
Activation of Magnesium Lignosulfonate and Kraft Lignin: Influence on the Properties of Phenolic Resin-Based Composites for Potential Applications in Abrasive Materials

 ,
, 




Abstract
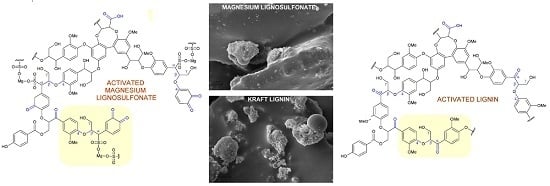
:
1. Introduction
2. Results and Discussion
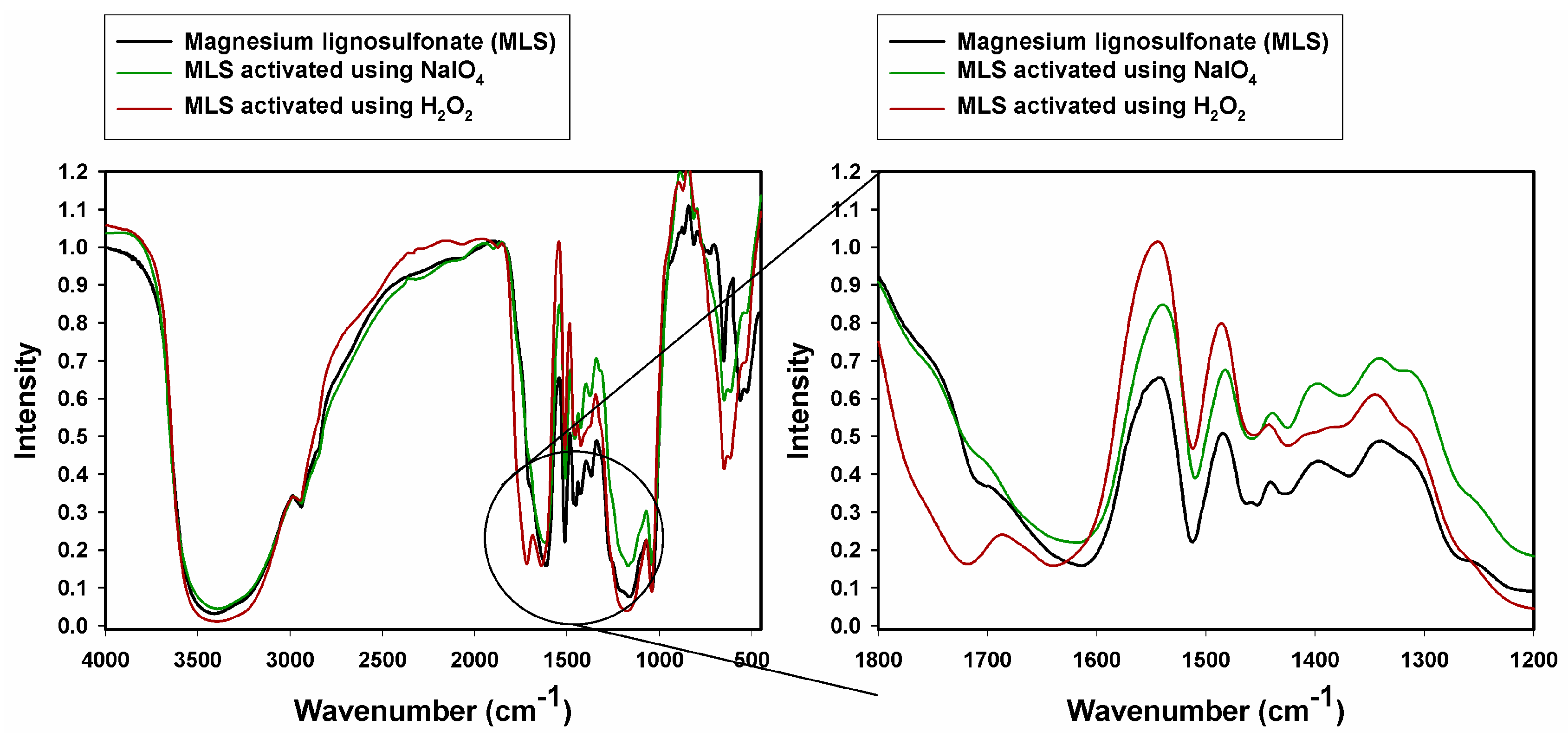
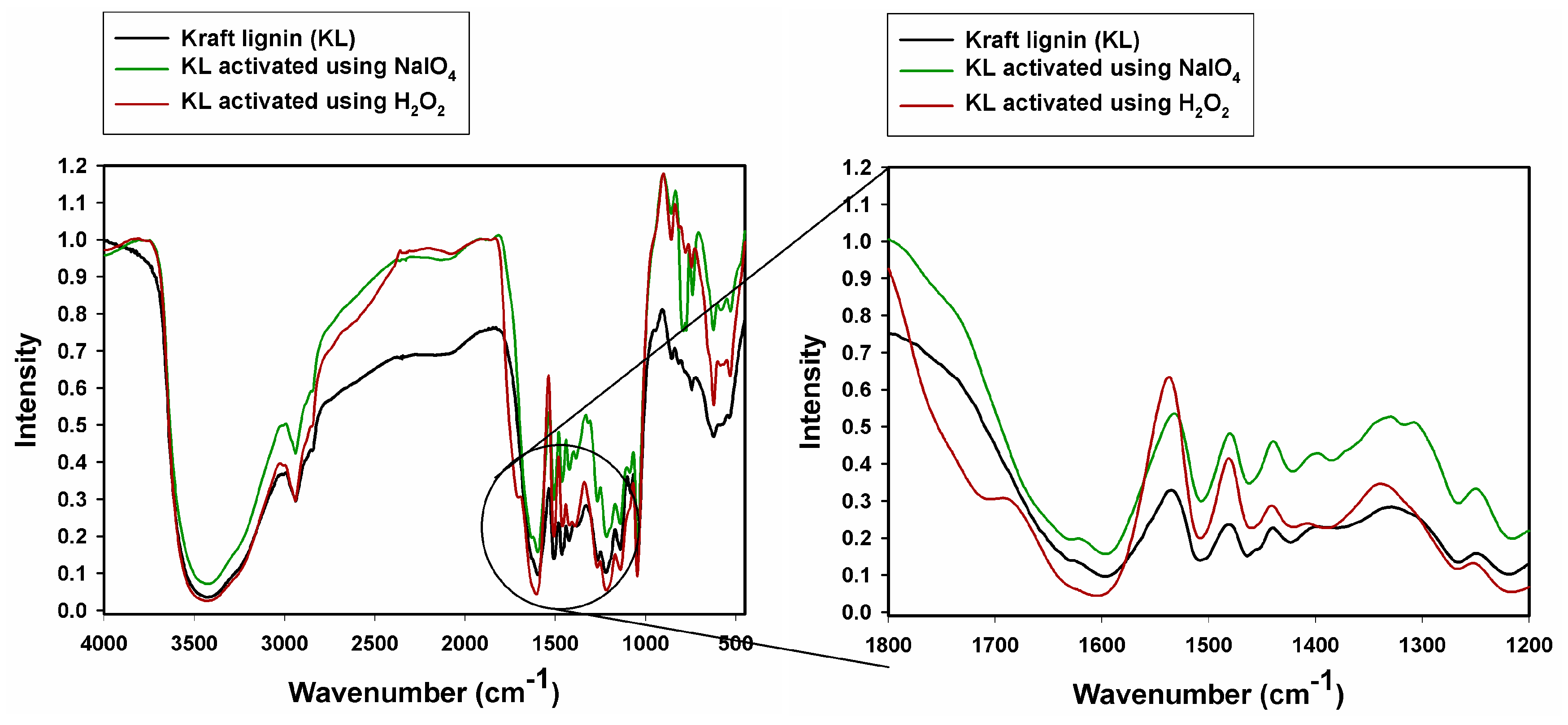
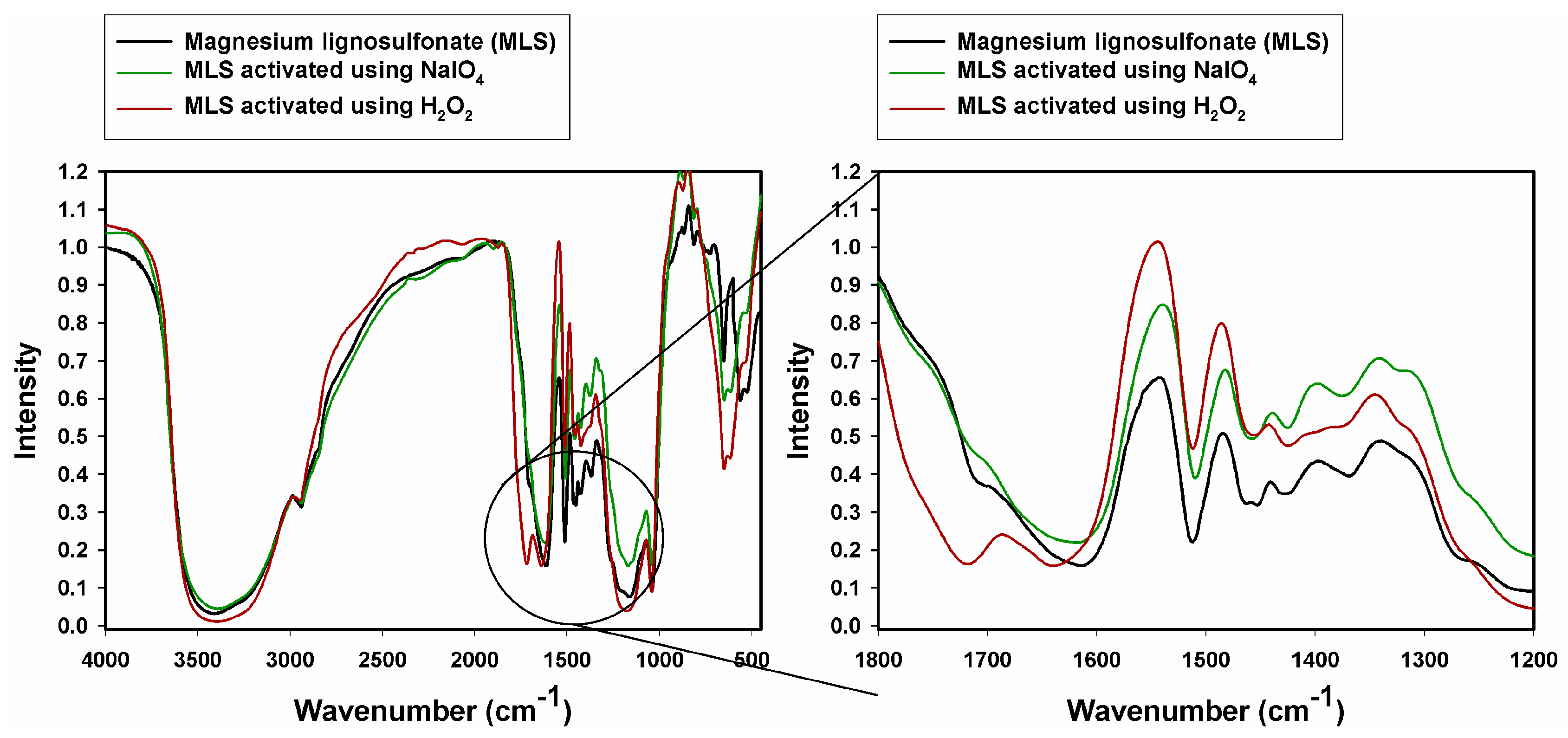
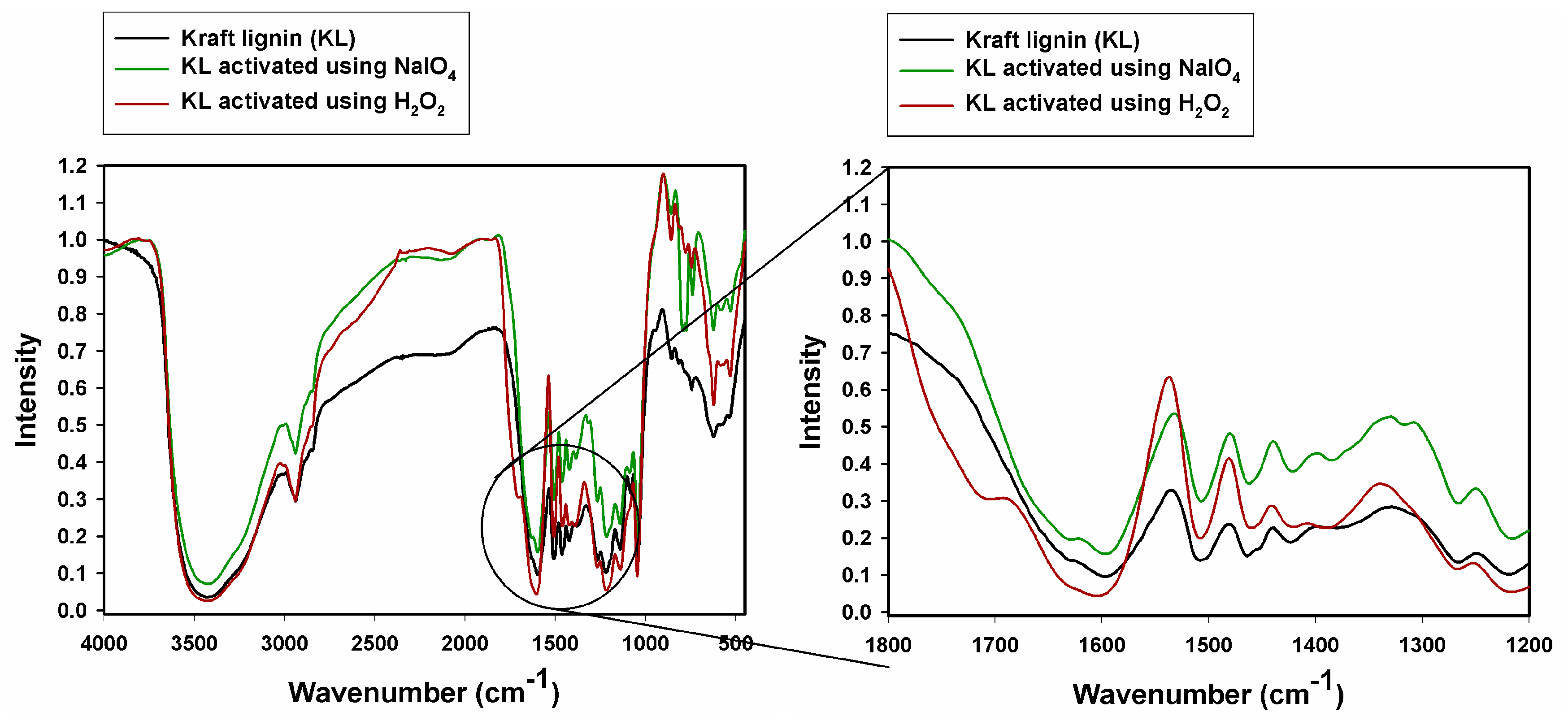
2.1. Fourier Transform Infrared Spectroscopy
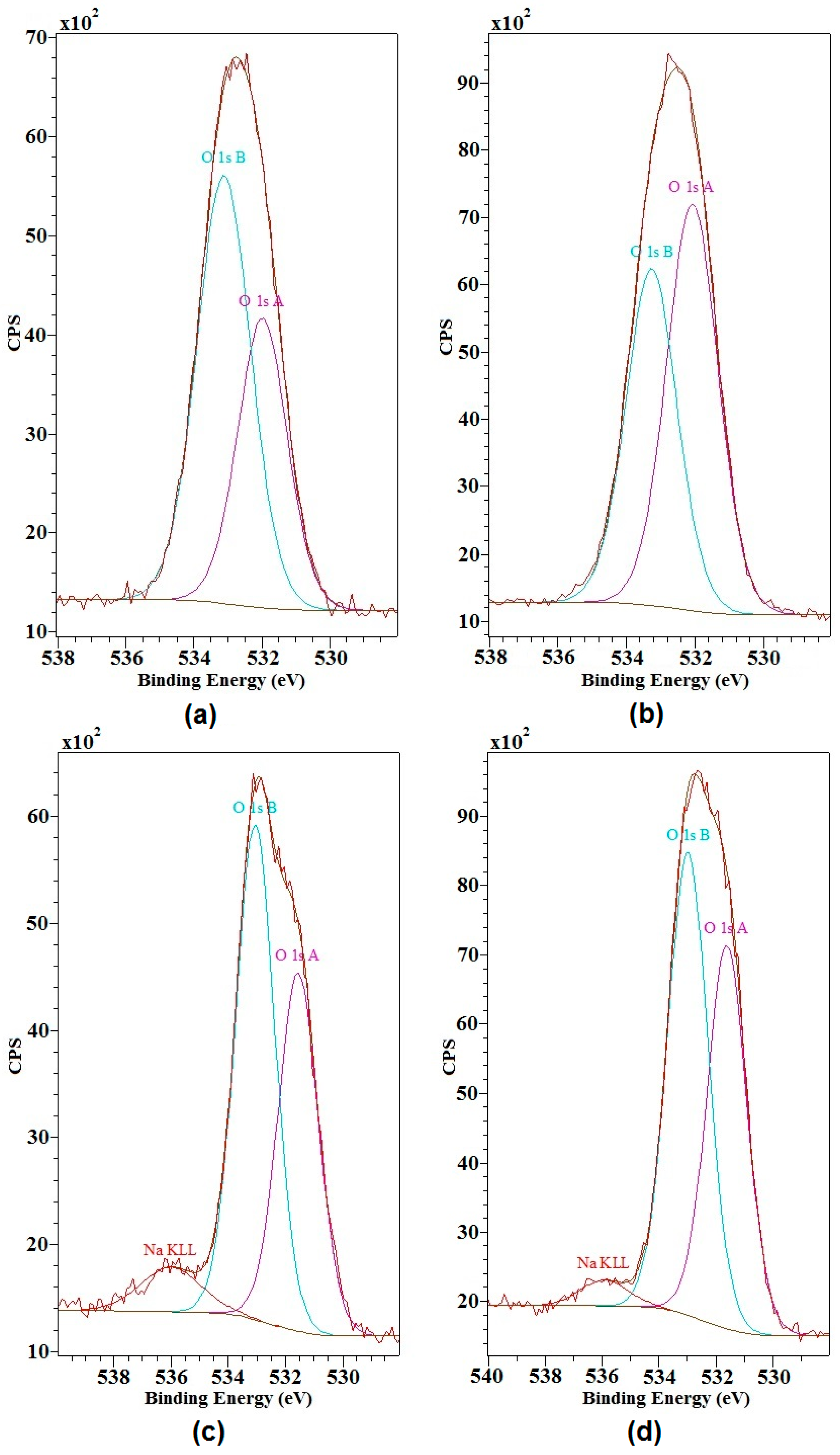
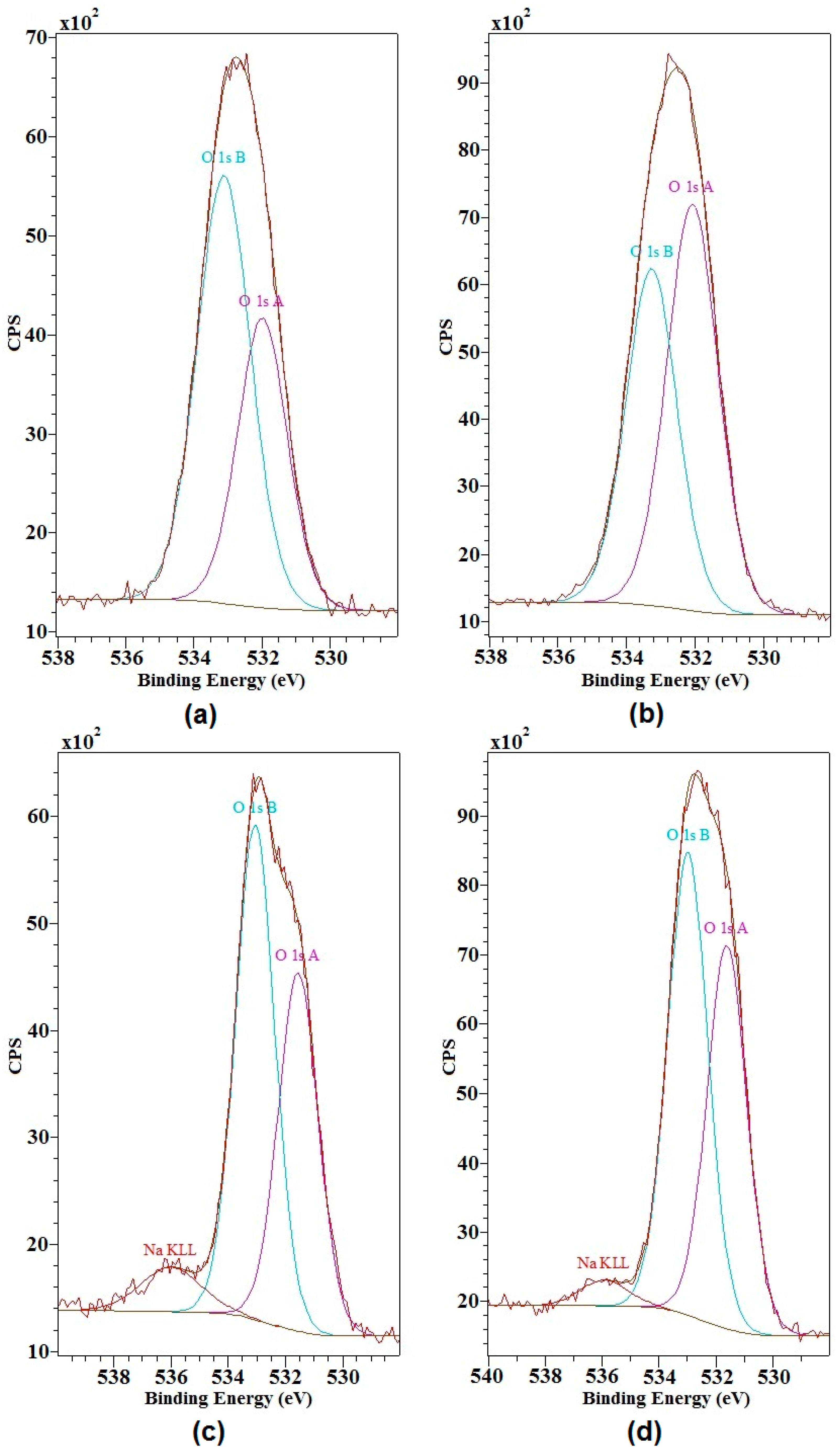
2.2. X-ray Photoelectron Spectroscopy
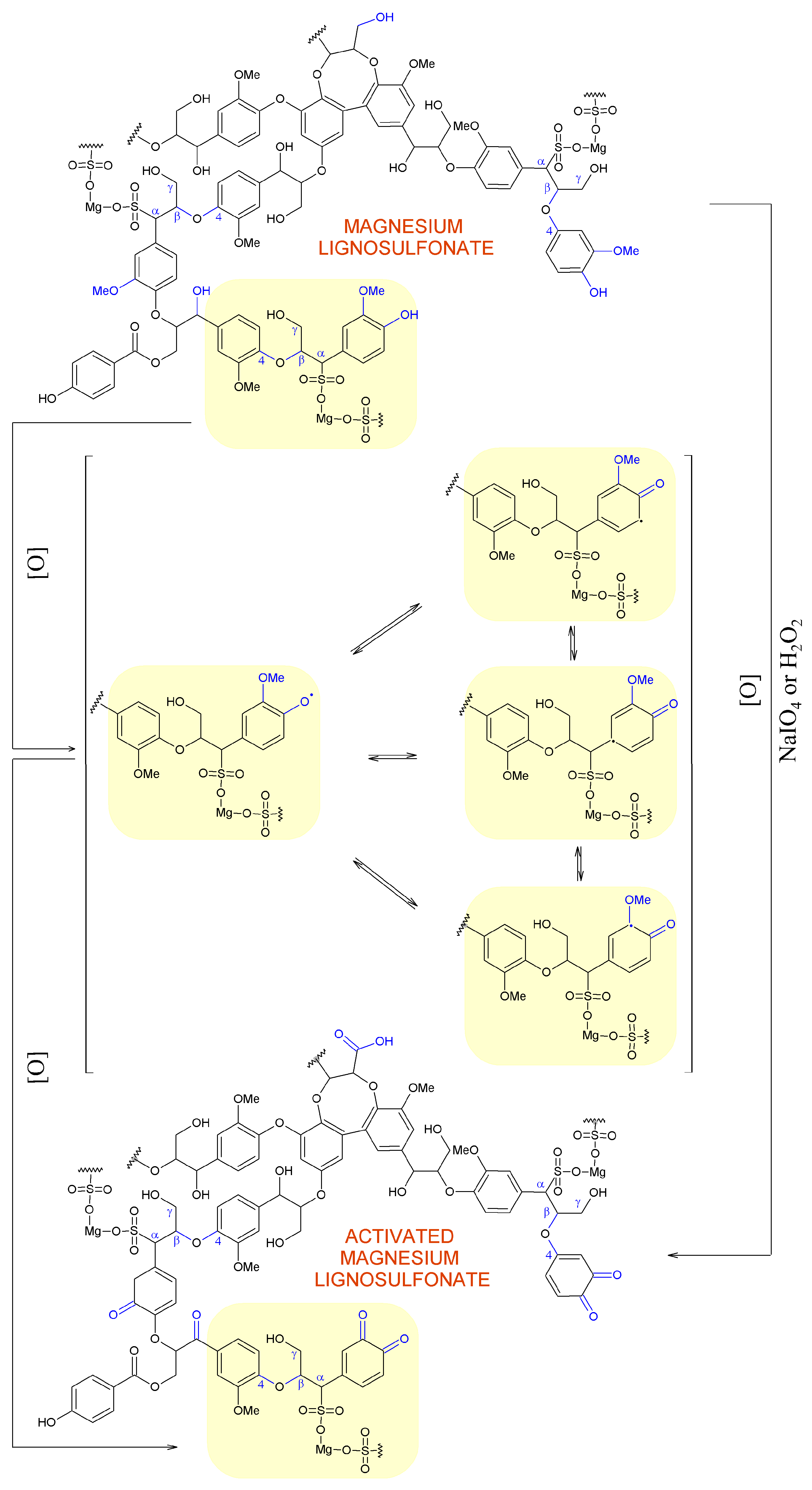
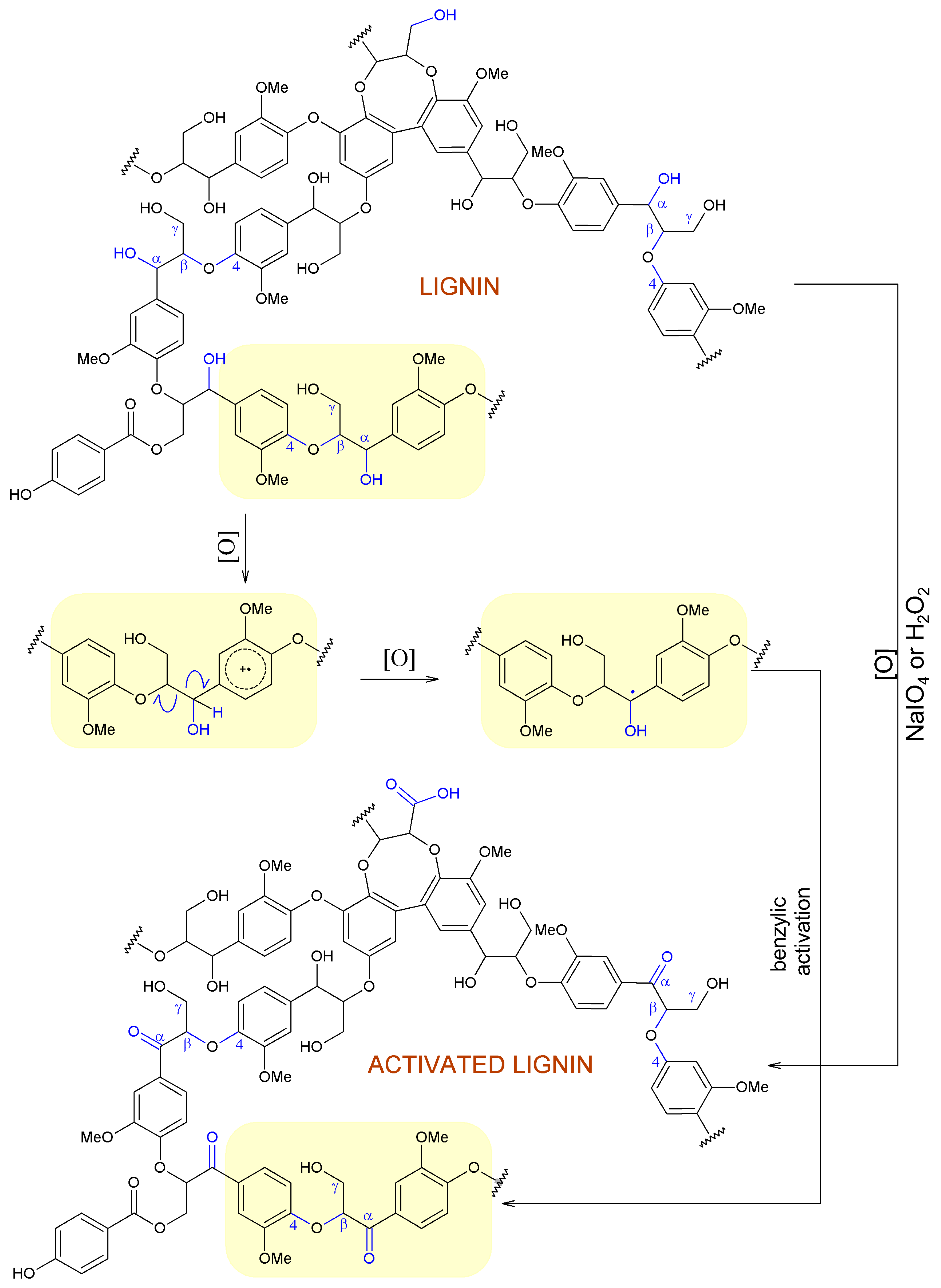
2.3. Hypothetical Mechanisms of Activation of Lignin and Lignosulfonate
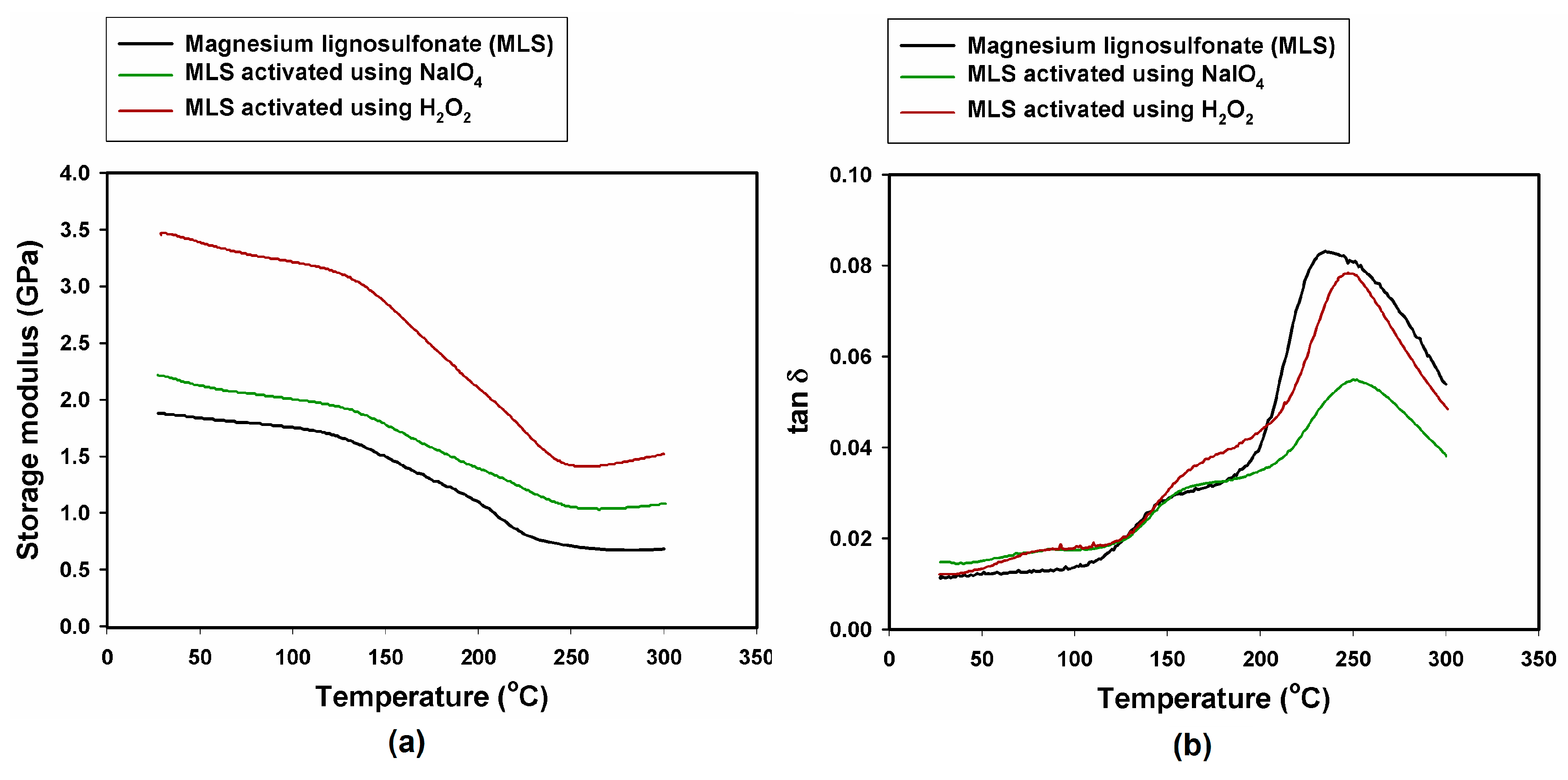
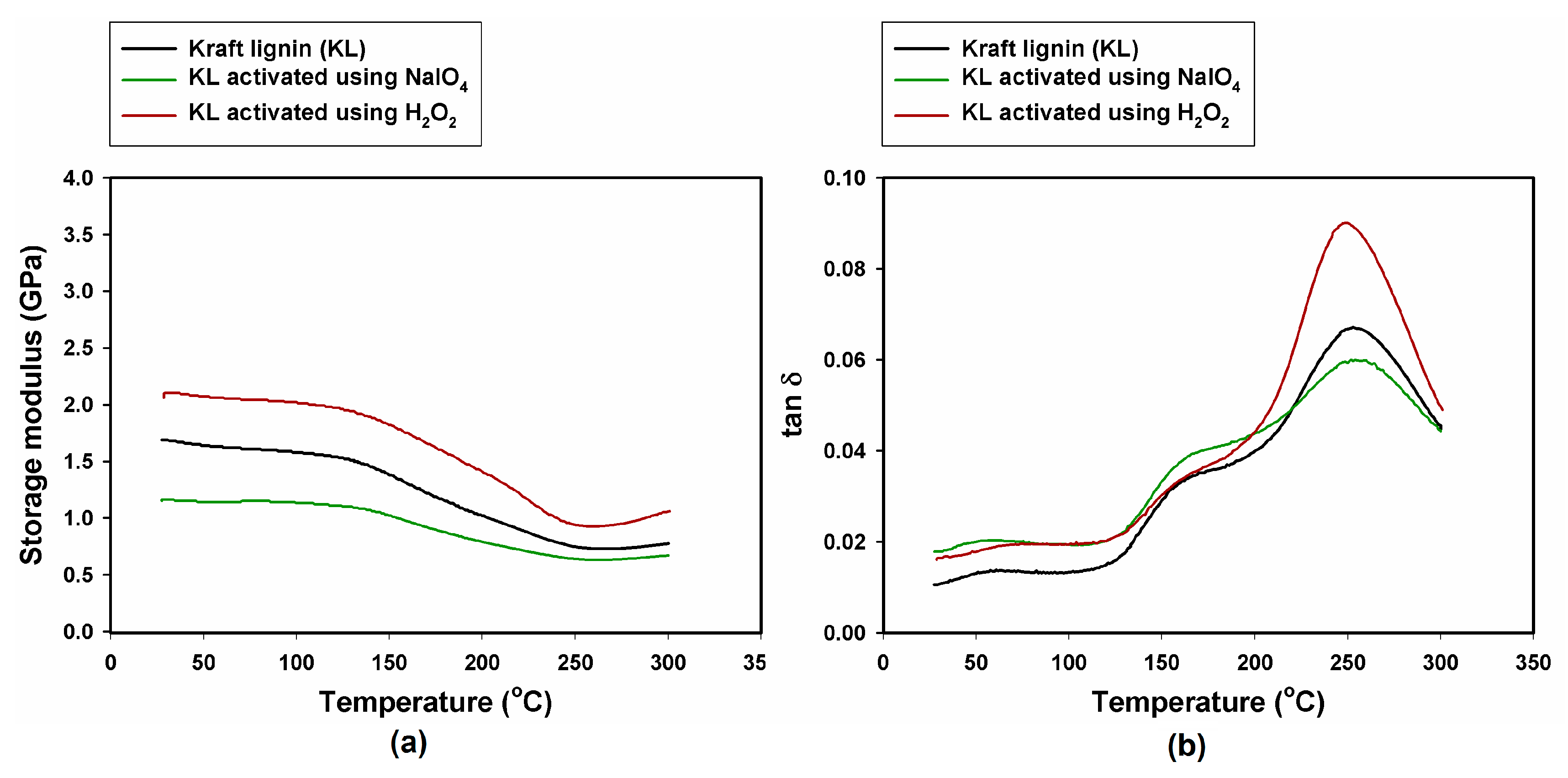
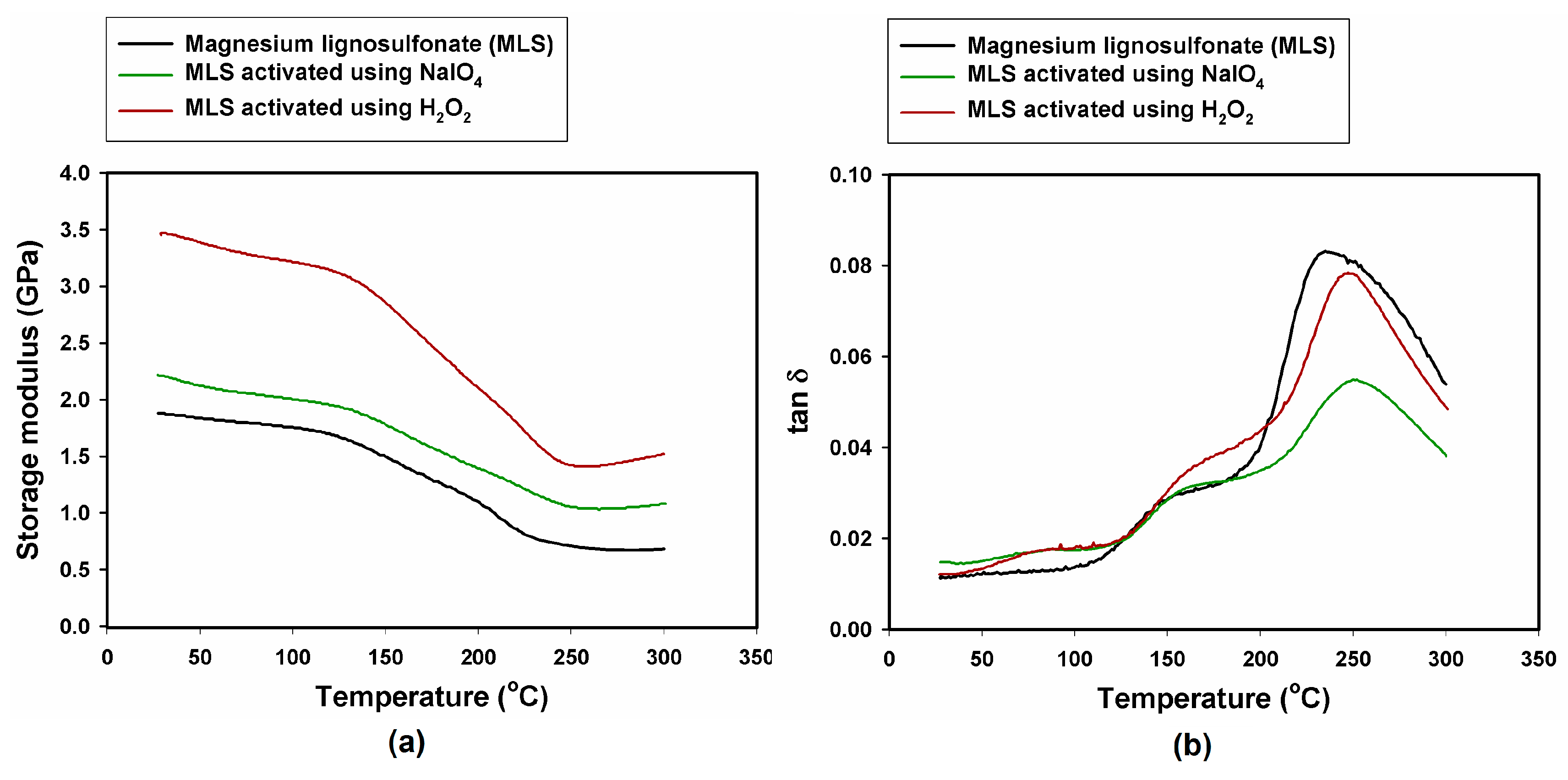
2.4. Dynamic Mechanical-Thermal Properties
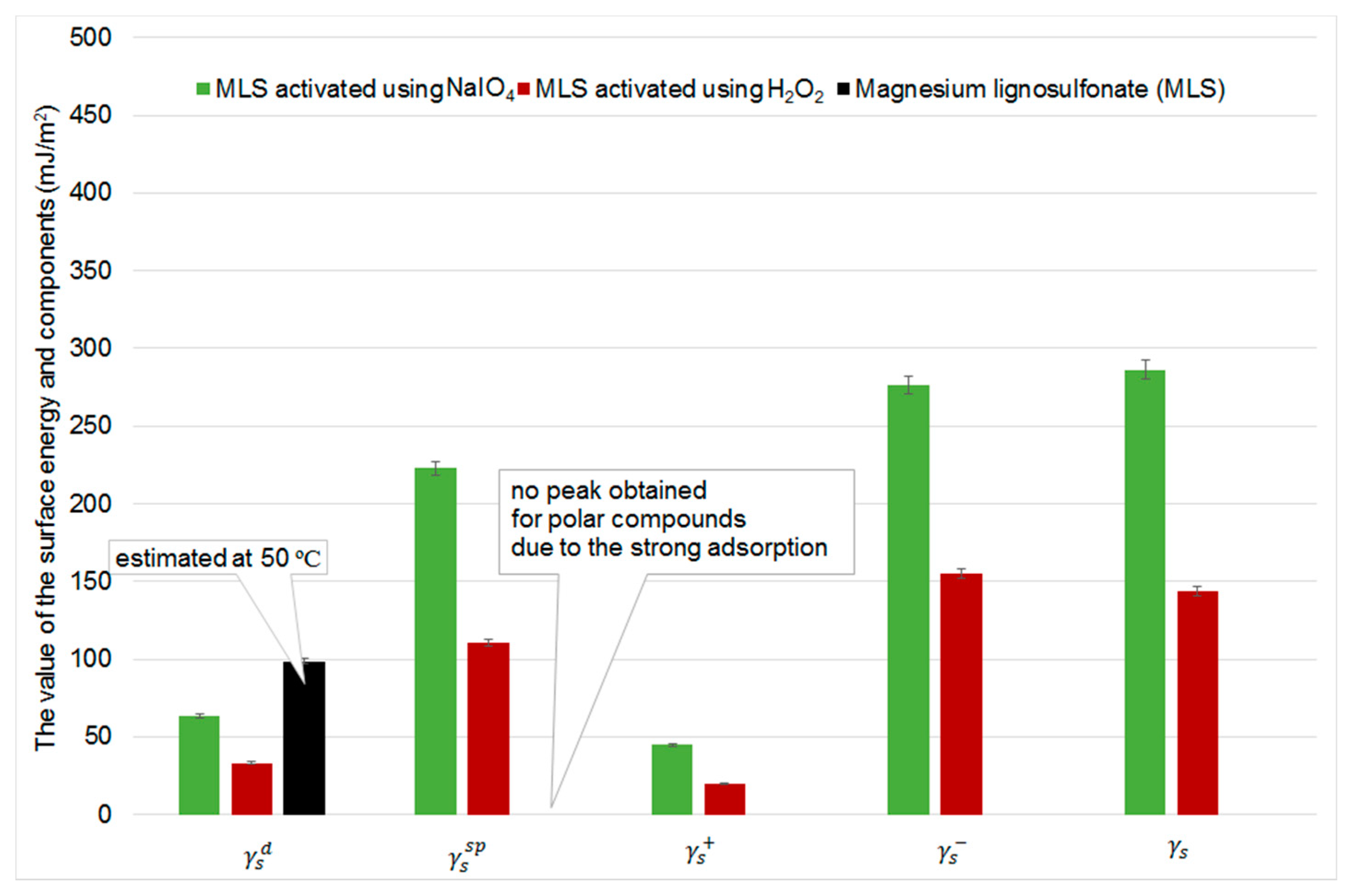
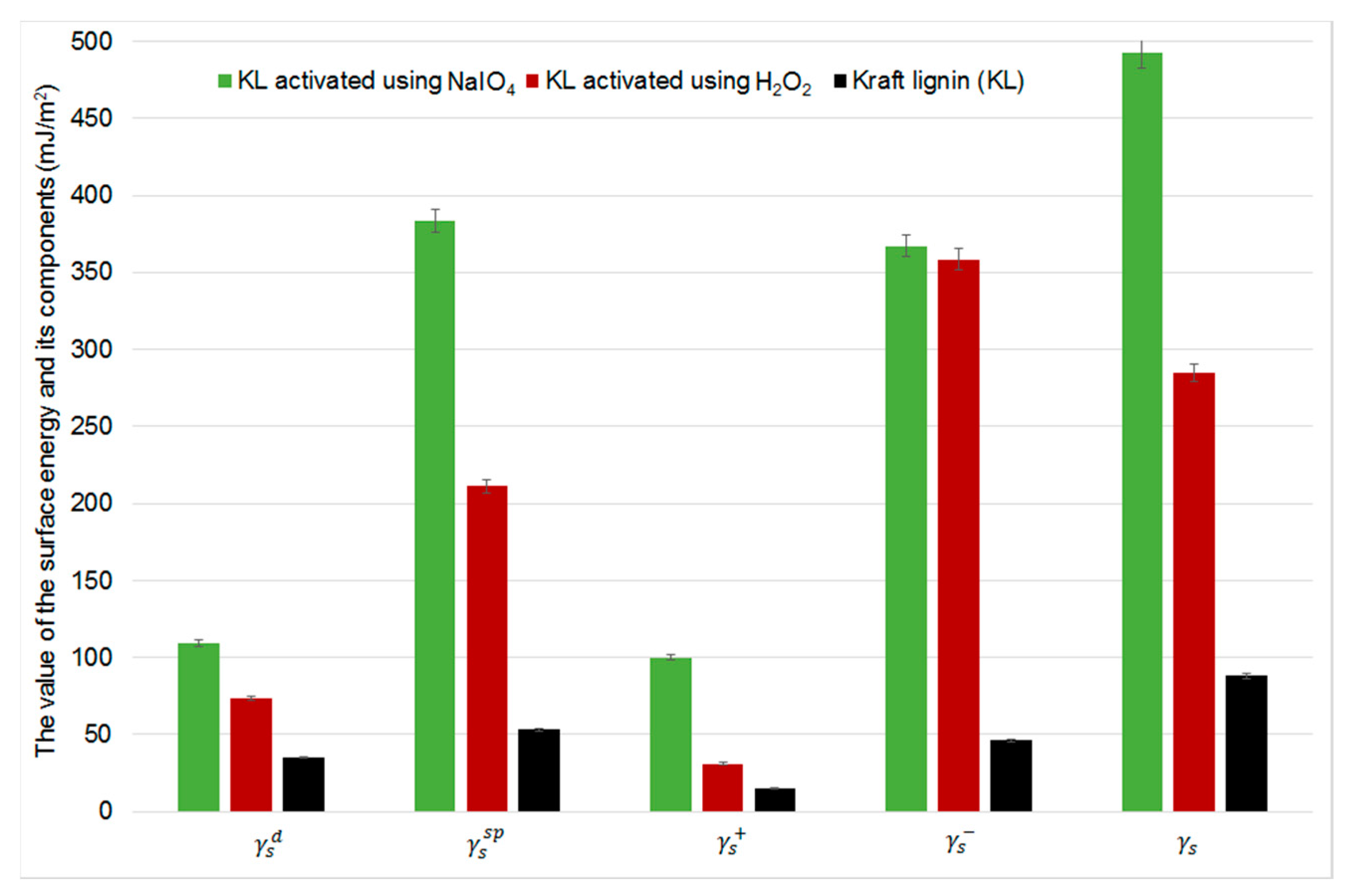
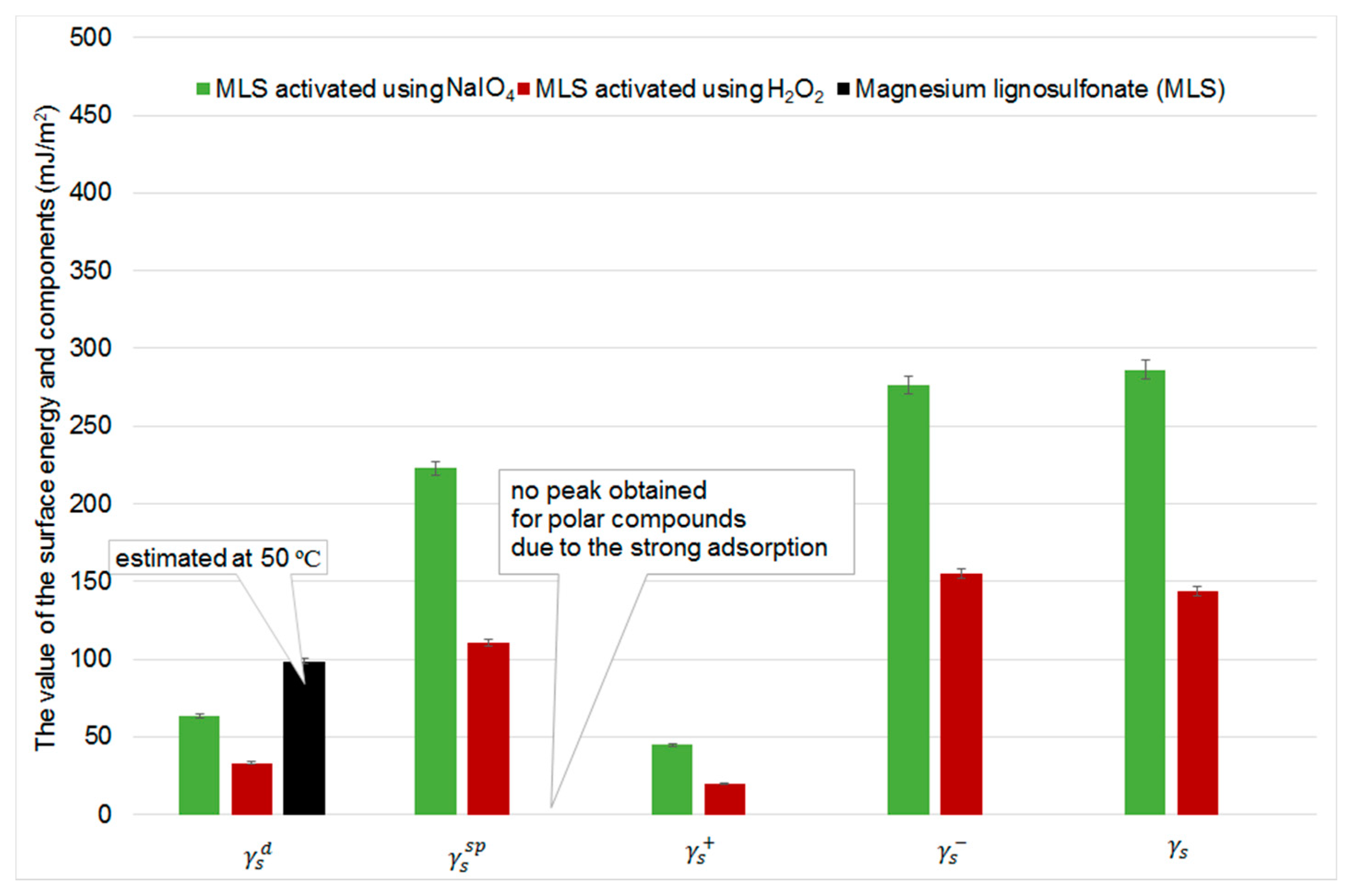
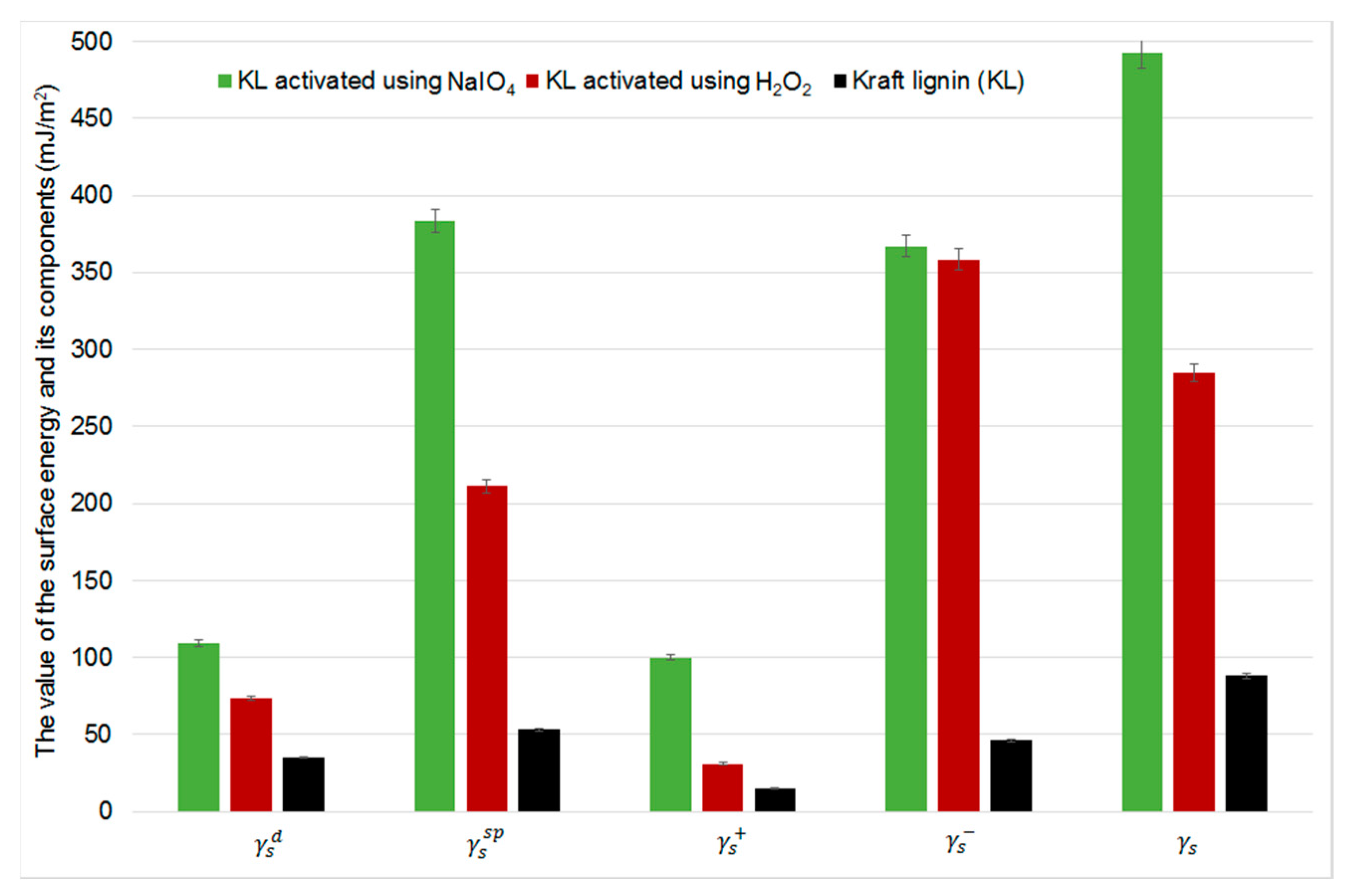
2.5. Inverse Gas Chromatography
2.6. Scanning Electron Microscopy
3. Materials and Methods
3.1. Modification of Kraft Lignin and Magnesium Lignosulfonate
3.2. Preparation of Composites
3.3. Material Analysis
3.3.1. Fourier Transform Infrared Spectroscopy
3.3.2. X-ray Photoelectron Spectroscopy
3.3.3. Dynamic Mechanical-Thermal Properties
3.3.4. Inverse Gas Chromatography
Determination of Dispersive Component of the Surface Free Energy
Determination of Specific Component of Surface Free Energy
3.3.5. Scanning Electron Microscopy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Calvo-Flores, F.G.; Dobado, J.A.; Isac-García, J.; Martín-Martínez, F.J. Lignin and Lignans as Renewable Raw Materials; Wiley: Chichester, UK, 2015. [Google Scholar]
- Biermann, C.J. Handbook of Pulping and Papermaking, 2nd ed.; Academic Press: London, UK, 1996. [Google Scholar]
- Fache, M.; Boutevin, B.; Caillol, S. Vanillin production from lignin and its use as a renewable chemical. ACS Sustain. Chem. Eng. 2016, 4, 35–46. [Google Scholar] [CrossRef]
- Waldron, K. Advances in Biorefineries: Biomass and Waste Supply Chain Exploitation; Woodhead Publishing: London, UK, 2014. [Google Scholar]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2016, 153, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Heitner, C.; Dimmel, D.R.; Schmidt, J.A. Lignin and Lignans: Advances in Chemistry; CRC Press: London, UK, 2010. [Google Scholar]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crop. Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Dilling, P. Sulfonation of Lignins. U.S. Patent A5049661, 19 January 1989. [Google Scholar]
- Lundquist, K.; Kirk, T.K. Acid degradation of lignin. IV. Analysis of lignin acidolysis products by gas chromatography, using trimethylsilyl derivatives. Acta Chem. Scand. 1971, 25, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, C.; Monties, B.; Rolando, C. Structural studies of lignins: Estimation of arylglycerol aryl ether bonds by thioacidolysis. C. R. Acad. Sci. 1984, 299, 441–444. [Google Scholar]
- Rowell, R.M. Acetylation of wood. For. Prod. J. 2006, 56, 4–12. [Google Scholar]
- Rowell, R.M. Chemical modification of wood: A short review. Wood Mater. Sci. Eng. 2006, 1, 29–33. [Google Scholar] [CrossRef]
- Siochi, E.J.; Ward, T.C.; Haney, M.A.; Mahn, B. The absolute molecular weight distribution of hydroxypropylated lignins. Macromolecules 1990, 23, 1420–1429. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, Y.; Liu, D.; Petrus, L. Qualitative analysis of products formed during the acid catalyzed liquefaction of bagasse in ethylene glycol. Bioresour. Technol. 2007, 98, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, F.; Dubois, M.; Charlet, K.; Frezet, L.; Béakou, A. Direct fluorination applied to wood flour used as a reinforcement for polymers. Carbohydr. Polym. 2013, 94, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Charlet, K.; Saulnier, F.; Dubois, M.; Béakou, A. Improvement of wood polymer composite mechanical properties by direct fluorination. Mater. Des. 2015, 74, 61–66. [Google Scholar] [CrossRef]
- Shao, Z.-Q.; Wang, W.-J.; Wang, F.-J.; Wang, J.-X.; Tau, H.-M. Nitration of wood cellulose in HNO3/organic solvent medium. J. Beijing Inst. Technol. 2006, 15, 111–114. [Google Scholar]
- Wang, F.-J.; Shao, Z.-Q.; Feng, L.; Wang, J.-X.; Wang, W.-J.; Li, Y.-H. Nitration of soft wood pulp board. Chin. J. Explos. Propel. 2010, 33, 69–72. [Google Scholar]
- Qin, Y.; Mo, W.; Yu, L.; Yang, D.; Qiu, X. A light-colored hydroxypropyl sulfonated alkali lignin for utilization as a dye dispersant. Holzforschung 2016, 70, 109–116. [Google Scholar] [CrossRef]
- Gierer, J.; Lindeberg, O. Reactions of lignin during sulfate pulping. Part XIX. Isolation and identification of new dimers from a spent sulfate liquor. Acta Chem. Scand. Ser. B 1980, 34, 161–170. [Google Scholar] [CrossRef]
- Travaini, R.; Martín-Juárez, J.; Lorenzo-Hernando, A.; Bolado-Rodríguez, S. Ozonolysis: An advantageus pretreatment for lignocellulosic biomass revisited. Bioresour. Technol. 2016, 199, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Negi, S.; Binod, P.; Larroche, C. Pretreatment of Biomass: Proceses and Technologies, 1st ed.; Elsevier: London, UK, 2015. [Google Scholar]
- Ohra-aho, T.; Gomes, F.J.B.; Colodette, J.L.; Tamminen, T. S/G ratio and lignin structure among Eucalyptus hybrids determined by Py-GC/MS and nitrobenzene oxidation. J. Anal. Appl. Pyrolysis 2013, 101, 166–171. [Google Scholar] [CrossRef]
- Min, D.; Xiang, Z.; Liu, J.; Jameel, H.; Chiang, V.; Jin, Y.; Chang, H.-M. Improved protocol for alkaline nitrobenzene oxidation of woody and non-woody biomass. J. Wood Chem. Technol. 2015, 35, 52–61. [Google Scholar] [CrossRef]
- Katahira, R.; Takano, T.; Kamitakahara, H.; Nakatsubo, F. Alkaline nitrobenzene oxidation of phenolic and non-phenolic dimeric, trimeric and oligomeric β-O-4 lignin model compounds. Cellul. Chem. Technol. 2007, 41, 529–536. [Google Scholar]
- Bose, S.K.; Wilson, K.L.; Francis, R.C.; Aoyama, M. Lignin analysis by permanganate oxidation. Part 1. Native spruce lignin. Holzforschung 1998, 52, 297–303. [Google Scholar] [CrossRef]
- Bose, S.K.; Wilson, K.L.; Hausch, D.L.; Francis, R.C. Lignin analysis by permanganate oxidation. Part 2. Lignins in acidic organosolv pulps. Holzforschung 1999, 53, 603–610. [Google Scholar] [CrossRef]
- Jolly, G.; Dupont, L.; Aplincourt, M.; Lambert, J. Improved Cu and Zn sorption on oxidized wheat lignocelluloses. Environ. Chem. Lett. 2006, 4, 219–223. [Google Scholar] [CrossRef]
- Hu, T.Q. Characterization of Lignocellulosic Materials; Wiley-Blackwell: Chichester, UK, 2008. [Google Scholar]
- Crestini, C.; Crucianelli, M.; Orlandi, M.; Saladino, R. Oxidative strategies in lignin chemistry: A new environmental friendly approach for the functionalisation of lignin and lignocellulosic fibers. Catal. Today 2010, 156, 8–22. [Google Scholar] [CrossRef]
- Li, M.; Foster, C.; Kelkar, S.; Pu, Y.; Holmes, D.; Ragauskas, A. Structural characterization of alkaline hydrogen peroxide pretreated grasses exhibiting diverse lignin phenotypes. Biotechnol. Biofuels 2012, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Neutelings, G. Lignin variability in plant cell walls: Contributions of new models. Plant Sci. 2011, 181, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.S. Structure and action mechanism of ligninolytic enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Xiao, L.; Meng, L.; Zhang, X.; Sun, R. Isolation and structural characterization of lignin from cotton stalk treated in an ammonia hydrothermal system. Int. J. Mol. Sci. 2012, 13, 15209–15226. [Google Scholar] [CrossRef] [PubMed]
- Gardziella, A.; Pilato, L.A.; Knop, A. Phenolic Resins: Chemistry, Applications, Standardization, Safety and Ecology, 2nd ed.; Springer: Berlin, Germany, 2000. [Google Scholar]
- Pilato, L.A. Phenolic Resins: A Century of Progress; Springer: Berlin, Germany, 2010. [Google Scholar]
- Guo, J.; Jiang, Y.; Hu, X.; Xu, Z. Compounds and metal leaching from composite products made from fiberglass-resin portion of printed circuit board waste. Environ. Sci. Technol. 2012, 46, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Gothwal, R.K.; Mohan, M.K.; Ghosh, P. Synthesis of low cost adhesives from pulp & paper industry waste. J. Sci. Ind. Res. 2010, 69, 390–395. [Google Scholar]
- Cavdar, D.A.; Kalaycioglu, H.; Hiziroglu, S. Some of the properties of oriented strand board manufactured using kraft lignin phenolic resin. J. Mater. Process. Technol. 2008, 202, 559–563. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Pizzi, A.; Salvado´, J. Lignin-based wood panel adhesives without formaldehyde. Holz Roh Werkst. 2007, 65, 65–70. [Google Scholar] [CrossRef]
- Silveira, J.V.W.; Bittencourt, E.; Aguila, Z.J. Thermal and dynamic investigations on brake pad composites produced with lignin-phenolformaldehyde resin. Mater. Sci. Forum 2013, 730–732, 390–394. [Google Scholar]
- Kuo, M.; Hse, C.Y.; Huang, D.H. Alkali treated kraft lignin as a component in flakeboard resins. Holzforschung 1991, 45, 47–54. [Google Scholar] [CrossRef]
- Poppius-Levlin, K. Lignin—new openings for applications. VTT Res. Highlights 2013, 5, 35–45. [Google Scholar]
- Grząbka-Zasadzińska, A.; Klapiszewski, Ł.; Bula, K.; Jesionowski, T.; Borysiak, S. Supermolecular structure and nucleation ability of polylactide-based composites with silica/lignin hybrid fillers. J. Therm. Anal. Calorim. 2016, 126, 263–275. [Google Scholar] [CrossRef]
- Bula, K.; Klapiszewski, Ł.; Jesionowski, T. A novel functional silica/lignin hybrid material as a potential bio-based polypropylene filler. Polym. Compos. 2015, 36, 913–922. [Google Scholar] [CrossRef]
- Klapiszewski, Ł.; Nowacka, M.; Milczarek, G.; Jesionowski, T. Physicochemical and electrokinetic properties of silica/lignin biocomposites. Carbohydr. Polym. 2013, 94, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Tomkinson, J.; Mao, F.C.; Sun, X.F. Physicochemical characterization of lignin from rice straw by hydrogen peroxide treatment. J. Appl. Polym. Sci. 2001, 79, 719–732. [Google Scholar] [CrossRef]
- Mancera, A.; Fierro, V.; Pizzi, A.; Dumarçay, S.; Gèrardin, P.; Velásquez, J.; Quintana, G.; Celzard, A. Physicochemical characterization of sugarcane bagasse lignin oxidized by hydrogen peroxide. Polym. Degrad. Stable 2010, 95, 470–476. [Google Scholar] [CrossRef]
- Ago, M.; Jakes, J.E.; Johansson, L.S.; Park, S.; Rojas, O.J. Interfacial properties of lignin-based electrospun nanofibers and films reinforced with cellulose nanocrystals. ACS Appl. Mater. Interfaces 2012, 4, 6849–6856. [Google Scholar] [CrossRef] [PubMed]
- Zdarta, J.; Klapiszewski, Ł.; Wysokowski, M.; Norman, M.; Kołodziejczak-Radzimska, A.; Moszyński, D.; Ehrlich, H.; Maciejewski, H.; Stelling, A.L.; Jesionowski, T. Chitin-lignin material as a novel matrix for enzyme immobilization. Mar. Drugs 2015, 13, 2424–2446. [Google Scholar] [CrossRef] [PubMed]
- Wysokowski, M.; Klapiszewski, Ł.; Moszyński, D.; Bartczak, P.; Szatkowski, T.; Majchrzak, I.; Siwińska-Stefańska, K.; Bazhenov, V.V.; Jesionowski, T. Modification of chitin with kraft lignin and development of new biosorbents for removal of cadmium(II) and nickel(II) ions. Mar. Drugs 2014, 12, 2245–2268. [Google Scholar] [CrossRef] [PubMed]
- Dueramae, I.; Jubsilp, C.; Takeichi, T.; Rimdusit, S. High thermal and mechanical properties enhancement obtained in highly filled polybenzoxazine nanocomposites with fumed silica. Compos. B 2014, 56, 197–206. [Google Scholar] [CrossRef]
- Sen, S.; Patila, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Jeong, H.; Park, J.; Kim, S.; Lee, J.; Ahn, N.; Roh, H.G. Preparation and characterization of thermoplastic polyurethanes using partially acetylated kraft lignin. Fiber Polym. 2013, 14, 1082–1093. [Google Scholar] [CrossRef]
- Das, A.; Sarkhel, G. Effect of stoichiometric ratios for synthesized epoxy phenolic novolac (EPN) resins on their physicochemical, thermomechanical and morphological properties. Pigment Resin Technol. 2016, 45, 265–279. [Google Scholar] [CrossRef]
- Sawpan, M.A.; Holdsworth, P.G.; Renshaw, P. Glass transitions of hygrothermal aged pultruded glass fibre reinforced polymer rebar by dynamic mechanical thermal analysis. Mater. Des. 2012, 42, 272–278. [Google Scholar] [CrossRef]
- Menard, K.P. Dynamic Mechanical Analysis a Practical Introduction, 2nd ed.; CRC Press: New York, NY, USA, 2008. [Google Scholar]
- Candan, Z.; Gardner, D.J.; Shaler, S.M. Dynamic mechanical thermal analysis (DMTA) of cellulose nanofibril/nanoclay/pMDI nanocomposites. Compos. B 2016, 90, 126–132. [Google Scholar] [CrossRef]
- Wolfrum, J.; Ehrenstein, G.W. Interdependence between the curing, structure, and the mechanical properties of phenolic resins. J. Appl. Polym. Sci. 1999, 74, 3173–3185. [Google Scholar] [CrossRef]
- Brostow, W.; Chiu, R.; Kalogeras, I.M.; Vassilikou-Dova, A. Prediction of glass transition temperatures: Binary blends and copolymers. Mater. Lett. 2008, 62, 3152–3155. [Google Scholar] [CrossRef]
- Gamelas, J.A.F.; Duarte, G.V.; Ferreira, P.J. Inverse gas chromatography and XPS of extracted kraft pulps. Holzforschung 2013, 67, 273–276. [Google Scholar] [CrossRef]
- Belgacem, M.N.; Blayo, A.; Gandini, A. Surface characterization of polysaccharides, lignins, printing ink pigments, and ink fillers by inverse gas chromatography. J. Colloid Interface Sci. 1996, 182, 431–436. [Google Scholar] [CrossRef]
- Gadhe, J.B.; Gupta, R.B.; Elder, T. Surface modification of lignocellulosic fibers using high-frequency ultrasound. Cellulose 2006, 13, 9–22. [Google Scholar] [CrossRef]
- Zhu, W.; Theliander, H. Precipitation of lignin softwood black liquor: An investigation of the equilibrium and molecular properties of lignin. BioResources 2015, 10, 1696–1714. [Google Scholar] [CrossRef]
- Öhman, F.; Theliander, H. Washing lignin precipitated from kraft black liquor. Paperi Ja Puu 2006, 88, 287–292. [Google Scholar]
- Öhman, F.; Theliander, H. Filtration properties of lignin precipitated from kraft black liquor. Tappi J. 2007, 6, 3–9. [Google Scholar]
- Voelkel, A. Physicochemical measurements by Inverse Gas Chromatography. In Gas Chromatography; Poole, C.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Strzemiecka, B.; Voelkel, A. Estimation of the work of adhesion by means of inverse gas chromatography for polymer complex systems. Int. J. Adhes. Adhes. 2012, 38, 84–88. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MLS | MLS NaIO4 | MLS H2O2 | KL | KL NaIO4 | KL H2O2 | Vibrational Assignment |
|---|---|---|---|---|---|---|
| 3425 | 3390 | 3400 | 3426 | 3424 | 3447 | O–H stretching |
| 2940 | 2945 | 2940 | 2940 | 2942 | 2947 | C–Hx stretching |
| - | - | 1725 | - | - | 1705 | C=O stretching |
| 1610 | 1617 | - | 1600 | 1603 | 1612 | C–C, C=C (aromatic skeleton), stretching |
| 1515 | 1513 | 1514 | 1509 | 1517 | 1512 | |
| 1465 | 1460 | 1430 | 1465 | 1454 | 1465 | C–H (CH3 + CH2), bending |
| 1427 | 1430 | - | 1421 | 1427 | - | C–C, C=C (aromatic skeleton), stretching |
| 1379 | 1379 | - | - | 1396 | ||
| - | - | - | 1271 | 1270 | 1272 | C–O (guaiacyl unit) stretching |
| - | - | - | 1219 | 1221 | 1224 | C–OH (phenolic OH) stretching |
| 1179 | 1170 | 1172 | 1143 | - | 1145 | Aromatic C–H (guaiacyl unit), stretching |
| 1044 | 1045 | 1035 | 1045 | 1050 | 1048 | C–OH + C–O–C (aliphatic OH + ether) stretching, sulfonic acids |
| - | - | - | 856 | - | 861 | Aromatic C–H (guaiacyl unit), bending |
| - | - | - | 777 | - | Aromatic C–H (guaiacyl unit), bending | |
| - | - | - | 744 | 743 | - | |
| 654 | 649 | 662 | - | 627 | 624 | CHx bending |
| Sample | Region | Name | Binding Energy (eV) | Atomic Concentration (%) | Bonds Assignment | Mass Concentration (%) |
|---|---|---|---|---|---|---|
| Magnesium lignosulfonate (MLS) | C 1s | C 1s A | 284.7 | 65.4 | C–C | 63.0 |
| C 1s B | 286.0 | 25.4 | C–O | |||
| C 1s C | 287.0 | 6.1 | C=O | |||
| C 1s D | 288.9 | 3.0 | O=C–O– | |||
| O 1s | O 1s A | 532.0 | 38.5 | C=O | 26.0 | |
| O 1s B | 533.1 | 61.5 | C–O | |||
| MLS activated using NaIO4 | C 1s | C 1s A | 284.7 | 50.1 | C–C | 47.4 |
| C 1s B | 286.1 | 30.3 | C–O | |||
| C 1s C | 287.2 | 7.6 | C=O | |||
| C 1s D | 289.0 | 12.1 | O=C–O– | |||
| O 1s | O 1s A | 532.1 | 54.7 | C=O | 38.9 | |
| O 1s B | 533.3 | 45.3 | C–O | |||
| MLS activated using H2O2 | C 1s | C 1s A | 284.7 | 59.3 | C–C | 45.4 |
| C 1s B | 286.0 | 28.8 | C–O | |||
| C 1s C | 287.1 | 7.2 | C=O | |||
| C 1s D | 288.9 | 4.7 | O=C–O– | |||
| O 1s | O 1s A | 532.3 | 65.6 | C=O | 29.4 | |
| O 1s B | 533.3 | 34.4 | C–O | |||
| Kraft lignin (KL) | C 1s | C 1s A | 284.7 | 60.1 | C–C | 56.3 |
| C 1s B | 286.3 | 35.7 | C–O | |||
| C 1s C | 288.2 | 4.2 | O=C–O– | |||
| O 1s | O 1s A | 531.5 | 42.6 | C=O | 30.7 | |
| O 1s B | 533.1 | 57.4 | C–O | |||
| KL activated using NaIO4 | C 1s | C 1s A | 284.7 | 50.9 | C–C | 55.8 |
| C 1s B | 286.2 | 40.2 | C–O | |||
| C 1s C | 288.6 | 8.9 | O=C–O– | |||
| O 1s | O 1s A | 531.6 | 45.3 | C=O | 33.0 | |
| O 1s B | 533.0 | 54.7 | C–O | |||
| KL activated using H2O2 | C 1s | C 1s A | 284.7 | 54.5 | C–C | 39.3 |
| C 1s B | 286.2 | 34.9 | C–O | |||
| C 1s C | 287.4 | 3.5 | C=O | |||
| C 1s D | 288.6 | 7.1 | O=C–O– | |||
| O 1s | O 1s A | 531.7 | 63.7 | C=O | 31.3 | |
| O 1s B | 533.1 | 36.3 | C-O |
| Sample, Model Abrasive Composite With: | G’ 25 °C (GPa) | G’ 50 °C (GPa) | G’ 300 °C (GPa) | tan δmax (−) | Tg (°C) |
|---|---|---|---|---|---|
| MLS | 1.88 | 1.84 | 0.68 | 0.083 | 235 |
| MLS activated with NaIO4 | 2.22 | 2.12 | 1.08 | 0.055 | 250 |
| MLS activated with H2O2 | 3.45 | 3.39 | 1.52 | 0.079 | 247 |
| KL | 1.69 | 1.64 | 0.78 | 0.067 | 252 |
| KL activated with NaIO4 | 1.15 | 1.14 | 0.67 | 0.060 | 254 |
| KL activated with H2O2 | 2.10 | 2.07 | 1.06 | 0.090 | 250 |
| Compound | Dispersive Component | Acidic Component | Basic Component |
|---|---|---|---|
| Dichloromethane | 24.5 | 5.2 | 0.0 |
| Ethyl acetate | 23.9 | 0.0 | 6.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klapiszewski, L.; Jamrozik, A.; Strzemiecka, B.; Matykiewicz, D.; Voelkel, A.; Jesionowski, T. Activation of Magnesium Lignosulfonate and Kraft Lignin: Influence on the Properties of Phenolic Resin-Based Composites for Potential Applications in Abrasive Materials. Int. J. Mol. Sci. 2017, 18, 1224. https://doi.org/10.3390/ijms18061224
Klapiszewski L, Jamrozik A, Strzemiecka B, Matykiewicz D, Voelkel A, Jesionowski T. Activation of Magnesium Lignosulfonate and Kraft Lignin: Influence on the Properties of Phenolic Resin-Based Composites for Potential Applications in Abrasive Materials. International Journal of Molecular Sciences. 2017; 18(6):1224. https://doi.org/10.3390/ijms18061224
Chicago/Turabian StyleKlapiszewski, Lukasz, Artur Jamrozik, Beata Strzemiecka, Danuta Matykiewicz, Adam Voelkel, and Teofil Jesionowski. 2017. "Activation of Magnesium Lignosulfonate and Kraft Lignin: Influence on the Properties of Phenolic Resin-Based Composites for Potential Applications in Abrasive Materials" International Journal of Molecular Sciences 18, no. 6: 1224. https://doi.org/10.3390/ijms18061224
APA StyleKlapiszewski, L., Jamrozik, A., Strzemiecka, B., Matykiewicz, D., Voelkel, A., & Jesionowski, T. (2017). Activation of Magnesium Lignosulfonate and Kraft Lignin: Influence on the Properties of Phenolic Resin-Based Composites for Potential Applications in Abrasive Materials. International Journal of Molecular Sciences, 18(6), 1224. https://doi.org/10.3390/ijms18061224