; White, A.J. Rituximab: A promising therapy in systemic lupus erythematosus. Autoimmun. Rev. 2006, 5, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Patin, P.; Huetz, F.; Descatoire, M.; Cagnard, N.; Bole-Feysot, C.; Le Gallou, S.; Khellaf, M.; Fain, O.; Boutboul, D.; et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J. Clin. Investig. 2013, 123, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Michel, M.; Vingert, B.; Moroch, J.; Boutboul, D.; Audia, S.; Cagnard, N.; Ripa, J.; Menard, C.; Tarte, K.; et al. Emergence of long-lived autoreactive plasma cells in the spleen of primary warm auto-immune hemolytic anemia patients treated with rituximab. J. Autoimmun. 2015, 62, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Samson, M.; Guy, J.; Janikashvili, N.; Fraszczak, J.; Trad, M.; Ciudad, M.; Leguy, V.; Berthier, S.; Petrella, T.; et al. Immunologic effects of rituximab on the human spleen in immune thrombocytopenia. Blood 2011, 118, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kapur, R.; Aslam, R.; Speck, E.R.; Zufferey, A.; Zhao, Y.; Kim, M.; Lazarus, A.H.; Ni, H.; Semple, J.W. CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell-mediated immune thrombocytopenia. Blood 2016, 127, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Vossenkämper, A.; Lutalo, P.M.; Spencer, J. Translational Mini-Review Series on B cell subsets in disease. Transitional B cells in systemic lupus erythematosus and Sjögren’s syndrome: Clinical implications and effects of B cell-targeted therapies. Clin. Exp. Immunol. 2012, 167, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Bumbaca, D.; Fielder, P.J.; Khawli, L.A. Compartmental tissue distribution of antibody therapeutics: Experimental approaches and interpretations. AAPS J. 2012, 14, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Nagengast, W.B.; de Vries, E.G.; Hospers, G.A.; Mulder, N.H.; de Jong, J.R.; Hollema, H.; Brouwers, A.H.; van Dongen, G.A.; Perk, L.R.; Lub-de Hooge, M.N. In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J. Nucl. Med. 2007, 48, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.H.; Peng, C.L.; Lee, S.Y.; Chiang, P.F.; Yao, C.J.; Lin, W.J.; Luo, T.Y.; Shieh, M.J. 111In-cetuximab as a diagnostic agent by accessible epidermal growth factor (EGF) receptor targeting in human metastatic colorectal carcinoma. Oncotarget 2015, 6, 16601–16610. [Google Scholar] [CrossRef] [PubMed]
- Terwisscha van Scheltinga, A.G.; Lub-de Hooge, M.N.; Abiraj, K.; Schröder, C.P.; Pot, L.; Bossenmaier, B.; Thomas, M.; Hölzlwimmer, G.; Friess, T.; Kosterink, J.G.; et al. ImmunoPET and biodistribution with human epidermal growth factor receptor 3 targeting antibody ⁸⁹Zr-RG7116. MAbs 2014, 6, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Wong, K.J.; Baidoo, K.E.; Nayak, T.K.; Regino, C.A.; Garmestani, K.; Brechbiel, M.W. Targeting HER2: A report on the in vitro and in vivo pre-clinical data supporting trastuzumab as a radioimmunoconjugate for clinical trials. MAbs 2010, 2, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Lobaugh, M.; Sandwall, P.; Glover, S.; Murry, M.; Dong, Z.; Spitz, H. Biodistribution of 212Pb conjugated trastuzumab in mice. Med. Appl. Phys. 2013, 296, 75–81. [Google Scholar] [CrossRef]
- Deng, R.; Bumbaca, D.; Pastuskovas, C.V.; Boswell, C.A.; West, D.; Cowan, K.J.; Chiu, H.; McBride, J.; Johnson, C.; Xin, Y.; et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs 2016, 8, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Han, T.H.; Zhao, B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab. Dispos. 2014, 42, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.A.; Flagella, K.; Beyer, J.; Tibbitts, J.; Kaur, S.; Saad, O.; Yi, J.H.; Girish, S.; Dybdal, N.; Reynolds, T. Preclinical safety profile of trastuzumab emtansine (T-DM1): Mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol. Appl. Pharmacol. 2013, 273, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Kosmin, M.; Makris, A.; Jawad, N.; Woolf, D.; Miles, D.; Padhani, A.R. Splenic Enlargement and Bone Marrow Hyperplasia in Patients Receiving Trastuzumab-Emtansine for Metastatic Breast Cancer. Target. Oncol. 2017, 12, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Dhimolea, E. The future of antibodies as cancer drugs. Drug Discov. Today 2012, 17, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Gerlowski, L.E.; Jain, R.K. Physiologically based pharmacokinetic modeling: Principles and applications. J. Pharm. Sci. 1983, 72, 1103–1127. [Google Scholar] [CrossRef] [PubMed]
- Covell, D.G.; Barbet, J.; Holton, O.D.; Black, C.D.; Parker, R.J.; Weinstein, J.N. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab’)2, and Fab’ in mice. Cancer Res. 1986, 46, 3969–3978. [Google Scholar] [PubMed]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.K.; DiMaio, V.J. Normal organ weights in men: Part II-the brain, lungs, liver, spleen, and kidneys. Am. J. Forensic Med. Pathol. 2012, 33, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Ferl, G.Z.; Wu, A.M.; DiStefano, J.J., 3rd. A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn). Ann. Biomed. Eng. 2005, 33, 1640–1652. [Google Scholar] [CrossRef] [PubMed]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Hamada, A.; Yoshida, M.; Shimma, S.; Hashimoto, J.; Yonemori, K.; Tani, H.; Miyakita, Y.; Kanayama, Y.; Wada, Y.; et al. 64Cu-DOTA-trastuzumab PET imaging and HER2 specificity of brain metastases in HER2-positive breast cancer patients. EJNMMI Res. 2015, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Duffus, J.H.; Nordberg, M.; Templeton, D.M. Glossary of terms used in toxicology, 2nd edition (IUPAC Recommendations 2007). Pure Appl. Chem. 2007, 79, 1153–1344. [Google Scholar] [CrossRef]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.J.; Juliano, R.L. Interactions of liposomes with the reticuloendothelial system effects of reticuloendothelial blockade on the clearance of large unilamellar vesicles. Biochim. Biophys. Acta 1981, 677, 453–461. [Google Scholar] [CrossRef]
- Varna, M.; Ratajczak, P.; Ferreira, I.; Leboeuf, C.; Bousquet, G.; Janin, A. In vivo Distribution of Inorganic Nanoparticles in Preclinical Models. J. Biomater. Nanobiotechnol. 2012, 3, 269–279. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Chiannilkulchai, N.; Ammoury, N.; Caillou, B.; Devissaguet, J.P.; Couvreur, P. Hepatic tissue distribution of doxorubicin-loaded nanoparticles after i.v. administration in reticulosarcoma M 5076 metastasis-bearing mice. Cancer Chemother. Pharmacol. 1990, 26, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Demoy, M.; Gibaud, S.; Andreux, J.P.; Weingarten, C.; Gouritin, B.; Couvreur, P. Splenic trapping of nanoparticles: Complementary approaches for in situ studies. Pharm. Res. 1997, 14, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Urrusuno, R.; Fattal, E.; Rodrigues, J.M., Jr.; Féger, J.; Bedossa, P.; Couvreur, P. Effect of polymeric nanoparticle administration on the clearance activity of the mononuclear phagocyte system in mice. J. Biomed. Mater. Res. 1996, 31, 401–408. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Porter, C.J.; Muir, I.S.; Illum, L.; Davis, S.S. Non-phagocytic uptake of intravenously injected microspheres in rat spleen: Influence of particle size and hydrophilic coating. Biochem. Biophys. Res. Commun. 1991, 177, 861–866. [Google Scholar] [CrossRef]
- Nie, S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine (Lond.) 2010, 5, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Hillaireau, H. Investigating interactions between nanoparticles and cells: Internalization and intracellular trafficking. In Polymer Nanoparticles for Nanomedicines: A Guide for their Design, Preparation and Development; Vauthier, C., Ponchel, G., Eds.; Springer: Cham, Switzerland, 2016; pp. 291–326. [Google Scholar]
- Owens, D.E., 3rd; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J.P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.M. Serum opsonins and liposomes: Their interaction and opsonophagocytosis. Crit. Rev. Ther. Drug Carr. Syst. 1992, 9, 39–90. [Google Scholar]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Demoy, M.; Andreux, J.P.; Weingarten, C.; Gouritin, B.; Guilloux, V.; Couvreur, P. In vitro evaluation of nanoparticles spleen capture. Life Sci. 1999, 64, 1329–1337. [Google Scholar] [CrossRef]
- Biewenga, J.; Van der Ende, M.; Krist, L.F.G.; Borst, A.; Ghufron, M.; Van Rooijen, N. Macrophage depletion in the rat after intraperitoneal administration of liposome-encapsulated clodronate: Depletion kinetics and accelerated repopulation of peritoneal and omental macrophages by administration of Freund’s adjuvant. Cell Tissue Res. 1995, 280, 189–196. [Google Scholar] [PubMed]
- Van Rooijen, N. Liposome-mediated elimination of macrophages. Res. Immunol. 1992, 143, 215–219. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Sanders, A. Liposome-mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hedeman, H.; Muir, I.S.; Illum, L.; Davis, S.S. An investigation of the filtration capacity and the fate of large filtered sterically-stabilized microspheres in rat spleen. Biochim. Biophys. Acta 1993, 1157, 233–240. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors controlling nanoparticle pharmacokinetics: An integrated analysis and perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Trubetskoy, V.S. Which polymers can make nanoparticulate drug carriers long-circulating? Adv. Drug Deliv. Rev. 1995, 16, 141–155. [Google Scholar] [CrossRef]
- Woodle, M.C. Controlling liposome blood clearance by surface grafted polymers. Adv. Drug Deliv. Rev. 1998, 32, 139–152. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Yeo, Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Romberg, B.; Hennink, W.E.; Storm, G. Sheddable coatings for long-circulating nanoparticles. Pharm. Res. 2008, 25, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harada, M.; Wang, X.Y.; Ichihara, M.; Irimura, K.; Kiwada, H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: Effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release 2005, 105, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Dams, E.T.; Laverman, P.; Oyen, W.J.; Storm, G.; Scherphof, G.L.; van Der Meer, J.W.; Corstens, F.H.; Boerman, O.C. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar] [PubMed]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ma, Y.; Zhao, Y.; She, Z.; Wang, L.; Li, J.; Wang, C.; Deng, Y. Accelerated drug release and clearance of PEGylated epirubicin liposomes following repeated injections: A new challenge for sequential low-dose chemotherapy. Int. J. Nanomed. 2013, 8, 1257–1268. [Google Scholar]
- Cheng, T.L.; Wu, P.Y.; Wu, M.F.; Chern, J.W.; Roffler, S.R. Accelerated clearance of polyethylene glycol-modified proteins by anti-polyethylene glycol IgM. Bioconjug. Chem. 1999, 10, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.L.; Chen, B.M.; Chern, J.W.; Wu, M.F.; Roffler, S.R. Efficient clearance of poly(ethylene glycol)-modified immunoenzyme with antiPEG monoclonal antibody for prodrug cancer therapy. Bioconjug. Chem. 2000, 11, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; Carstens, M.G.; Boerman, O.C.; Dams, E.T.; Oyen, W.J.; van Rooijen, N.; Corstens, F.H.; Storm, G. Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection. J. Pharmacol. Exp. Ther. 2001, 298, 607–612. [Google Scholar] [PubMed]
- Ishida, T.; Ichihara, M.; Wang, X.; Kiwada, H. Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J. Control. Release 2006, 115, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Wang, X.; Shimizu, T.; Nawata, K.; Kiwada, H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J. Control. Release 2007, 122, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Koide, H.; Asai, T.; Hatanaka, K.; Akai, S.; Ishii, T.; Kenjo, E.; Ishida, T.; Kiwada, H.; Tsukada, H.; Oku, N. T cell-independent B cell response is responsible for ABC phenomenon induced by repeated injection of PEGylated liposomes. Int. J. Pharm. 2010, 392, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Mond, J.J.; Lees, A.; Snapper, C.M. T cell-independent antigens type 2. Annu. Rev. Immunol. 1995, 13, 655–692. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ishida, T.; Kiwada, H. Transport of PEGylated liposomes from the splenic marginal zone to the follicle in the induction phase of the accelerated blood clearance phenomenon. Immunobiology 2013, 218, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Yahuafai, J.; Asai, T.; Nakamura, G.; Fukuta, T.; Siripong, P.; Hyodo, K.; Ishihara, H.; Kikuchi, H.; Oku, N. Suppression in mice of immunosurveillance against PEGylated liposomes by encapsulated doxorubicin. J. Control. Release 2014, 192, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ichihara, M.; Hyodo, K.; Yamamoto, E.; Ishida, T.; Kiwada, H.; Ishihara, H.; Kikuchi, H. Accelerated blood clearance of PEGylated liposomes containing doxorubicin upon repeated administration to dogs. Int. J. Pharm. 2012, 436, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, Q.; Wang, L.; Zhou, X.; Zhao, Y.; Deng, Y. Repeated injections of PEGylated liposomal topotecan induces accelerated blood clearance phenomenon in rats. Eur. J. Pharm. Sci. 2012, 45, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Abu Lila, A.S.; Ishida, T.; Kiwada, H. Abrogation of the accelerated blood clearance phenomenon by SOXL regimen: Promise for clinical application. Int. J. Pharm. 2013, 441, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Abu Lila, A.S.; Ichihara, M.; Shimizu, T.; Ishida, T.; Kiwada, H. Ex Vivo/in-vitro anti-polyethylene glycol (PEG) immunoglobulin M production from murine splenic B cells stimulated by PEGylated liposome. Biol. Pharm. Bull. 2013, 36, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, R.; Shi, Y.; Zhang, Z.; Zhang, X. Zwitterionic poly(carboxybetaine)-based cationic liposomes for effective delivery of small interfering RNA therapeutics without accelerated blood clearance phenomenon. Theranostics 2015, 5, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Bu, L.L.; Xu, J.H.; Cai, B.; Yu, G.T.; Yu, X.; He, Z.; Huang, Q.; Li, A.; Guo, S.S.; et al. Red Blood Cell Membrane as a Biomimetic Nanocoating for Prolonged Circulation Time and Reduced Accelerated Blood Clearance. Small 2015, 11, 6225–6236. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M. Mechanisms of splenic clearance of blood cells and particles: Towards development of new splenotropic agents. Adv. Drug Deliv. Rev. 1995, 17, 103–115. [Google Scholar] [CrossRef]
- Peracchia, M.T.; Fattal, E.; Desmaële, D.; Besnard, M.; Noël, J.P.; Gomis, J.M.; Appel, M.; d’Angelo, J.; Couvreur, P. Stealth PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J. Control. Release 1999, 60, 121–128. [Google Scholar] [CrossRef]
- Fattal, E.; Youssef, M.; Couvreur, P.; Andremont, A. Treatment of experimental salmonellosis in mice with ampicillin-bound nanoparticles. Antimicrob. Agents Chemother. 1989, 33, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.; Fattal, E.; Alonso, M.J.; Roblot-Treupel, L.; Sauzières, J.; Tancrède, C.; Omnès, A.; Couvreur, P.; Andremont, A. Effectiveness of nanoparticle-bound ampicillin in the treatment of Listeria monocytogenes infection in athymic nude mice. Antimicrob. Agents Chemother. 1988, 32, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Fattal, E.; Rojas, J.; Youssef, M.; Couvreur, P.; Andremont, A. Liposome-entrapped ampicillin in the treatment of experimental murine listeriosis and salmonellosis. Antimicrob. Agents Chemother. 1991, 35, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Pandey, R.; Sharma, S.; Khuller, G.K. Novel chemotherapy for tuberculosis: Chemotherapeutic potential of econazole- and moxifloxacin-loaded PLG nanoparticles. Int. J. Antimicrob. Agents 2008, 31, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, P.; Mishra, B. Mannose-conjugated chitosan nanoparticles loaded with rifampicin for the treatment of visceral leishmaniasis. Carbohydr. Polym. 2014, 101, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Imbuluzqueta, E.; Gamazo, C.; Lana, H.; Campanero, M.Á.; Salas, D.; Gil, A.G.; Elizondo, E.; Ventosa, N.; Veciana, J.; Blanco-Prieto, M.J. Hydrophobic gentamicin-loaded nanoparticles are effective against Brucella melitensis infection in mice. Antimicrob. Agents Chemother. 2013, 57, 3326–3333. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Sharma, S.; Shafiq, N.; Pandhi, P.; Khuller, G.K.; Malhotra, S. Pharmacokinetics and tissue distribution studies of orally administered nanoparticles encapsulated ethionamide used as potential drug delivery system in management of multi-drug resistant tuberculosis. Drug Deliv. 2011, 18, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lecaroz, C.; Gamazo, C.; Blanco-Prieto, M.J. Nanocarriers with gentamicin to treat intracellular pathogens. J. Nanosci. Nanotechnol. 2006, 6, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, K.D.; Yadav, T.P.; Prajapati, V.K.; Kumar, S.; Rai, M.; Dube, A.; Srivastava, O.N.; Sundar, S. Antileishmanial activity of nano-amphotericin B deoxycholate. J. Antimicrob. Chemother. 2008, 62, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Seleem, M.N.; Jain, N.; Pothayee, N.; Ranjan, A.; Riffle, J.S.; Sriranganathan, N. Targeting Brucella melitensis with polymeric nanoparticles containing streptomycin and doxycycline. FEMS Microbiol. Lett. 2009, 294, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Veerareddy, P.R.; Vobalaboina, V.; Ali, N. Antileishmanial activity, pharmacokinetics and tissue distribution studies of mannose-grafted amphotericin B lipid nanospheres. J. Drug Target. 2009, 17, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Jain, N.K. Targeting potential and anti-HIV activity of lamivudine loaded mannosylated poly (propyleneimine) dendrimer. Biochim. Biophys. Acta 2007, 1770, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Gajbhiye, V.; Ganesh, N.; Barve, J.; Jain, N.K. Synthesis, characterization and targeting potential of zidovudine loaded sialic acid conjugated-mannosylated poly(propyleneimine) dendrimers. Eur. J. Pharm. Sci. 2013, 48, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Ravi, P.R.; Vats, R.; Balija, J.; Adapa, S.P.; Aditya, N. Modified pullulan nanoparticles for oral delivery of lopinavir: Formulation and pharmacokinetic evaluation. Carbohydr. Polym. 2014, 110, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Skinner, A.L.; Araínga, M.A.; Puligujja, P.; Palandri, D.L.; Baldridge, H.M.; Edagwa, B.J.; McMillan, J.M.; Mosley, R.L.; Gendelman, H.E. Cellular Responses and Tissue Depots for Nanoformulated Antiretroviral Therapy. PLoS ONE 2015, 10, e0145966. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Kourembanas, S. Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. 2015, 77, 13–27. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Hung, M.E.; Breakefield, X.O.; Leonard, J.N. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439–464. [Google Scholar] [CrossRef] [PubMed]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hwang do, W.; Choi, H.; Jang, S.C.; Yoo, M.Y.; Park, J.Y.; Choi, N.E.; Oh, H.J.; Ha, S.; Lee, Y.S.; Jeong, J.M.; et al. Noninvasive imaging of radiolabeled exosome-mimetic nanovesicle using (99m)Tc-HMPAO. Sci. Rep. 2015, 5, 15636. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Kato, K.; Yamashita, T.; Imai, T.; Saji, H.; Takakura, Y. Quantitative analysis of tissue distribution of the B16BL6-derived exosomes using a streptavidin-lactadherin fusion protein and iodine-125-labeled biotin derivative after intravenous injection in mice. J. Pharm. Sci. 2015, 104, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takahashi, Y.; Nishikawa, M.; Kato, K.; Morishita, M.; Yamashita, T.; Matsumoto, A.; Charoenviriyakul, C.; Takakura, Y. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J. Extracell. Vesicles 2015, 4, 26238. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Turkes, A.; Dewitt, S.; Steadman, R.; Mason, M.D.; Hallett, M.B. Adhesion and signaling by B cell-derived exosomes: The role of integrins. FASEB J. 2004, 18, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Végh, P.; Prechl, J.; Kerekes, K.; Kovács, J.; Csikós, G.; Bajtay, Z.; Erdei, A.B. lymphocytes and macrophages release cell membrane deposited C3-fragments on exosomes with T cell response-enhancing capacity. Mol. Immunol. 2008, 45, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Barrès, C.; Blanc, L.; Bette-Bobillo, P.; André, S.; Mamoun, R.; Gabius, H.J.; Vidal, M. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Saunderson, S.C.; Dunn, A.C.; Crocker, P.R.; McLellan, A.D. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014, 123, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [PubMed]
- Tarantino, G.; Savastano, S.; Capone, D.; Colao, A. Spleen: A new role for an old player? World J. Gastroenterol. 2011, 17, 3776–3784. [Google Scholar] [CrossRef] [PubMed]
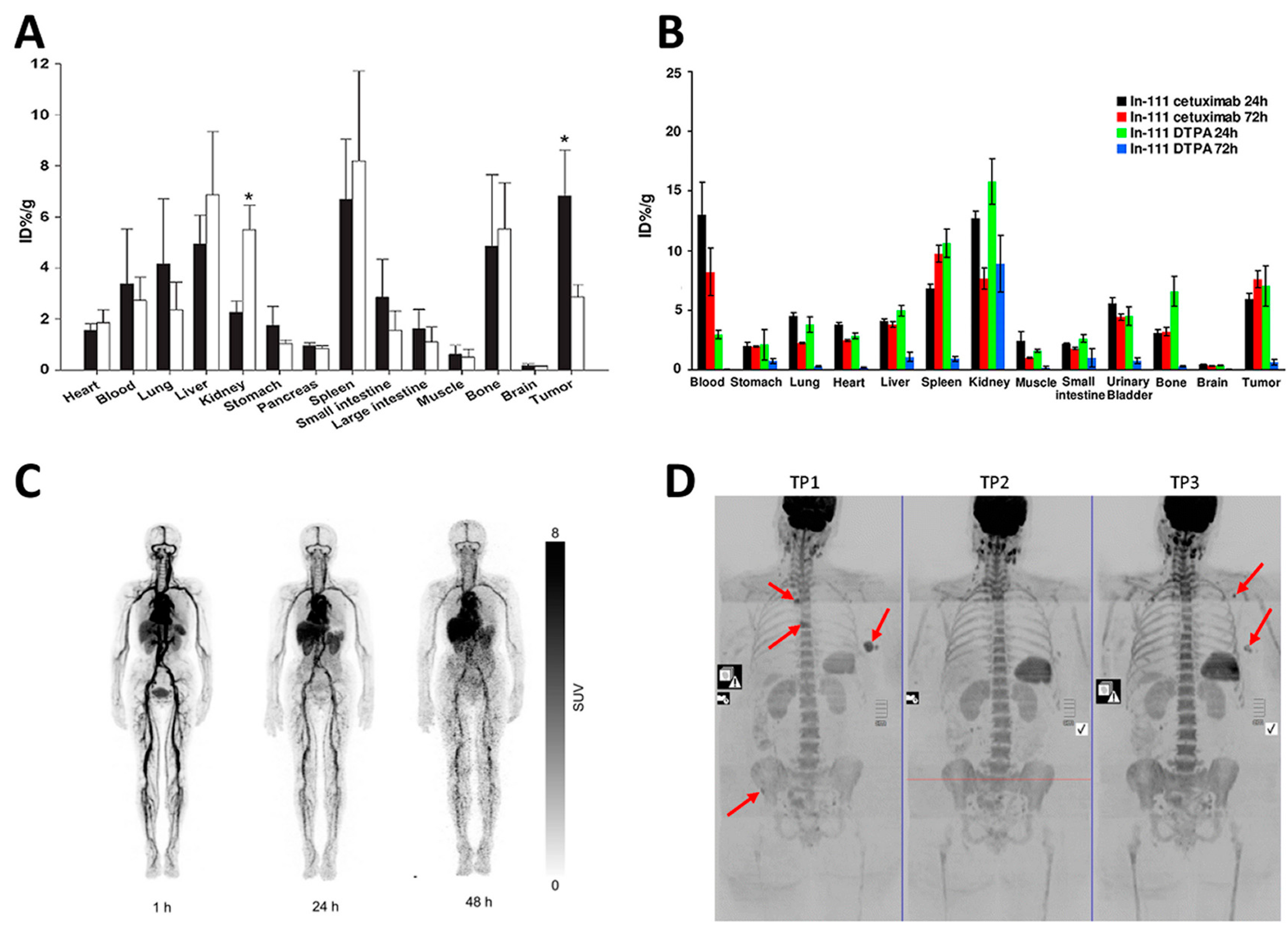
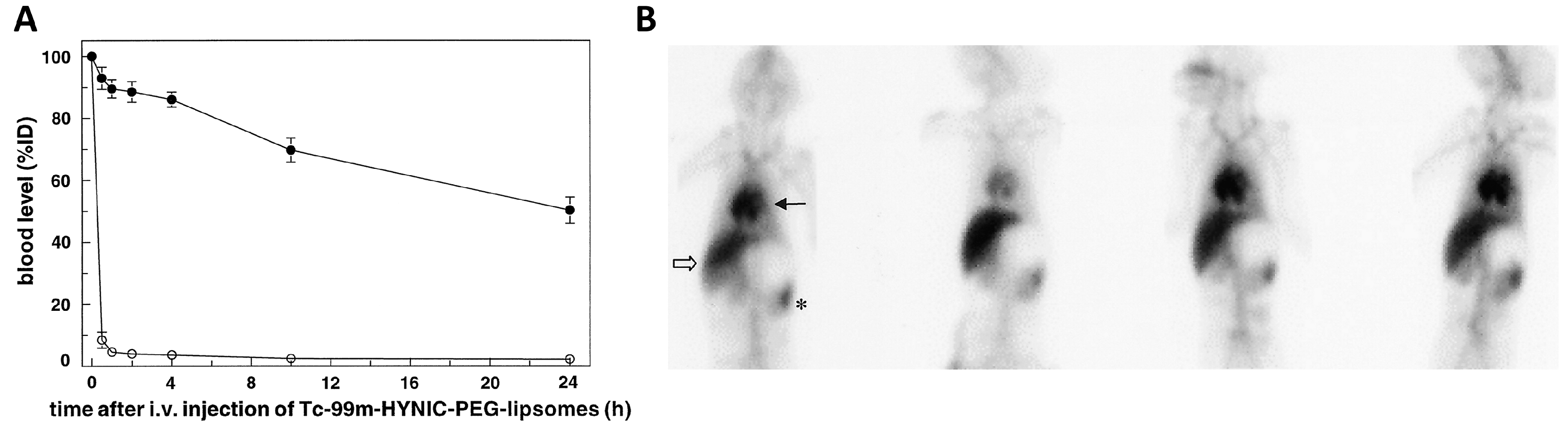

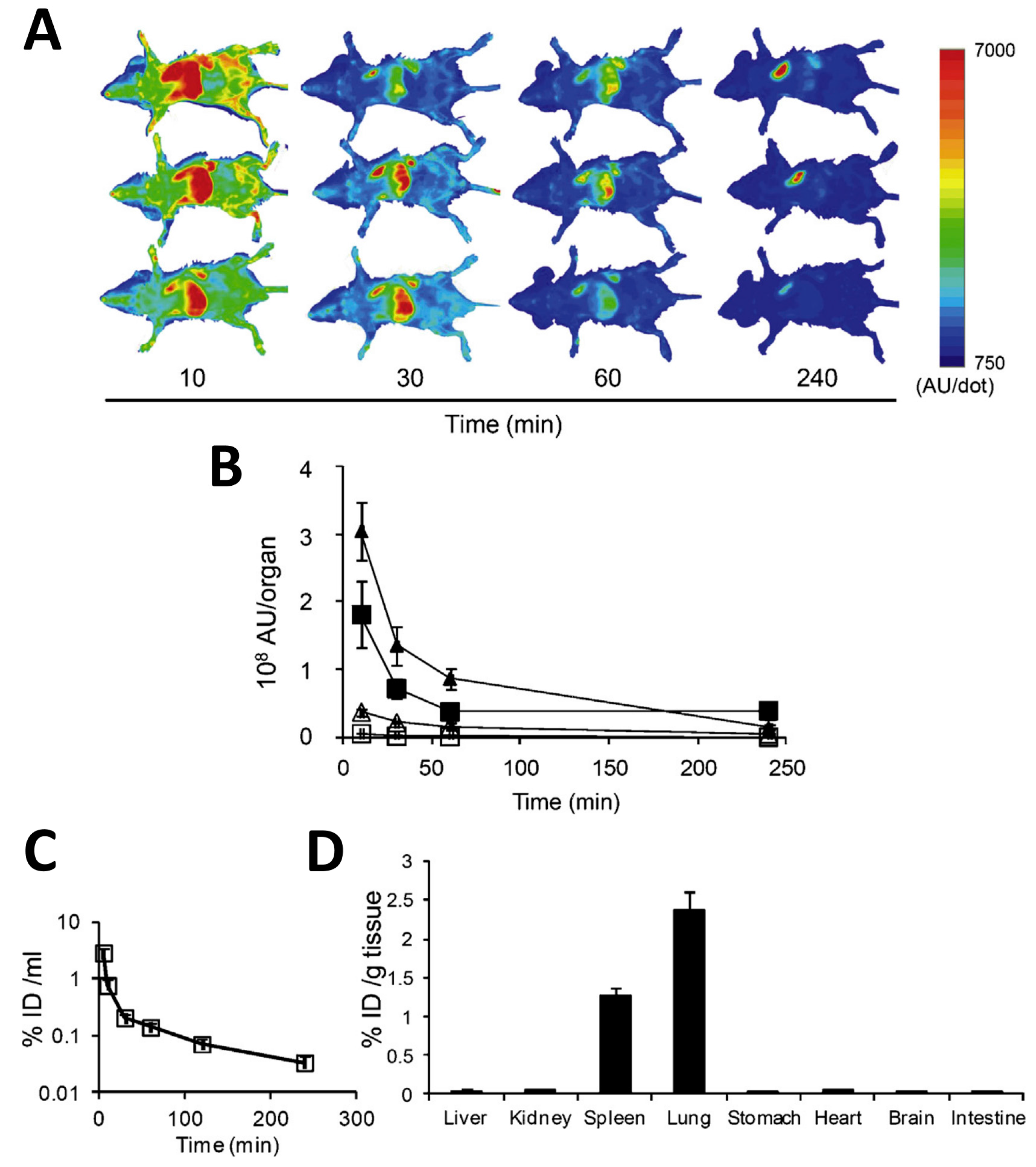
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cataldi, M.; Vigliotti, C.; Mosca, T.; Cammarota, M.; Capone, D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int. J. Mol. Sci. 2017, 18, 1249. https://doi.org/10.3390/ijms18061249
Cataldi M, Vigliotti C, Mosca T, Cammarota M, Capone D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. International Journal of Molecular Sciences. 2017; 18(6):1249. https://doi.org/10.3390/ijms18061249
Chicago/Turabian StyleCataldi, Mauro, Chiara Vigliotti, Teresa Mosca, MariaRosaria Cammarota, and Domenico Capone. 2017. "Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes" International Journal of Molecular Sciences 18, no. 6: 1249. https://doi.org/10.3390/ijms18061249
APA StyleCataldi, M., Vigliotti, C., Mosca, T., Cammarota, M., & Capone, D. (2017). Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. International Journal of Molecular Sciences, 18(6), 1249. https://doi.org/10.3390/ijms18061249