
Gene Therapy for Pancreatic Cancer: Specificity, Issues and Hopes

 ,
, 
Abstract
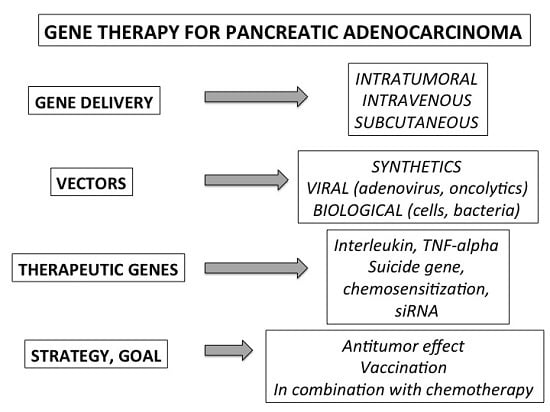
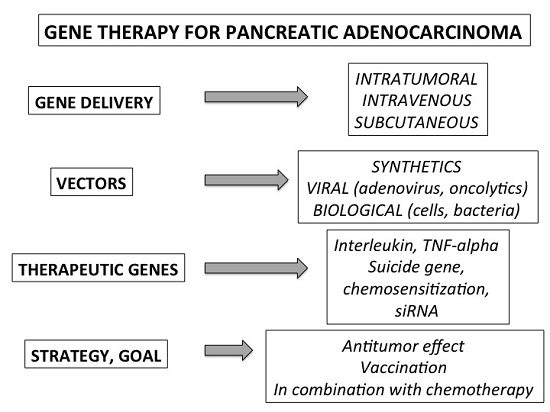
1. Introduction
2. Specificity and Tools of the Gene Therapy Applied to Pancreatic Cancer
2.1. General Strategy and Experimental Models
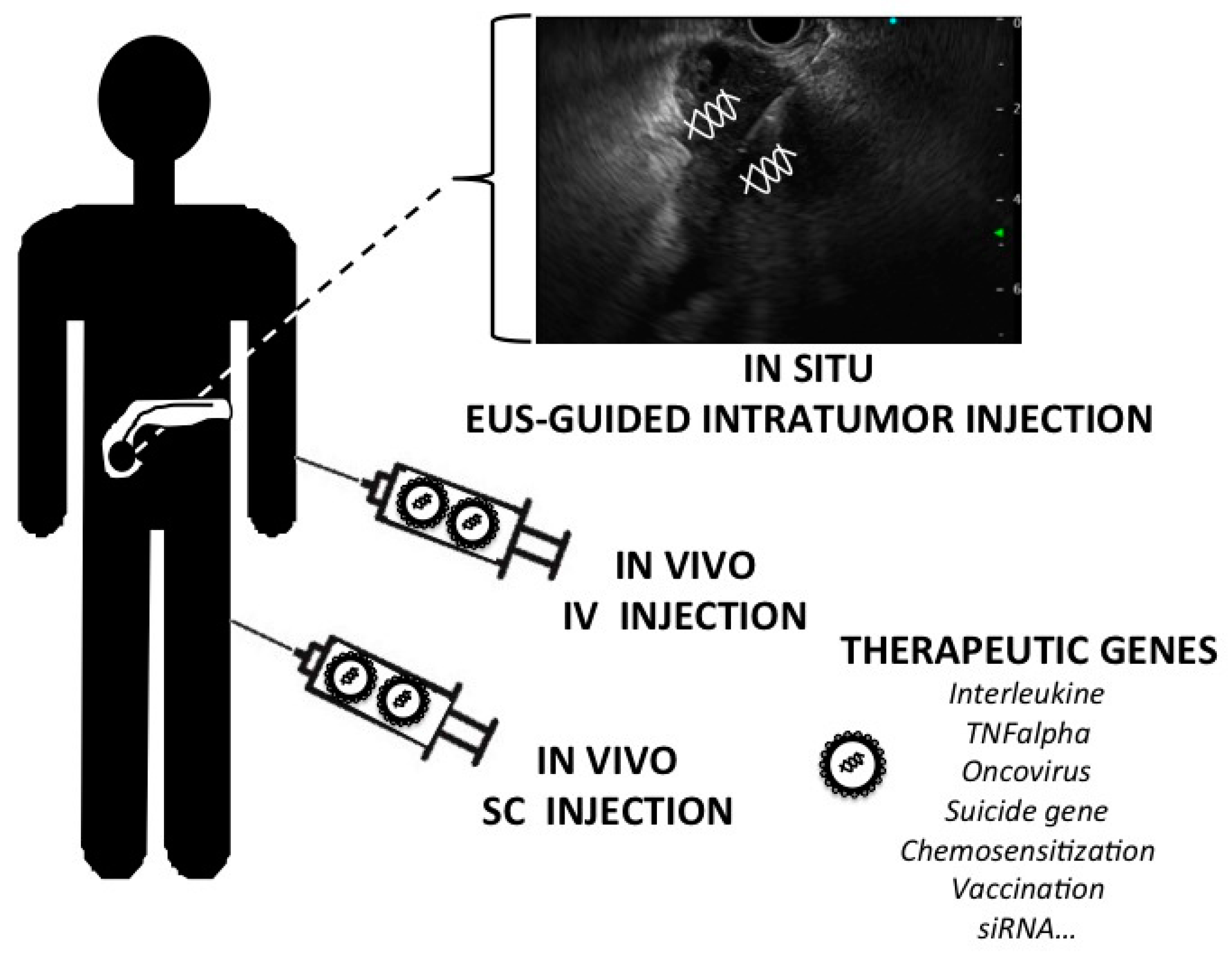
2.2. Methods of Gene Delivery
2.3. Vectors
3. Therapeutic Genes and Pre-Clinical Strategies
3.1. Tumor Suppressor Genes
3.2. Antiangiogenic and Proapototic Genes
3.3. Suicide Genes
3.4. Small Non-Coding mRNA, Interferent mRNA and Antisense Therapy
3.5. Immunotherapy and Vaccination
4. Oncolytic Virotheray
5. Clinical Trials
6. Conclusions
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic Ductal Adenocarcinoma |
| EUS | Endoscopic Ultrasound |
| CAR | Chimeric Antigen Receptors |
| VEGF | Vascular endothetial Growth Factor |
| EGF | Epidermal Growth Factor |
| MUC-1 | Mucin 1 |
| HSV-TK | Herpes Virus Thymidine Kinase |
| TNF | Tumor Necrosis Factor |
| CAR | Chimeric Antigen Receptor |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
References
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Boige, V.; Malka, D. Treatment of advanced pancreatic cancer. Semin. Oncol. 2007, 34, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Mulligan, E.A.; Vong, W.T.; Thomas, H.D.; Kahn, S.; Kyle, S.; Mukhopadhyay, A.; Los, G.; Hostomsky, Z.; Plummer, E.R.; et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J. Natl. Cancer Inst. 2011, 103, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiao, Y.; dal Molin, M.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 21188–21193. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Faure, P.; Bournet, B.; Selves, J.; Escourrou, J. Interventional endoscopic ultrasound in pancreatic diseases. Pancreatology 2006, 6, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Koorstra, J.B.M.; Hustinx, S.R.; Offerhaus, G.J.A.; Maitra, A. Pancreatic carcinogenesis. Pancreatol. Off. J. Int. Assoc. Pancreatol. IAP Al 2008, 8, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schmiegel, W.H. Recent discoveries in cancer genetics of exocrine pancreatic neoplasia. Digestion 1998, 59, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Delpu, Y.; Hanoun, N.; Lulka, H.; Sicard, F.; Selves, J.; Buscail, L.; Torrisani, J.; Cordelier, P. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr. Genom. 2011, 12, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed]
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer Oxf. Engl. 1990 2015, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Rochaix, P.; Delesque, N.; Estève, J.P.; Saint-Laurent, N.; Voight, J.J.; Vaysse, N.; Susini, C.; Buscail, L. Gene therapy for pancreatic carcinoma: Local and distant antitumor effects after somatostatin receptor sst2 gene transfer. Hum. Gene Ther. 1999, 10, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Vernejoul, F.; Faure, P.; Benali, N.; Calise, D.; Tiraby, G.; Pradayrol, L.; Susini, C.; Buscail, L. Antitumor Effect of in Vivo Somatostatin Receptor Subtype 2 Gene Transfer in Primary and Metastatic Pancreatic Cancer Models. Cancer Res. 2002, 62, 6124–6131. [Google Scholar] [PubMed]
- Benali, N.; Cordelier, P.; Calise, D.; Pages, P.; Rochaix, P.; Nagy, A.; Esteve, J.P.; Pour, P.M.; Schally, A.V.; Vaysse, N.; et al. Inhibition of growth and metastatic progression of pancreatic carcinoma in hamster after somatostatin receptor subtype 2 (SST2) gene expression and administration of cytotoxic somatostatin analog AN-238. Proc. Natl. Acad. Sci. USA 2000, 97, 9180–9185. [Google Scholar] [CrossRef] [PubMed]
- Thorne, B.; Takeya, R.; Vitelli, F.; Swanson, X. Gene Therapy. Adv. Biochem. Eng. Biotechnol. 2017. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Doody, A.M.; Korley, J.N.; Dang, K.P.; Zawaneh, P.N.; Putnam, D. Characterizing the structure/function parameter space of hydrocarbon-conjugated branched polyethylenimine for DNA delivery in vitro. J. Control. Release 2006, 116, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.L.; Meister, G.E.; Koerber, J.T.; Pack, D.W. Partial acetylation of polyethylenimine enhances in vitro gene delivery. Pharm. Res. 2004, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Klibanov, A.M. Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14640–14645. [Google Scholar] [CrossRef] [PubMed]
- Oskuee, R.K.; Dehshahri, A.; Shier, W.T.; Ramezani, M. Alkylcarboxylate grafting to polyethylenimine: A simple approach to producing a DNA nanocarrier with low toxicity. J. Gene Med. 2009, 11, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C.; Yaouanc, J.J.; Jaffrès, P.A. Chemical vectors for gene delivery: A current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, E.; Gonçalves, C.; Billiet, L.; Gomez, J.P.; Pichon, C.; Cheradame, H.; Midoux, P.; Guégan, P. Histidinylated linear PEI: A new efficient non-toxic polymer for gene transfer. Chem. Commun. Camb. Engl. 2011, 47, 12547–12549. [Google Scholar] [CrossRef] [PubMed]
- Nayerossadat, N.; Maedeh, T.; Ali, P.A. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Touchefeu, Y.; Harrington, K.J.; Galmiche, J.P.; Vassaux, G. Review article: Gene therapy, recent developments and future prospects in gastrointestinal oncology. Aliment. Pharmacol. Ther. 2010, 32, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Xia, Z.S.; Zhong, Y.Q. Gene therapy in pancreatic cancer. World J. Gastroenterol. 2014, 20, 13343–13368. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, N.; Gayral, M.; Pointreau, A.; Buscail, L.; Cordelier, P. Initial Characterization of Integrase-Defective Lentiviral Vectors for Pancreatic Cancer Gene Therapy. Hum. Gene Ther. 2016, 27, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Vassaux, G.; Nitcheu, J.; Jezzard, S.; Lemoine, N.R. Bacterial gene therapy strategies. J. Pathol. 2006, 208, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.K.; Vermerris, W. Recent Advances in Nanomaterials for Gene Delivery-A Review. Nanomater. Basel Switz. 2017, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Shirasawa, H.; Sashiyama, H.; Kawahira, H.; Kaneko, K.; Asano, T.; Ochiai, T. P16INK4a expression adenovirus vector to suppress pancreas cancer cell proliferation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 4182–4185. [Google Scholar]
- Joshi, U.S.; Dergham, S.T.; Chen, Y.Q.; Dugan, M.C.; Crissman, J.D.; Vaitkevicius, V.K.; Sarkar, F.H. Inhibition of pancreatic tumor cell growth in culture by p21WAF1 recombinant adenovirus. Pancreas 1998, 16, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Craig, C.; Kim, M.; Ohri, E.; Wersto, R.; Katayose, D.; Li, Z.; Choi, Y.H.; Mudahar, B.; Srivastava, S.; Seth, P.; et al. Effects of adenovirus-mediated p16INK4A expression on cell cycle arrest are determined by endogenous p16 and Rb status in human cancer cells. Oncogene 1998, 16, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Rabb, M.; Madureira, P.A.; Clements, D.; Gujar, S.A.; Waisman, D.M.; Giacomantonio, C.A.; Lee, P.W.K. Gemcitabine-mediated tumour regression and p53-dependent gene expression: Implications for colon and pancreatic cancer therapy. Cell Death Dis. 2013, 4, e791. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sathishkumar, S.; Ahmed, M.M. Influence of cell cycle checkpoints and p53 function on the toxicity of temozolomide in human pancreatic cancer cells. Pancreatol. Off. J. Int. Assoc. Pancreatol. IAP Al 2010, 10, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Philip, P.A.; Aboukameel, A.; Wang, Z.; Banerjee, S.; Zafar, S.F.; Goustin, A.S.; Almhanna, K.; Yang, D.; Sarkar, F.H.; et al. Reactivation of p53 by novel MDM2 inhibitors: Implications for pancreatic cancer therapy. Curr. Cancer Drug Targets 2010, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Tao, G.Q.; Li, D.C.; Zhu, X.G.; Bai, X.; Cai, B. Inhibition of pancreatic carcinoma cell growth in vitro by DPC4 gene transfection. World J. Gastroenterol. 2008, 14, 6254–6260. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Farnebo, F.; Yu, E.Y.; Christofferson, R.; Swearingen, R.A.; Carter, R.; von Recum, H.A.; Yuan, J.; Kamihara, J.; Flynn, E.; et al. Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc. Natl. Acad. Sci. USA 2001, 98, 4605–4610. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Takayama, K.; Takakura, N.; Kitano, S.; Ueno, H. Anti-tumor angiogenesis therapy using soluble receptors: Enhanced inhibition of tumor growth when soluble fibroblast growth factor receptor-1 is used with soluble vascular endothelial growth factor receptor. Cancer Gene Ther. 2002, 9, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, J.; Lawler, J.; Terwilliger, E.; Parangi, S. Adeno-associated virus-mediated antiangiogenic gene therapy with thrombospondin-1 type 1 repeats and endostatin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 3968–3976. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Fang, Y.; Wang, X.; Jin, R.; Zhang, Q.; Andersson, R. Experimental studies on treatment of pancreatic cancer with double-regulated duplicative adenovirus AdTPHre-hEndo carrying human endostatin gene. Pancreatol. Off. J. Int. Assoc. Pancreatol. IAP Al 2013, 13, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Tysome, J.R.; Briat, A.; Alusi, G.; Cao, F.; Gao, D.; Yu, J.; Wang, P.; Yang, S.; Dong, Z.; Wang, S.; et al. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 2009, 16, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Rigg, A.S.; Lemoine, N.R. Adenoviral delivery of TIMP1 or TIMP2 can modify the invasive behavior of pancreatic cancer and can have a significant antitumor effect in vivo. Cancer Gene Ther. 2001, 8, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.H.; Spivack, D.E.; Takimoto, S.; Fang, B.; Burton, D.W.; Moossa, A.R.; Hoffman, R.M.; Bouvet, M. Gene therapy of pancreatic cancer with green fluorescent protein and tumor necrosis factor-related apoptosis-inducing ligand fusion gene expression driven by a human telomerase reverse transcriptase promoter. Ann. Surg. Oncol. 2003, 10, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, X.X.; Chen, D.Z.; Li, S.F.; Zhang, L.S. Herpes simplex virus thymidine kinase and ganciclovir suicide gene therapy for human pancreatic cancer. World J. Gastroenterol. 2004, 10, 400–403. [Google Scholar] [PubMed]
- Fogar, P.; Greco, E.; Basso, D.; Habeler, W.; Navaglia, F.; Zambon, C.F.; Tormen, D.; Gallo, N.; Cecchetto, A.; Plebani, M.; et al. Suicide gene therapy with HSV-TK in pancreatic cancer has no effect in vivo in a mouse model. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2003, 29, 721–730. [Google Scholar] [CrossRef]
- Greco, E.; Fogar, P.; Basso, D.; Stefani, A.L.; Navaglia, F.; Zambon, C.F.; Mazza, S.; Gallo, N.; Piva, M.G.; Scarpa, A.; Pedrazzoli, S.; et al. Retrovirus-mediated herpes simplex virus thymidine kinase gene transfer in pancreatic cancer cell lines: An incomplete antitumor effect. Pancreas 2002, 25, e21–e29. [Google Scholar] [CrossRef] [PubMed]
- Erbs, P.; Regulier, E.; Kintz, J.; Leroy, P.; Poitevin, Y.; Exinger, F.; Jund, R.; Mehtali, M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000, 60, 3813–3822. [Google Scholar] [PubMed]
- Fogar, P.; Navaglia, F.; Basso, D.; Greco, E.; Zambon, C.F.; Fadi, E.; Falda, A.; Stranges, A.; Vannozzi, F.; Danesi, R.; et al. Suicide gene therapy with the yeast fusion gene cytosine deaminase/uracil phosphoribosyltransferase is not enough for pancreatic cancer. Pancreas 2007, 35, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.I.; Searle, P.F.; Weedon, S.J.; Green, N.K.; McNeish, I.A.; Kerr, D.J. Virus-directed enzyme prodrug therapy using CB1954. Anticancer. Drug Des. 1999, 14, 461–472. [Google Scholar] [PubMed]
- Green, N.K.; Youngs, D.J.; Neoptolemos, J.P.; Friedlos, F.; Knox, R.J.; Springer, C.J.; Anlezark, G.M.; Michael, N.P.; Melton, R.G.; Ford, M.J.; et al. Sensitization of colorectal and pancreatic cancer cell lines to the prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) by retroviral transduction and expression of the E. coli nitroreductase gene. Cancer Gene Ther. 1997, 4, 229–238. [Google Scholar] [PubMed]
- Carrió, M.; Visa, J.; Cascante, A.; Estivill, X.; Fillat, C. Intratumoral activation of cyclophosphamide by retroviral transfer of the cytochrome P450 2B1 in a pancreatic tumor model. Combination with the HSVtk/GCV system. J. Gene Med. 2002, 4, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M.; Müller, P.; Karle, P.; Stange, J.; Mitzner, S.; Jesnowski, R.; Nizze, H.; Nebe, B.; Liebe, S.; Salmons, B.; et al. Targeted chemotherapy by intratumour injection of encapsulated cells engineered to produce CYP2B1, an ifosfamide activating cytochrome P450. Gene Ther. 1998, 5, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Sicard, F.; Gayral, M.; Lulka, H.; Buscail, L.; Cordelier, P. Targeting miR-21 for the therapy of pancreatic cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002, 2, 243–247. [Google Scholar] [CrossRef]
- Réjiba, S.; Wack, S.; Aprahamian, M.; Hajri, A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci. 2007, 98, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Morioka, C.Y.; Machado, M.C.C.; Saito, S.; Nakada, Y.; Matheus, A.S.; Jukemura, J.; Bacchella, T.; Takahara, T.; Watanabe, A. Suppression of invasion of a hamster pancreatic cancer cell line by antisense oligonucleotides mutation-matched to K-ras gene. Vivo Athens Greece 2005, 19, 535–538. [Google Scholar]
- Shen, Y.M.; Yang, X.C.; Yang, C.; Shen, J.K. Enhanced therapeutic effects for human pancreatic cancer by application K-ras and IGF-IR antisense oligodeoxynucleotides. World J. Gastroenterol. 2008, 14, 5176–5185. [Google Scholar] [CrossRef] [PubMed]
- Alberts, S.R.; Schroeder, M.; Erlichman, C.; Steen, P.D.; Foster, N.R.; Moore, D.F.; Rowland, K.M.; Nair, S.; Tschetter, L.K.; Fitch, T.R. Gemcitabine and ISIS-2503 for patients with locally advanced or metastatic pancreatic adenocarcinoma: A North Central Cancer Treatment Group phase II trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.W.; Mali, P.; Wu, E.Y.; Ng, A.H.M.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Song, M. The CRISPR/Cas9 system: Their delivery, in vivo and ex vivo applications and clinical development by startups. Biotechnol. Prog. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mout, R.; Ray, M.; Lee, Y.W.; Scaletti, F.; Rotello, V.M. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjug. Chem. 2017, 28, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Tomitaka, A.; Raymond, A.; Nair, M. Current application of CRISPR/Cas9 gene-editing technique to eradication of HIV/AIDS. Gene Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Winters, I.P.; Wang, J.; Naranjo, S.; Dudgeon, C.; Tamburini, F.B.; Brady, J.J.; Yang, D.; Grüner, B.M.; Chuang, C.H.; et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015, 29, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; van Eijck, C.H.; Peppelenbosch, M.P.; Sleijfer, S.; Bruno, M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat Rev. 2014, 40, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Liu, L.; Xu, H.X.; Wu, C.T.; Xiang, J.F.; Xu, J.; Liu, C.; Long, J.; Ni, Q.X.; Yu, X.J. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg. 2016, 103, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.D.; Pillarisetty, V.G. T-cell programming in pancreatic adenocarcinoma: A review. Cancer Gene Ther. 2017, 24, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A., III; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Andersson, R.; Bauden, M.; Hammes, S.; Holdenrieder, S.; Ansari, D. Immune checkpoint therapy for pancreatic cancer. World J. Gastroenterol. 2016, 22, 9457. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tagawa, M.; Takenaga, K.; Kondo, F.; Yamaguchi, T.; Saisho, H.; Nakagawara, A.; Sakiyama, S. Loss of tumorigenicity of human pancreatic carcinoma cells engineered to produce interleukin-2 or interleukin-4 in nude mice: A potentiality for cancer gene therapy. Cancer Lett. 1998, 128, 47–53. [Google Scholar] [CrossRef]
- Liu, L.; Meng, J.; Zhang, C.; Duan, Y.; Zhao, L.; Wang, S.; Shan, B. Effects on apoptosis and cell cycle arrest contribute to the antitumor responses of interleukin-27 mediated by retrovirus in human pancreatic carcinoma cells. Oncol. Rep. 2012, 27, 1497–1503. [Google Scholar] [PubMed]
- Ravet, E.; Lulka, H.; Gross, F.; Casteilla, L.; Buscail, L.; Cordelier, P. Using lentiviral vectors for efficient pancreatic cancer gene therapy. Cancer Gene Ther. 2010, 17, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Davydova, J.; Brown, E.; Han, J.; Yamamoto, M.; Vickers, S.M. Delivery of interferon alpha using a novel Cox2-controlled adenovirus for pancreatic cancer therapy. Surgery 2012, 152, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, S.R.; King, C.R.; Osborn, R.; Fairweather, W.R.; O’Reilly, E.M.; Thornton, M.O.; Wei, L.L. Combination of human tumor necrosis factor-α (hTNF-α) gene delivery with gemcitabine is effective in models of pancreatic cancer. Cancer Gene Ther. 2009, 16, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Lortal, B.; Gross, F.; Peron, J.M.; Pénary, M.; Berg, D.; Hennebelle, I.; Favre, G.; Couderc, B. Preclinical study of an ex vivo gene therapy protocol for hepatocarcinoma. Cancer Gene Ther. 2009, 16, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.; Stashwick, C.; Haas, A.R.; Tanyi, J.L. Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy 2016, 8, 449–460. [Google Scholar] [CrossRef] [PubMed]
- DeSelm, C.J.; Tano, Z.E.; Varghese, A.M.; Adusumilli, P.S. CAR T-cell therapy for pancreatic cancer. J. Surg. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Kudrin, A. Overview of cancer vaccines: Considerations for development. Hum. Vaccines Immunother. 2012, 8, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
- Plate, J.M.D. Advances in therapeutic vaccines for pancreatic cancer. Discov. Med. 2012, 14, 89–94. [Google Scholar] [PubMed]
- Maki, R.G.; Livingston, P.O.; Lewis, J.J.; Janetzki, S.; Klimstra, D.; Desantis, D.; Srivastava, P.K.; Brennan, M.F. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Dig. Dis. Sci. 2007, 52, 1964–1972. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Qin, X.; Jin, D.; Lou, W.; Wu, L.; Wang, D.; Wu, W.; Ni, X.; Mao, Z.; Kuang, T.; et al. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin. Exp. Med. 2012, 12, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, S.L.; Gjertsen, M.K.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Meo, M.; Buanes, T.; Gaudernack, G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br. J. Cancer 2006, 95, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Obrist, F.; Eggermont, A.; Galon, J.; Hervé Fridman, W.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: DNA vaccines for cancer therapy. Oncoimmunology 2014, 3, e28185. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Obrist, F.; Eggermont, A.; Galon, J.; Cremer, I.; Erbs, P.; Limacher, J.M.; Preville, X.; Zitvogel, L.; et al. Trial Watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2014, 3, e28694. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, B.R.; Wu, Y.Q.; Wang, F.C.; Zhang, J.; Wang, Y.G. Oncolytic viruses against cancer stem cells: A promising approach for gastrointestinal cancer. World J. Gastroenterol. 2016, 22, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Husseini, F.; Delord, J.P.; Fournel-Federico, C.; Guitton, J.; Erbs, P.; Homerin, M.; Halluard, C.; Jemming, C.; Orange, C.; Limacher, J.M.; et al. Vectorized gene therapy of liver tumors: Proof-of-concept of TG4023 (MVA-FCU1) in combination with flucytosine. Ann. Oncol. 2017, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.; Li, S.; McFadden, G. Oncolytic virotherapy for pancreatic cancer. Expert Rev. Mol. Med. 2011, 13, e18. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, H.; Su, C.; Li, Z. Viral therapy for pancreatic cancer: Tackle the bad guys with poison. Cancer Lett. 2013, 333, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Francis, J.; Eddouadi, A.; Lemoine, N.R.; Halldén, G. An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine. Cancer Gene Ther. 2011, 18, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Bedford, R.; Abbruzzese, J.L.; Lahoti, S.; Reid, T.R.; Soetikno, R.M.; Kirn, D.H.; Freeman, S.M. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin. Cancer Res. 2003, 9, 555–561. [Google Scholar] [PubMed]
- Lucas, T.; Benihoud, K.; Vigant, F.; Schmidt, C.Q.; Schmidt, C.Q.A.; Wortmann, A.; Bachem, M.G.; Simmet, T.; Kochanek, S. Hexon modification to improve the activity of oncolytic adenovirus vectors against neoplastic and stromal cells in pancreatic cancer. PLoS ONE 2015, 10, e0117254. [Google Scholar] [CrossRef] [PubMed]
- Ilkow, C.S.; Marguerie, M.; Batenchuk, C.; Mayer, J.; Ben Neriah, D.; Cousineau, S.; Falls, T.; Jennings, V.A.; Boileau, M.; Bellamy, D.; et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015, 21, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Gayral, M.; Lulka, H.; Hanoun, N.; Biollay, C.; Sèlves, J.; Vignolle-Vidoni, A.; Berthommé, H.; Trempat, P.; Epstein, A.L.; Buscail, L.; et al. Targeted oncolytic herpes simplex virus type 1 eradicates experimental pancreatic tumors. Hum. Gene Ther. 2015, 26, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Aprahamian, M.; Grekova, S.P.; Hajri, A.; Leuchs, B.; Giese, N.A.; Dinsart, C.; Herrmann, A.; Balboni, G.; Rommelaere, J.; et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin. Cancer Res. 2009, 15, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Réjiba, S.; Bigand, C.; Parmentier, C.; Masmoudi, A.; Hajri, A. Oncosuppressive suicide gene virotherapy “PVH1-yCD/5-FC” for pancreatic peritoneal carcinomatosis treatment: NFκB and Akt/PI3K involvement. PLoS ONE 2013, 8, e70594. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Huesing, J.; Rommelaere, J.; Schlehofer, J.R.; Leuchs, B.; Dahm, M.; Krebs, O.; von Knebel Doeberitz, M.; Huber, B.; Hajda, J. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 2012, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Leoni, A.L.; Pohlmeyer-Esch, G.; Loebhard, S.; Leuchs, B.; Hoefer, C.; Jochims, K.; Dahm, M.; Huber, B.; Rommelaere, J.; et al. Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp. Med. 2015, 65, 36–45. [Google Scholar] [PubMed]
- Breitbach, C.J.; Lichty, B.D.; Bell, J.C. Oncolytic Viruses: Therapeutics with an Identity Crisis. EBioMedicine 2016, 9, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Vassaux, G.; Angelova, A.; Baril, P.; Midoux, P.; Rommelaere, J.; Cordelier, P. The Promise of Gene Therapy for Pancreatic Cancer. Hum. Gene Ther. 2016, 27, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Gilly, F.N.; Beaujard, A.; Bienvenu, J.; Trillet Lenoir, V.; Glehen, O.; Thouvenot, D.; Malcus, C.; Favrot, M.; Dumontet, C.; Lombard-Bohas, C.; et al. Gene therapy with Adv-IL-2 in unresectable digestive cancer: Phase I-II study, intermediate report. Hepatogastroenterology 1999, 46, 1268–1273. [Google Scholar] [PubMed]
- Mulvihill, S.; Warren, R.; Venook, A.; Adler, A.; Randlev, B.; Heise, C.; Kirn, D. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: A phase I trial. Gene Ther. 2001, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M.; Hoffmeyer, A.; Kröger, J.; Freund, M.; Hain, J.; Holle, A.; Karle, P.; Knöfel, W.T.; Liebe, S.; Müller, P.; et al. Microencapsulated cell-mediated treatment of inoperable pancreatic carcinoma. Lancet Lond. Engl. 2001, 357, 1591–1592. [Google Scholar] [CrossRef]
- Salmons, B.; Löhr, M.; Günzburg, W.H. Treatment of inoperable pancreatic carcinoma using a cell-based local chemotherapy: Results of a phase I/II clinical trial. J. Gastroenterol. 2003, 38 (Suppl. S15), 78–84. [Google Scholar] [PubMed]
- Pecher, G.; Häring, A.; Kaiser, L.; Thiel, E. Mucin gene (MUC1) transfected dendritic cells as vaccine: Results of a phase I/II clinical trial. Cancer Immunol. Immunother. CII 2002, 51, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Cornelio, G.H.; Lorenzo, C.C.; Levy, J.P.; Reed, R.A.; Liu, L.; Hall, F.L. First clinical experience using a “pathotropic” injectable retroviral vector (Rexin-G) as intervention for stage IV pancreatic cancer. Int. J. Oncol. 2004, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Mazzolini, G.; Ruiz, J.; Herraiz, M.; Quiroga, J.; Herrero, I.; Benito, A.; Larrache, J.; Pueyo, J.; Subtil, J.C.; et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Mani, S.; Rosemurgy, A.; Nemunaitis, J.; Cunningham, C.; Guha, C.; Bayol, N.; Gillen, M.; Chu, K.; Rasmussen, C.; et al. TNFerade biologic, an adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene: A phase I study in patients with solid tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, G.; Alfaro, C.; Sangro, B.; Feijoó, E.; Ruiz, J.; Benito, A.; Tirapu, I.; Arina, A.; Sola, J.; Herraiz, M.; et al. Intratumoral injection of dendritic cells engineered to secrete interleukin-12 by recombinant adenovirus in patients with metastatic gastrointestinal carcinomas. J. Clin. Oncol. 2005, 23, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim-Schulze, S.; Manson, K.; DeRaffele, G.; Mitcham, J.; Seo, K.S.; Kim, D.W.; Marshall, J. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J. Transl. Med. 2007, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Carlson, S.K.; Foster, N.R.; Lowe, V.; Quevedo, F.; McWilliams, R.R.; Grothey, A.; Jatoi, A.; Alberts, S.R.; Rubin, J. Phase I trial of a pathotropic retroviral vector expressing a cytocidal cyclin G1 construct (Rexin-G) in patients with advanced pancreatic cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: A pilot study of safety, feasibility, and immune activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; Chua, V.S.; Fernandez, L.; Quon, D.; Blackwelder, W.C.; Gordon, E.M.; Hall, F.L. Advanced phase I/II studies of targeted gene delivery in vivo: Intravenous Rexin-G for gemcitabine-resistant metastatic pancreatic cancer. Mol. Ther. 2010, 18, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Kasuya, H.; Sahin, T.T.; Nomura, N.; Kanzaki, A.; Misawa, M.; Shirota, T.; Yamada, S.; Fujii, T.; Sugimoto, H.; et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [PubMed]
- Kubuschok, B.; Pfreundschuh, M.; Breit, R.; Hartmann, F.; Sester, M.; Gärtner, B.; König, J.; Murawski, N.; Held, G.; Zwick, C.; et al. Mutated Ras-transfected, EBV-transformed lymphoblastoid cell lines as a model tumor vaccine for boosting T-cell responses against pancreatic cancer: A pilot trial. Hum. Gene Ther. 2012, 23, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Ohana, P.; Konikoff, F.M.; Leichtmann, G.; Hubert, A.; Appelbaum, L.; Kopelman, Y.; Czerniak, A.; Hochberg, A. Phase 1/2a, dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene Ther. 2012, 19, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Farrell, J.J.; Senzer, N.; Nemunaitis, J.; Rosemurgy, A.; Chung, T.; Hanna, N.; Chang, K.J.; Javle, M.; Posner, M.; et al. EUS or percutaneously guided intratumoral TNFerade biologic with 5-fluorouracil and radiotherapy for first-line treatment of locally advanced pancreatic cancer: A phase I/II study. Gastrointest. Endosc. 2012, 75, 332. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A Live-attenuated Listeria Vaccine (ANZ-100) and a Live-attenuated Listeria Vaccine Expressing Mesothelin (CRS-207) for Advanced Cancers: Phase 1 Studies of Safety and Immune Induction. Clin. Cancer Res. 2012, 18, 858. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of Ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. Hagerstown Md 1997 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: A phase 2 study. J. Gastrointest. Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.M.; Wild, A.T.; Wang, H.; Tran, P.T.; Chang, K.J.; Taylor, G.E.; Donehower, R.C.; Pawlik, T.M.; Ziegler, M.A.; Cai, H.; et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: Final results. J. Clin. Oncol. 2013, 31, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Löhr, J.M.; Haas, S.L.; Kröger, J.C.; Friess, H.M.; Höft, R.; Goretzki, P.E.; Peschel, C.; Schweigert, M.; Salmons, B.; Gunzburg, W.H. Encapsulated cells expressing a chemotherapeutic activating enzyme allow the targeting of subtoxic chemotherapy and are safe and efficacious: Data from two clinical trials in pancreatic cancer. Pharmaceutics 2014, 6, 447–466. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, L.K.; Shirley, L.A.; Chung, V.M.; Marsh, C.L.; Walker, J.; Coyle, W.; Marx, H.; Bekaii-Saab, T.; Lesinski, G.B.; Swanson, B.; et al. Gene-mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol. Immunother. CII 2015, 64, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Vernejoul, F.; Cambois, G.; Lulka, H.; Hanoun, N.; Dufresne, M.; Meulle, A.; Vignolle-Vidoni, A.; Ligat, L.; et al. First-in-man Phase 1 Clinical Trial of Gene Therapy for Advanced Pancreatic Cancer: Safety, Biodistribution, and Preliminary Clinical Findings. Mol. Ther. 2015, 23, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized Phase 2 Trial of the Oncolytic Virus Pelareorep (Reolysin) in Upfront Treatment of Metastatic Pancreatic Adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Virus | Insertion Capacity | Target Cells | Delivery | Transgene Expression | Level of Expression | Pre-existant Immunity | Bio-Safety |
|---|---|---|---|---|---|---|---|
| AdV | 35 kb | Dividing or non dividing | Ex vivo or In situ | Transient | High | Yes | Immunogenic, inflammation No integration |
| AAV | 4.8 kb | Dividing or non dividing | Ex vivo or In situ | Stable | Moderate | Yes | Mutational integration |
| Retrov | 8 kb | Dividing | Ex vivo or In situ | Stable | Moderate | No | Mutational integration |
| LentiV | 10 kb | Dividing or non dividing | Ex vivo or In situ | Stable | High | No | Mutational integration, recombination with WT HIV |
| HSV | 30 kb | Dividing or non dividing | Ex vivo or In situ | Transient | High | Yes | Mutational integration, neurotoxicity |
| Pox | 25 kb | Dividing or non dividing | In vivo or In situ | Stable | High | No | Immunogenic, Adjuvant to vaccination |
| SV40 | 5 kb | Dividing or non dividing | Ex vivo or In situ | Stable | Moderate | No | Mutational integration |
| Strategy | Detailed Genes |
|---|---|
| Gene transfer | Tumor suppressor genes (P16, p21, pRb, p53, Smad4/DPC4…) Ani-angiogenic genes (endostatin, thrombospondin-1, angiostatin, Matrix metalloproteinase inhibitors, somatostatin receptor sst2 subtype, arrestin…) Apoptosis related genes (TRAIL, TNF) Suicide genes |
| Gene invalidation | antisens therapy (Kras, HER-2/ErbB-2, MDR-1) MicroRNA (MIR-21) |
| Active immunotherapy | Interleukin expression Cytokine expression |
| Vaccination | Pulsed dendritic cells DNA, peptide, engineered cells and bacteria |
| Adoptive immunotherapy | CAR-T cells (targeting mesothelin or MUC-1) |
| Author, Year [Reference] | Phase, (Patient Number-StAge III–IV) | Route | Vector/Strategy | Results |
|---|---|---|---|---|
| Gilly et al., 1999 [120] | I/II (7) | IT, during surgery | AdV/Interleukin 2 | Well tolerated, 1 tumor regression |
| Mulvihill et al., 2001 [121] | I (3) | IT (CT, surgery) | AdV/ONYX-015 | No objective response |
| Löhr et al., 2001 Salmons et al.,2003 [122,123] | I/II (14) | angiography | Lipofectamine/Cyto. P450 (*) | Well tolerated, 2 PR, 12 SD, OS survival: 10 months |
| Pecher et al., 2002 [124] | I/II (10) | SC | Cationic liposome/dendritic cells transfected with MUC1 cDNA | No side effects, 9 PD, 1 SD |
| Hecht et al., 2003 [107] | I/II (21) | IT, EUS-guided | AdV/ONYX-015 + gemcitabine | 2 duodenal perforations, 4 PR, 6 SD, 11 PD |
| Gordon et al., 2003 [125] | I (3) | Intravenous | Rv/Rexin-G | Well tolerated 2 SD (4, 5 months) 1 PD (21 months) |
| Sangro et al., 2004 [126] | I (7) | IT, EUS-guided | AdV/Interleukin 12 | Well tolerated, SD |
| Senzer et al., 2004 [127] | I (30) | IT EUS-and CT-guided | TNFerade | 22% fever, 19% chills 5 complete responses and 16 PR |
| Mazzolini et al., 2005 [128] | I (11) | IT, EUS-guided | Autologous dendritic cells transfected with an Adv encoding interleukin-12 gene | Well tolerated, 1 PR, 2 SD, 8 PD |
| Kaufman et al., 2007 [129] | 1 (10) | ID | Vaccination with Vaccinia and Pox virus expressing CEA MUC-1) and co-stimulatory molecules | 10/10 Antibody responses against vaccinia virus 6, 3 months of OS |
| Galanis et al., 2008 [130] | I (12) | IV | Rv/Rexin-G | Well tolerated, no evidence of anti-tumor activity |
| Laheru et al., 2008 [131] | I (50) | ID | Plasmid/GVAX Arm A: GVAX alone Arm B: GVAX with cyclophosphamide | Well tolerated, Median survival : A: 2.3 months B: 4.3 months |
| Chawla et al., 2010 [132] | I/II (9) | IV | Rv/Rexin-G at two dosages | Well tolerated, 8 SD, 1 PR, Median overall survival between 4.3 and 9.2 months |
| Nakao et al., 2011 [133] | I (6) | IT during surgery | HF10 oncolytic herpes virus | Well tolerated, 3 SD, 1 PR, 2 PD |
| Lutz et al., 2011 [134] | II (60) | ID | Plasmid/GVAX with chemoradiation | Well tolerated Median disease free survival 17.3 months (resected tumors) |
| Kubusho et al., 2012 [135] | I (7 with mutated KRAS) | SC | EBV/Peripheral blood lymphocytes genetically modified with an episomal EBV expressing Ras mutant | No acute EBV infection 4 SD, 6 T cell response |
| Hanna et al., 2012 [136] | I/Iia (9) | IT EUS- and CT-guided | Plasmid / expression of diphtheria-toxin gene | Well tolerated, 3 PR |
| Hecht et al., 2012 [137] | I (50) | IT EUS- and CT-guided | AdV/TNFerade with chemoradiation (5 FU) | 21 mild adverse events 1 complete response, 7 PR, 12 SD, 19 PD Median OS: 10 months |
| Le et al., 2012 [138] | I (26) | IV (GI metastatic cancers included) | Attenuated listeria vaccine expressing (17) or not (9) mesotheline | Immune response, OS > 15 months in 10 patients |
| Le et al., 2013 [139] | SC | A: Ipilimumab alone B: GVAX + Ipilimumab | 20% Grade 3/4 immune-relate adverse events A and B : SD OS: 3.2 vs 5.7 (NS) 1 year survival 7 Vas 27% | |
| Hardacre et al., 2013 [140] | II (70 after tumor resection) | SC | Algenpantucel-L + gemcitabine + 5FU | 12% induration at the injection site one year OS: 86% |
| Herman et al., 2013 [141] | III (304) | IT EUS- and CT-guided | AdV/TNFerade + Chemoradiation Vs chemoradiation | More grade 1 to 2 AE in the TNFerade + chemoradiation arm OS: 10 months, both arms |
| Löhr et al., 2014 [142] | II (13) | angiography | Lipofectamine/Cyto. P450 (*) | Well tolerated 4 PR, 4 PD, 5 SD OS: 9.5 months |
| Aguilar et al., 2015 [143] | I (24) | IT EUS- and CT-guided | AdV/HSV thymidine kinase Arm A borderline: HSK −TK + valacyclovir Arm B non resectable: HSV − TK + valacyclovir + chemoradiation | Well tolerated Median OS: 10 months in Arm A and 12 months in arm B with 25% of RECIST response. |
| Buscail et al., 2015 [144] | I (22) | IT EUS- guided | Complexed plasmid/CYL-02 + gemcitabine | Well tolerated, 12 SD, OS in non metastatic patients 12.6 months |
| Golan et al., 2015 [145] | I/IIa (15) | IT EUS- and CT-guided | IT placement of SiG12-LODER® + gemcitabine | 90% minimal AE 2 PR, 10 SD, 2 PR OS: 15 months |
| Le et al., 2015 [119] | II (90) | SC | GVAX + CRS 2017 (Arm A) vs GVAX alone (Arm B) | Local reactions 77%, general minor AE 53–62% OS: 6.1 months in arm A Vs 3.9 months in arm B |
| Noonan et al., 2016 [146] | II (73) | IV | Arm A: Reolysin + paclitaxel + carboplatin Arm B: paclitaxel + carboplatin | Well tolerated No difference I term of PFS and OS between the two arms |
| Identification Number Date of Start Trial Stage | Phase, (Patient Number) | Route | Vector/Strategy |
|---|---|---|---|
| NCT00711997 August 2009 Completed | I/II (9) | IT CT- or US- or EUS-guided | jetPEI/DTA-H19 |
| NCT00669734 February 2010 Active, not recruiting | I (18) | IT or SC | Lowlpox and Vaccinia virus/PANVAC-V+ PANVAC-F + GM-CSF) |
| NCT01088789 April 2010 Recruiting | II (72) | ID | PANC 10.05 pcDNA-1/GM-Neo and PANC 6.03 pcDNA-1 neo vaccine (allogenic pancreatic tumor cell vaccine transfected with the GM-CSF gene) +/− cyclophosphamide |
| NCT01191684 October 2011 Completed | I (2) | SC | Vaccinia virus/Vaccinia virus ankara vaccine expressing p53 |
| NCT01583686 April 2012 Recruiting | I/II (136) | IV | Rv/Anti-mesothelin CAR-T cell |
| NCT01836432 May 2013 Active, not recruiting | III (302) | ID | Rv/Algenpantucel-L +/− chemotherapy (Folfirinox or Gemcitabine + Nab-paclitaxel) |
| NCT02239861 September 2014 Recruiting | I (18) | IV | Tumor-associated antigen (TAA)-specific cytotoxic T lymphocytes 5 common TAAs: NY-ESO-1, MAGEA4, PRAME, Survivin and SSX. |
| NCT02340117 January 2015 Recruiting | II (28) | IV | Cationic liposome/SGT-53 + Gemcitabine + Nab-Paclitaxel |
| NCT02416466 April 2015 Active, not recruiting | I (8) | Percutan-eous hepatic artery infusion | SIR-Spheres microspheres/Anti-CEA CAR-T cells |
| NCT02465983 May 2015 Active, not recruiting | I (12) | IV | CART-meso-19 T cells + Cyclophosphamide |
| NCT02432963 November 2015 Recruiting | I (12) | ID | Vaccinia Ankara/Modified Vaccinia Ankara vector expressing full length wild type human p53 + Pembrolizumab |
| NCT02806687 June 2016 Recruiting | II (100) | IT EUS-guided | JetPEI/CYL-02 + Gemcitabine |
| NCT02576665 July 2016 Recruiting | I (26) | IV or IT | Rv/Toca 511 + Toca FC |
| NCT02894944 August 2016 Recruiting | I (9) | AdV/Theragene + Chemotherapy | |
| NCT02705196 November 2016 Not yet recruiting | I/II (26) | IT EUS- or US-guided | AdV/LOAd703 + Gemcitabine + Nab-paclitaxel |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rouanet, M.; Lebrin, M.; Gross, F.; Bournet, B.; Cordelier, P.; Buscail, L. Gene Therapy for Pancreatic Cancer: Specificity, Issues and Hopes. Int. J. Mol. Sci. 2017, 18, 1231. https://doi.org/10.3390/ijms18061231
Rouanet M, Lebrin M, Gross F, Bournet B, Cordelier P, Buscail L. Gene Therapy for Pancreatic Cancer: Specificity, Issues and Hopes. International Journal of Molecular Sciences. 2017; 18(6):1231. https://doi.org/10.3390/ijms18061231
Chicago/Turabian StyleRouanet, Marie, Marine Lebrin, Fabian Gross, Barbara Bournet, Pierre Cordelier, and Louis Buscail. 2017. "Gene Therapy for Pancreatic Cancer: Specificity, Issues and Hopes" International Journal of Molecular Sciences 18, no. 6: 1231. https://doi.org/10.3390/ijms18061231
APA StyleRouanet, M., Lebrin, M., Gross, F., Bournet, B., Cordelier, P., & Buscail, L. (2017). Gene Therapy for Pancreatic Cancer: Specificity, Issues and Hopes. International Journal of Molecular Sciences, 18(6), 1231. https://doi.org/10.3390/ijms18061231