Gene-Diet Interactions in Type 2 Diabetes: The Chicken and Egg Debate


Abstract
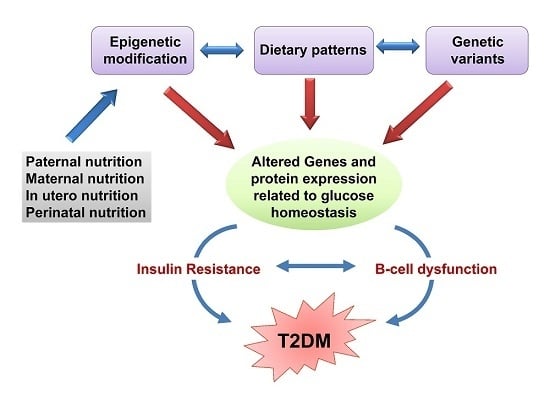
1. Introduction
2. Effects of Nutrients on Gene Expression in the Pathogenesis of T2DM
2.1. Polyphenol-Gene Interactions in T2DM Pathogenesis
2.1.1. Flavonoid-Gene Interactions in DM Pathogenesis
2.1.2. Phenolic Acid-Gene Interactions in T2DM Pathogenesis
2.1.3. Other Bioactive Compound-Gene Interactions in T2DM Pathogenesis (Polyphenols)
2.2. Vitamin-Gene Interactions in T2DM Pathogenesis
2.3. Amino Acid-Gene Interactions in T2DM Pathogenesis
2.4. Dietary Fat-Gene Interactions and Their Role in T2DM
3. Diet-Gene Interaction and Risk of T2DM
3.1. Gene Variants Associated with Insulin Stimulus-Secretion Coupling and T2DM
3.2. Gene Variants Related to IR and T2DM
3.3. Other Genetic Variants and T2DM
4. The Role of Epigenetics in the Onset of T2DM
4.1. Epigenetic Modifications as Biomarkers of T2DM
4.2. Fetal Developmental Memory
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| T2DM | Type 2 diabetes Mellitus |
| STZ | Streptozotocin |
| PI3K | Endothelial phosphatidylinositol 3-kinase |
| EGCG | Epigallocatechin gallate |
| Ins | Insulin |
| IR | Insulin resistance |
| IRS | Insulin receptor substrate |
| AMPK | AMP-activated protein kinase |
| GLUT | Glucose transporter |
| Ppp1r15a | Protein phosphatase 1, regulatory subunit 15A |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A |
| ACO-1 | Acyl CoA oxidase-1 |
| CPT-1 | Carnitine palmitoyl transferase-1 |
| HbA1c | Glycated hemoglobin |
| PPAR | Peroxisome proliferator-activated receptor |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| HFD | High-fat diet |
| HSP | Heat shock proteins |
| NO | Nitric oxide |
| NF-κB | Nuclear factor kappa B |
| CRP | C-reactive protein |
| IFN-γ | Interferon-gamma |
| IL | Interleukin |
| G6Pase | Glucose-6-phosphatase |
| RBP | Retinol binding protein |
| Pdx1 | Duodenal homeobox factor transcription factor |
| Hnf | Hepatocyte nuclear factor |
| Gk | Glucokinase |
| Cacna1d | Calcium Voltage-Gated Channel Subunit Alpha1 D |
| MafA | v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog A |
| BCAAs | Branched-chain amino acids |
| SFAs | Saturated fatty acids |
| ROS | Reactive oxygen species |
| MUFAs | Monounsaturated fatty acids |
| PUFAs | Polyunsaturated fatty acids |
| EPA | Eicosapentaenoic acid |
| DHA | Docosahexaenoic acid |
| GWAS | Genome-wide association studies |
| IRS | Insulin receptor substrate |
| FTO | Fat mass and obesity-associated |
| SNPs | Single nucleotide polymorphisms |
| CLOCK | Circadian Locomotor Output Cycles Kaput |
| MC4R | Melanocortin-4 receptor |
| PLIN | Perilipin |
| FABP | Fatty acid-binding protein |
| miRNAs | microRNAs |
| LncRNAs | Long non-coding RNAs |
| Pax | Paired box gene |
| Arx | Arista-less-related gene homeobox |
| GLP | Glucagon-like peptide |
| IUGR | Intrauterine growth retardation |
| LDL | Low-density lipoprotein |
| HDL | High-density lipoprotein |
References
- International Diabetes Federation. IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015. [Google Scholar]
- Schulze, M.B.; Hu, F.B. Primary prevention of diabetes: What can be done and how much can be prevented? Annu. Rev. Public Health 2005, 26, 445–467. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.; Golson, M.L.; Kaestner, K.H. Epigenetic control of β-cell function and failure. Diabetes Res. Clin. Pract. 2017, 123, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Stančáková, A.; Laakso, M. Genetics of Type 2 diabetes. Endocr. Dev. 2016, 31, 203–220. [Google Scholar] [PubMed]
- Berná, G.; Oliveras-López, M.J.; Jurado-Ruíz, E.; Tejedo, J.; Bedoya, F.; Soria, B.; Martín, F. Nutrigenetics and nutrigenomics insights into diabetes etiopathogenesis. Nutrients 2014, 6, 5338–5369. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Kumar, S.; Rathore, N.; Padwad, Y.; Bhushana, S. Nutrigenomics and its impact on life style associated metabolic diseases. Curr. Genom. 2016, 17, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Sales, N.M.R.; Pelegrini, P.B.; Goersch, M.C. Nutrigenomics: Definitions and advances of this new science. J. Nutr. Metab. 2014, 2014, 202759. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [PubMed]
- Xiao, J.B.; Högger, P. Dietary polyphenols and Type 2 diabetes: Current insights and future perspectives. Curr. Med. Chem. 2015, 22, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Shidfar, F.; Heydari, I.; Hajimiresmaiel, S.J.; Hosseini, S.; Shidfar, S.; Amiri, F. The effects of cranberry juice on serum glucose, apoB, apoA-I, Lp(a), and paraoxonase-1 activity in type 2 diabetic male patients. J. Res. Med. Sci. 2012, 17, 355–360. [Google Scholar] [PubMed]
- Lin, D.; Xiao, M.; Zhao, J.; Li, Z.; Xing, B.; Li, X.; Kong, M.; Li, L.; Zhang, Q.; Liu, Y.; et al. An overview of plant phenolic compounds and their importance in human nutrition and management of type 2 diabetes. Molecules 2016, 21, 1374. [Google Scholar] [CrossRef] [PubMed]
- Hanhineva, K.; Törrönen, R.; Bondia-Pons, I.; Pekkinen, J.; Kolehmainen, M.; Mykkänen, H.; Poutanen, K. Impact of dietary polyphenols on carbohydrate metabolism. Int. J. Mol. Sci. 2010, 11, 1365–1402. [Google Scholar] [CrossRef] [PubMed]
- Kerimi, A.; Williamson, G. At the interface of antioxidant signalling and cellular function: Key polyphenol effects. Mol. Nutr. Food Res. 2016, 60, 1770–1788. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Keogh, J.; Clifton, P. Polyphenols and glycemic control. Nutrients 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Babu, P.V.A.; Liu, D.; Gilbert, E.R. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J. Nutr. Biochem. 2013, 24, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wedick, N.M.; Tworoger, S.S.; Pan, A.; Townsend, M.K.; Cassidy, A.; Franke, A.A.; Rimm, E.B.; Hu, F.B.; van Dam, R.M. Urinary excretion of select dietary polyphenol metabolites is associated with a lower risk of Type 2 diabetes in proximate but not remote follow-up in a prospective investigation in 2 cohorts of US women. J. Nutr. 2015, 145, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Manson, J.E.; Buring, J.E.; Sesso, H.D.; Liu, S. Associations of dietary flavonoids with risk of Type 2 diabetes, and markers of insulin resistance and systemic inflammation in women: A prospective study and cross-sectional analysis. J. Am. Coll. Nutr. 2005, 24, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Wedick, N.M.; Pan, A.; Cassidy, A.; Rimm, E.B.; Sampson, L.; Rosner, B.; Willett, W.; Hu, F.B.; Sun, Q.; van Dam, R.M. Dietary flavonoid intakes and risk of Type 2 diabetes in US men and women. Am. J. Clin. Nutr. 2012, 95, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; Harnack, L.J.; Scrafford, C.G.; Mink, P.J.; Barraj, L.M.; Jacobs, D.R. Dietary flavonoids and flavonoid-rich foods are not associated with risk of Type 2 diabetes in postmenopausal women. J. Nutr. 2006, 136, 3039–3045. [Google Scholar] [PubMed]
- Bhardwaj, P.; Khanna, D.; Balakumar, P. Catechin averts experimental diabetes mellitus-induced vascular endothelial structural and functional abnormalities. Cardiovasc. Toxicol. 2014, 14, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Cai, E.P.; Lin, J.-K. Epigallocatechin gallate (EGCG) and rutin suppress the glucotoxicity through activating IRS2 and AMPK signalling in rat pancreatic beta cells. J. Agric. Food Chem. 2009, 57, 9817–9827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Li, Q.; Liang, J.; Dai, X.Q.; Ding, Y.; Wang, J.B.; Li, Y. Epigallocatechin-3-O-gallate (EGCG) protects the insulin sensitivity in rat L6 muscle cells exposed to dexamethasone condition. Phytomedicine 2010, 17, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Ueda-Wakagi, M.; Mukai, R.; Fuse, N.; Mizushina, Y.; Ashida, H. 3-O-Acyl-epicatechins increase glucose uptake activity and GLUT4 translocation through activation of PI3K signalling in skeletal muscle cells. Int. J. Mol. Sci. 2015, 16, 16288–16299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ding, Y.; Dai, X.; Wang, J.; Li, Y. Epigallocatechin-3-gallate protects pro-inflammatory cytokine induced injuries in insulin-producing cells through the mitochondrial pathway. Eur. J. Pharmacol. 2011, 670, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Ortsäter, H.; Grankvist, N.; Wolfram, S.; Kuehn, N.; Sjöholm, A. Diet supplementation with green tea extract epigallocatechin gallate prevents progression to glucose intolerance in db/db mice. Nutr. Metab. 2012, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, S.; Raederstorff, D.; Preller, M.; Wang, Y.; Teixeira, S.R.; Riegger, C.; Weber, P. Epigallocatechin gallate supplementation alleviates diabetes in rodents. J. Nutr. 2006, 136, 2512–2518. [Google Scholar] [PubMed]
- Tsuneki, H.; Ishizuka, M.; Terasawa, M.; Wu, J.-B.; Sasaoka, T.; Kimura, I. Effect of green tea on blood glucose levels and serum proteomic patterns in diabetic (db/db) mice and on glucose metabolism in healthy humans. BMC Pharmacol. 2004, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, T.; Leary, L.; Brooks, W.B. The effect of an extract of green and black tea on glucose control in adults with Type 2 diabetes mellitus: Double-blind randomized study. Metabolism 2007, 56, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-X.; Xu, Y.-L.; Li, S.-H.; Hui, R.; Wu, Y.-J.; Huang, X.-H. Effects of green tea catechins with or without caffeine on glycemic control in adults: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2013, 97, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Lee, M.-K.; Park, Y.B.; Kang, M.A.; Choi, M.-S. Effect of citrus flavonoids on lipid metabolism and glucose-regulating enzyme mRNA levels in type-2 diabetic mice. Int. J. Biochem. Cell Biol. 2006, 38, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Bharti, S.; Ojha, S.; Bhatia, J.; Kumar, N.; Ray, R.; Kumari, S.; Arya, D.S. Up-regulation of PPARγ, heat shock protein-27 and -72 by naringin attenuates insulin resistance, β-cell dysfunction, hepatic steatosis and kidney damage in a rat model of Type 2 diabetes. Br. J. Nutr. 2011, 106, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Coskun, O.; Kanter, M.; Korkmaz, A.; Oter, S. Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and beta-cell damage in rat pancreas. Pharmacol. Res. 2005, 51, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liang, X.; Zhang, H.; Wu, Q.; Qu, L.; Sun, Q. Quercetin protects rat dorsal root ganglion neurons against high glucose-induced injury in vitro through Nrf-2/HO-1 activation and NF-κB inhibition. Acta Pharmacol. Sin. 2013, 34, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.F.; Hassan, N.A.; El Bassossy, H.M.; Fahmy, A. Quercetin protects against diabetes-induced exaggerated vasoconstriction in rats: Effect on low grade inflammation. PLoS ONE 2013, 8, e63784. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Takahashi, Y. Dietary quercetin alleviates diabetic symptoms and reduces streptozotocin-induced disturbance of hepatic gene expression in mice. Mol. Nutr. Food Res. 2009, 53, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.K.; Soileau, J.L.; Ribnicky, D.; Wang, Z.Q.; Raskin, I.; Poulev, A.; Majewski, M.; Cefalu, W.T.; Gettys, T.W. Quercetin transiently increases energy expenditure but persistently decreases circulating markers of inflammation in C57BL/6J mice fed a high-fat diet. Metabolism 2008, 57, S39–S46. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.; Morón, R.; Sánchez, M.; Zarzuelo, A.; Galisteo, M. Quercetin ameliorates metabolic syndrome and improves the inflammatory status in obese Zucker rats. Obesity 2008, 16, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-K.; Kwon, K.-B.; Song, M.-Y.; Han, M.-J.; Lee, J.-H.; Lee, Y.-R.; Lee, J.-H.; Ryu, D.-G.; Park, B.-H.; Park, J.-W. Flavonoids protect against cytokine-induced pancreatic β-cell damage through suppression of nuclear factor κB activation. Pancreas 2007, 35, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Noriega-López, L.; Tovar, A.R.; Gonzalez-Granillo, M.; Hernández-Pando, R.; Escalante, B.; Santillán-Doherty, P.; Torres, N. Pancreatic insulin secretion in rats fed a soy protein high fat diet depends on the interaction between the amino acid pattern and isoflavones. J. Biol. Chem. 2007, 282, 20657–20666. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Zhang, W.; Zhen, W.; Lum, H.; Nadler, J.; Bassaganya-Riera, J.; Jia, Z.; Wang, Y.; Misra, H.; Liu, D. Genistein induces pancreatic beta-cell proliferation through activation of multiple signalling pathways and prevents insulin-deficient diabetes in mice. Endocrinology 2010, 151, 3026–3037. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Costantini, B.; Suriano, R.; Perri, C.; Macrì, F.; Ricciardi, L.; Panunzi, S.; Lanzone, A. The differential effect of the phytoestrogen genistein on cardiovascular risk factors in postmenopausal women: Relationship with the metabolic status. J. Clin. Endocrinol. Metab. 2009, 94, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kang, M.; Xie, Q.; Xu, B.; Sun, C.; Chen, K.; Wu, Y. Anthocyanins from Chinese bayberry extract protect β cells from oxidative stress-mediated injury via HO-1 upregulation. J. Agric. Food Chem. 2011, 59, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Nishimura, N.; Hoshino, H.; Isa, Y.; Kadowaki, M.; Ichi, T.; Tanaka, A.; Nishiumi, S.; Fukuda, I.; Ashida, H.; et al. Cyanidin 3-glucoside ameliorates hyperglycemia and insulin sensitivity due to downregulation of retinol binding protein 4 expression in diabetic mice. Biochem. Pharmacol. 2007, 74, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, M.; Inoue, S.; Horio, F.; Tsuda, T. Dietary anthocyanin-rich bilberry extract ameliorates hyperglycemia and insulin sensitivity via activation of AMP-activated protein kinase in diabetic mice. J. Nutr. 2010, 140, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Bhupathiraju, S.N.; Chen, M.; van Dam, R.M.; Hu, F.B. Caffeinated and decaffeinated coffee consumption and risk of Type 2 diabetes: A systematic review and a dose-response meta-analysis. Diabetes Care 2014, 37, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, D.; Jiang, W. Coffee and caffeine intake and incidence of Type 2 diabetes mellitus: A meta-analysis of prospective studies. Eur. J. Nutr. 2014, 53, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Van Dieren, S.; Uiterwaal, C.S.P.M.; van der Schouw, Y.T.; van der A, D.L.; Boer, J.M.A.; Spijkerman, A.; Grobbee, D.E.; Beulens, J.W.J. Coffee and tea consumption and risk of Type 2 diabetes. Diabetologia 2009, 52, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mao, Q.-X.; Xu, H.-X.; Ma, X.; Zeng, C.-Y. Tea consumption and risk of Type 2 diabetes mellitus: A systematic review and meta-analysis update. BMJ Open 2014, 4, e005632. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-S.; Wang, W.-Y.; Fan, W.-Y.; Deng, Q.; Wang, X. Tea consumption and risk of Type 2 diabetes: A dose–response meta-analysis of cohort studies. Br. J. Nutr. 2014, 111, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.M.; Nanri, A.; Kochi, T.; Kuwahara, K.; Tsuruoka, H.; Kurotani, K.; Akter, S.; Kabe, I.; Sato, M.; Hayabuchi, H.; et al. Coffee and green tea consumption is associated with insulin resistance in Japanese adults. Metabolism 2014, 63, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Arnlöv, J.; Vessby, B.; Risérus, U. Coffee consumption and insulin sensitivity. JAMA 2004, 291, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Yarmolinsky, J.; Mueller, N.T.; Duncan, B.B.; Bisi Molina, M.D.C.; Goulart, A.C.; Schmidt, M.I. Coffee Consumption, Newly Diagnosed Diabetes, and Other Alterations in Glucose Homeostasis: A Cross-Sectional Analysis of the Longitudinal Study of Adult Health (ELSA-Brasil). PLoS ONE 2015, 10, e0126469. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, R.M.; Dekker, J.M.; Nijpels, G.; Stehouwer, C.D.A.; Bouter, L.M.; Heine, R.J. Hoorn study Coffee consumption and incidence of impaired fasting glucose, impaired glucose tolerance, and Type 2 diabetes: The Hoorn Study. Diabetologia 2004, 47, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Agunloye, O.M.; Adefegha, S.A.; Akinyemi, A.J.; Ademiluyi, A.O. Caffeic and chlorogenic acids inhibit key enzymes linked to Type 2 diabetes (in vitro): A comparative study. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, R.; Funakoshi-Tago, M.; Fujiwara, Y.; Tamura, H. Coffee inhibits adipocyte differentiation via inactivation of PPARγ. Biol. Pharm. Bull. 2014, 37, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chang, C.; Zhang, L.; Liu, Y.; Huang, X.; Chen, Z. Chlorogenic acid improves late diabetes through adiponectin receptor signalling pathways in db/db mice. PLoS ONE 2015, 10, e0120842. [Google Scholar]
- Narasimhan, A.; Chinnaiyan, M.; Karundevi, B. Ferulic acid exerts its antidiabetic effect by modulating insulin-signalling molecules in the liver of high-fat diet and fructose-induced type-2 diabetic adult male rat. Appl. Physiol. Nutr. Metab. 2015, 40, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Chinnaiyan, M.; Karundevi, B. Ferulic acid regulates hepatic GLUT2 gene expression in high fat and fructose-induced type-2 diabetic adult male rat. Eur. J. Pharmacol. 2015, 761, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Bagul, P.K.; Banerjee, S.K. Application of resveratrol in diabetes: Rationale, strategies and challenges. Curr. Mol. Med. 2015, 15, 312–330. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T.; Szkudelska, K. Resveratrol and diabetes: From animal to human studies. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Zagotta, I.; Dimova, E.Y.; Debatin, K.-M.; Wabitsch, M.; Kietzmann, T.; Fischer-Posovszky, P. Obesity and inflammation: Reduced cytokine expression due to resveratrol in a human in vitro model of inflamed adipose tissue. Front. Pharmacol. 2015, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Kim, T.T.; Denou, E.; Soltys, C.-L.M.; Hamza, S.M.; Byrne, N.J.; Masson, G.; Park, H.; Wishart, D.S.; Madsen, K.L.; et al. Improved glucose homeostasis in obese mice treated with resveratrol is associated with alterations in the gut microbiome. Diabetes 2017, 66, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; de Ligt, M.; Phielix, E.; van de Weijer, T.; Hansen, J.; Moonen-Kornips, E.; Schaart, G.; Kunz, I.; Hesselink, M.K.C.; Schrauwen-Hinderling, V.B.; et al. Resveratrol as add-on therapy in subjects with well-controlled Type 2 diabetes: A randomized controlled trial. Diabetes Care 2016, 39, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Bo, S.; Ponzo, V.; Ciccone, G.; Evangelista, A.; Saba, F.; Goitre, I.; Procopio, M.; Pagano, G.F.; Cassader, M.; Gambino, R. Six months of resveratrol supplementation has no measurable effect in type 2 diabetic patients. A randomized, double blind, placebo-controlled trial. Pharmacol. Res. 2016, 111, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Crandall, J.P.; Oram, V.; Trandafirescu, G.; Reid, M.; Kishore, P.; Hawkins, M.; Cohen, H.W.; Barzilai, N. Pilot study of resveratrol in older adults with impaired glucose tolerance. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, J.K.; Thomas, S.; Nanjan, M.J. Resveratrol supplementation improves glycemic control in Type 2 diabetes mellitus. Nutr. Res. 2012, 32, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Brasnyó, P.; Molnár, G.A.; Mohás, M.; Markó, L.; Laczy, B.; Cseh, J.; Mikolás, E.; Szijártó, I.A.; Mérei, A.; Halmai, R.; et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br. J. Nutr. 2011, 106, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Rouse, M.; Younès, A.; Egan, J.M. Resveratrol and curcumin enhance pancreatic β-cell function by inhibiting phosphodiesterase activity. J. Endocrinol. 2014, 223, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Chuengsamarn, S.; Rattanamongkolgul, S.; Luechapudiporn, R.; Phisalaphong, C.; Jirawatnotai, S. Curcumin extract for prevention of Type 2 diabetes. Diabetes Care 2012, 35, 2121–2127. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu. Rev. Nutr. 2010, 30, 173–199. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Chu, A.; Go, V.L.W.; Saad, M.F. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am. J. Clin. Nutr. 2004, 79, 820–825. [Google Scholar] [PubMed]
- Oh, Y.S.; Jun, H.-S.; Jun, H.-S. Role of bioactive food components in diabetes prevention: Effects on Beta-cell function and preservation. Nutr. Metab. Insights 2014, 7, 51–59. [Google Scholar] [PubMed]
- Cade, C.; Norman, A.W. Rapid normalization/stimulation by 1,25-dihydroxyvitamin D3 of insulin secretion and glucose tolerance in the vitamin D-deficient rat. Endocrinology 1987, 120, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Maestro, B.; Molero, S.; Bajo, S.; Dávila, N.; Calle, C. Transcriptional activation of the human insulin receptor gene by 1,25-dihydroxyvitamin D3. Cell Biochem. Funct. 2002, 20, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, T.W.; Väisänen, S.; Frank, C.; Molnár, F.; Sinkkonen, L.; Carlberg, C. The human peroxisome proliferator-activated receptor delta gene is a primary target of 1α,25-dihydroxyvitamin D3 and its nuclear receptor. J. Mol. Biol. 2005, 349, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; van Etten, E.; Overbergh, L.; Stoffels, K.; Bouillon, R.; Mathieu, C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. Diabetes Res. Clin. Pract. 2007, 77, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Wolden-Kirk, H.; Overbergh, L.; Christesen, H.T.; Brusgaard, K.; Mathieu, C. Vitamin D and diabetes: Its importance for beta cell and immune function. Mol. Cell. Endocrinol. 2011, 347, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Bojesen, S.E.; Nordestgaard, B.G. Low 25-hydroxyvitamin D and risk of Type 2 diabetes: A prospective cohort study and metaanalysis. Clin. Chem. 2013, 59, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.K.; Swaminathan, B.; Hanley, A.J.; Connelly, P.W.; Sermer, M.; Zinman, B.; Retnakaran, R. Prospective associations of vitamin D status with β-Cell function, insulin sensitivity, and glycemia: The impact of parathyroid hormone status. Diabetes 2014, 63, 3868–3879. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; González-Clemente, J.M.; Blanco-Vaca, F.; Mauricio, D. Role of vitamin D in the pathogenesis of Type 2 diabetes mellitus. Diabetes Obes. Metab. 2008, 10, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Leung, P. The potential protective action of vitamin d in hepatic insulin resistance and pancreatic islet dysfunction in Type 2 diabetes Mellitus. Nutrients 2016, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Tabesh, M.; Azadbakht, L.; Faghihimani, E.; Tabesh, M.; Esmaillzadeh, A. Effects of calcium-vitamin D co-supplementation on metabolic profiles in vitamin D insufficient people with Type 2 diabetes: A randomised controlled clinical trial. Diabetologia 2014, 57, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Seida, J.C.; Mitri, J.; Colmers, I.N.; Majumdar, S.R.; Davidson, M.B.; Edwards, A.L.; Hanley, D.A.; Pittas, A.G.; Tjosvold, L.; Johnson, J.A. Clinical review: Effect of vitamin D3 supplementation on improving glucose homeostasis and preventing diabetes: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, 3551–3560. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Yokoyama, K.; Yokoo, T.; Urashima, M. Role of vitamin D in diabetes mellitus and chronic kidney disease. World J. Diabetes 2016, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.A.; Rhoten, W.B.; Driscoll, H.K.; Chertow, B.S. Vitamin A deficiency impairs fetal islet development and causes subsequent glucose intolerance in adult rats. J. Nutr. 2004, 134, 1958–1963. [Google Scholar] [PubMed]
- Delaspre, F.; Massumi, M.; Salido, M.; Soria, B.; Ravassard, P.; Savatier, P.; Skoudy, A. Directed pancreatic acinar differentiation of mouse embryonic stem cells via embryonic signalling molecules and exocrine transcription factors. PLoS ONE 2013, 8, e54243. [Google Scholar] [CrossRef] [PubMed]
- Trasino, S.E.; Benoit, Y.D.; Gudas, L.J. Vitamin A deficiency causes hyperglycemia and loss of pancreatic β-cell mass. J. Biol. Chem. 2015, 290, 1456–1473. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Ramos, R.; Guadarrama-López, A.L.; Martínez-Carrillo, B.E.; Benítez-Arciniega, A.D. Vitamins and Type 2 diabetes mellitus. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and Type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.-A.; Smith, U.; et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 2006, 354, 2552–2563. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Nakamura, M.; Ogawa, K.; Ikoma, Y.; Yano, M. High-serum carotenoids associated with lower risk for developing Type 2 diabetes among Japanese subjects: Mikkabi cohort study. BMJ Open Diabetes Res. Care 2015, 3, e000147. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, S.; Manson, J.E.; Gaziano, J.M.; Buring, J.E.; Sesso, H.D. The consumption of lycopene and tomato-based food products is not associated with the risk of Type 2 diabetes in women. J. Nutr. 2006, 136, 620–625. [Google Scholar] [PubMed]
- Sena, C.M.; Nunes, E.; Gomes, A.; Santos, M.S.; Proença, T.; Martins, M.I.; Seiça, R.M. Supplementation of coenzyme Q10 and α-tocopherol lowers glycated hemoglobin level and lipid peroxidation in pancreas of diabetic rats. Nutr. Res. 2008, 28, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Wu, J.H.Y.; Clarke, M.W.; Puddey, I.B.; Burke, V.; Croft, K.D.; Hodgson, J.M. The effect of vitamin E on blood pressure in individuals with Type 2 diabetes: A randomized, double-blind, placebo-controlled trial. J. Hypertens. 2007, 25, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, S.; Tao, A.; Chen, G.; Zhang, M. Influence of vitamin E supplementation on glycaemic control: A meta-analysis of randomised controlled trials. PLoS ONE 2014, 9, e95008. [Google Scholar] [CrossRef] [PubMed]
- Lazo de la Vega-Monroy, M.L.; Larrieta, E.; German, M.S.; Baez-Saldana, A.; Fernandez-Mejia, C. Effects of biotin supplementation in the diet on insulin secretion, islet gene expression, glucose homeostasis and beta-cell proportion. J. Nutr. Biochem. 2013, 24, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Sahin, K.; Tuzcu, M.; Orhan, C.; Sahin, N.; Kucuk, O.; Ozercan, I.H.; Juturu, V.; Komorowski, J.R. Anti-diabetic activity of chromium picolinate and biotin in rats with Type 2 diabetes induced by high-fat diet and streptozotocin. Br. J. Nutr. 2013, 110, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, B.; Gönül, B.; Erdogan, D.; Elbeg, S. Effects of limited food intake and vitamin C supplementation on pancreatic glucagon and insulin in guinea pigs. Eur. J. Histochem. 2007, 51, 137–144. [Google Scholar] [PubMed]
- Sinclair, A.J.; Taylor, P.B.; Lunec, J.; Girling, A.J.; Barnett, A.H. Low plasma ascorbate levels in patients with Type 2 diabetes mellitus consuming adequate dietary vitamin C. Diabet. Med. 1994, 11, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Wells, W.W.; Dou, C.Z.; Dybas, L.N.; Jung, C.H.; Kalbach, H.L.; Xu, D.P. Ascorbic acid is essential for the release of insulin from scorbutic guinea pig pancreatic islets. Proc. Natl. Acad. Sci. USA 1995, 92, 11869–11873. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.-H.; Wareham, N.J.; Bingham, S.A.; Khaw, K.; Luben, R.; Welch, A.; Forouhi, N.G. Plasma vitamin C level, fruit and vegetable consumption, and the risk of new-onset Type 2 diabetes mellitus: the European prospective investigation of cancer–norfolk prospective study. Arch. Intern. Med. 2008, 168, 1493. [Google Scholar] [CrossRef] [PubMed]
- Bergsten, P.; Moura, A.S.; Atwater, I.; Levine, M. Ascorbic acid and insulin secretion in pancreatic islets. J. Biol. Chem. 1994, 269, 1041–1045. [Google Scholar] [PubMed]
- Eriksson, J.; Kohvakka, A. Magnesium and ascorbic acid supplementation in diabetes mellitus. Ann. Nutr. Metab. 1995, 39, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Balbi, V.; Volpe, C.; Varricchio, G.; Gambardella, A.; Saccomanno, F.; Ammendola, S.; Varricchio, M.; D’Onofrio, F. Metabolic benefits deriving from chronic vitamin C supplementation in aged non-insulin dependent diabetics. J. Am. Coll. Nutr. 1995, 14, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Karne, R.J.; Hall, G.; Campia, U.; Panza, J.A.; Cannon, R.O.; Wang, Y.; Katz, A.; Levine, M.; Quon, M.J. High-dose oral vitamin C partially replenishes vitamin C levels in patients with Type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H137–H145. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004, 80, 1194–1200. [Google Scholar] [PubMed]
- Alam, M.M.; Iqbal, S.; Naseem, I. Ameliorative effect of riboflavin on hyperglycemia, oxidative stress and DNA damage in type-2 diabetic mice: Mechanistic and therapeutic strategies. Arch. Biochem. Biophys. 2015, 584, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-S.; Li, D.; Zhou, Y.-M.; Sun, W.-P.; Liu, Q.-G. B-vitamin consumption and the prevalence of diabetes and obesity among the US adults: Population based ecological study. BMC Public Health 2010, 10, 746. [Google Scholar] [CrossRef] [PubMed]
- Cobianchi, L.; Fornoni, A.; Pileggi, A.; Molano, R.D.; Sanabria, N.Y.; Gonzalez-Quintana, J.; Bocca, N.; Marzorati, S.; Zahr, E.; Hogan, A.R.; et al. Riboflavin inhibits IL-6 expression and p38 activation in islet cells. Cell Transplant. 2008, 17, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Soria, B.; Roche, E.; Berná, G.; León-Quinto, T.; Reig, J.A.; Martín, F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 2000, 49, 157–162. [Google Scholar] [CrossRef] [PubMed]
- León-Quinto, T.; Jones, J.; Skoudy, A.; Burcin, M.; Soria, B. In vitro directed differentiation of mouse embryonic stem cells into insulin-producing cells. Diabetologia 2004, 47, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Vaca, P.; Berná, G.; Araujo, R.; Carneiro, E.M.; Bedoya, F.J.; Soria, B.; Martín, F. Nicotinamide induces differentiation of embryonic stem cells into insulin-secreting cells. Exp. Cell Res. 2008, 314, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.Z.; Tai, M.-H.; Linning, K.D.; Szabo, C.; Olson, L.K. MafA expression and insulin promoter activity are induced by nicotinamide and related compounds in INS-1 pancreatic beta-cells. Diabetes 2006, 55, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Soria, B. Amino acid-induced [Ca2+]i oscillations in single mouse pancreatic islets of Langerhans. J. Physiol. 1995, 361–371. [Google Scholar] [CrossRef]
- Bolea, S.; Pertusa, J.A.; Martín, F.; Sanchez-Andrés, J.V.; Soria, B. Regulation of pancreatic beta-cell electrical activity and insulin release by physiological amino acid concentrations. Pflugers Arch. 1997, 433, 699–704. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.A.M.; Latorraca, M.Q.; de Mello, M.A.R.; Carneiro, E.M. Mechanisms of insulin secretion in malnutrition: Modulation by amino acids in rodent models. Amino Acids 2011, 40, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.B.; Kastenmüller, G.; Shin, S.-Y.; Petersen, A.-K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for Type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013, 62, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Imam, M.U.; Ismail, M. Nutrigenomic effects of germinated brown rice and its bioactives on hepatic gluconeogenic genes in type 2 diabetic rats and HEPG2 cells. Mol. Nutr. Food Res. 2013, 57, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Azzout-Marniche, D.; Gaudichon, C.; Tomé, D. Dietary protein and blood glucose control. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Healy, N.P.; Kirwan, A.M.; McArdle, M.A.; Holohan, K.; Nongonierma, A.B.; Keane, D.; Kelly, S.; Celkova, L.; Lyons, C.L.; McGillicuddy, F.C.; et al. A casein hydrolysate protects mice against high fat diet induced hyperglycemia by attenuating NLRP3 inflammasome-mediated inflammation and improving insulin signalling. Mol. Nutr. Food Res. 2016, 60, 2421–2432. [Google Scholar] [CrossRef] [PubMed]
- Gannon, M.C.; Nuttall, J.A.; Damberg, G.; Gupta, V.; Nuttall, F.Q. Effect of Protein Ingestion on the Glucose Appearance Rate in People with Type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Manders, R.J.F.; Hansen, D.; Zorenc, A.H.G.; Dendale, P.; Kloek, J.; Saris, W.H.M.; van Loon, L.J.C. Protein co-ingestion strongly increases postprandial insulin secretion in Type 2 diabetes patients. J. Med. Food 2014, 17, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, L.; Ding, E.L.; Malik, V.S.; de Goede, J.; Geleijnse, J.M.; Soedamah-Muthu, S.S. Consumption of dairy foods and diabetes incidence: A dose-response meta-analysis of observational studies. Am. J. Clin. Nutr. 2016, 103, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Ning, N.; Wang, C.; Wang, Y.; Li, Q.; Meng, Z.; Liu, Y.; Li, Q. Dairy products consumption and risk of Type 2 diabetes: Systematic review and dose-response meta-analysis. PLoS ONE 2013, 8, e73965. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.B.; Pasin, G. Emerging evidence for the importance of dietary protein source on glucoregulatory markers and Type 2 diabetes: Different effects of dairy, meat, fish, egg, and plant protein foods. Nutrients 2016, 8, 446. [Google Scholar] [CrossRef] [PubMed]
- Bifari, F.; Nisoli, E. Branched-chain amino acids differently modulate catabolic and anabolic states in mammals: A pharmacological point of view. Br. J. Pharmacol. 2017, 174, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Lian, K.; Du, C.; Liu, Y.; Zhu, D.; Yan, W.; Zhang, H.; Hong, Z.; Liu, P.; Zhang, L.; Pei, H.; et al. Impaired adiponectin signalling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes 2015, 64, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.-G.; Fritsche, A.; Haring, H.-U.; Hrabe de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of Type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Watkins, S.M.; Lorenzo, C.; Wagenknecht, L.E.; Il’yasova, D.; Chen, Y.-D.I.; Haffner, S.M.; Hanley, A.J. Branched-chain amino acids and insulin metabolism: The insulin resistance atherosclerosis study (IRAS). Diabetes Care 2016, 39, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Giesbertz, P.; Daniel, H. Branched-chain amino acids as biomarkers in diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Batch, B.C.; Hyland, K.; Svetkey, L.P. Branch chain amino acids: Biomarkers of health and disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, C.; Savage, D.B. An amino acid profile to predict diabetes? Nat. Med. 2011, 17, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.; Meininger, C.J.; Wu, G. l-Leucine and NO-mediated cardiovascular function. Amino Acids 2015, 47, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Kwon, G.; Marshall, C.A.; Pappan, K.L.; Remedi, M.S.; McDaniel, M.L. Signalling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 2004, 53 (Suppl. S3), S225–S232. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Kwon, G.; Cruz, W.S.; Marshall, C.A.; McDaniel, M.L. Metabolic regulation by leucine of translation initiation through the mTOR-signalling pathway by pancreatic beta-cells. Diabetes 2001, 50, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Dickson, L.M.; Rhodes, C.J. Pancreatic beta-cell growth and survival in the onset of Type 2 diabetes: A role for protein kinase B in the Akt? AJP Endocrinol. Metab. 2004, 287, E192–E198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jeppesen, P.B.; Gregersen, S.; Chen, X.; Hermansen, K. Dose- and glucose-dependent effects of amino acids on insulin secretion from isolated mouse islets and clonal INS-1E beta-cells. Rev. Diabet. Stud. 2008, 5, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-S. The Emerging Role of Branched-chain amino acids in insulin resistance and metabolism. Nutrients 2016, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, G.; Cerasi, E.; Ketzinel-Gilad, M. The role of mTOR in the adaptation and failure of beta-cells in Type 2 diabetes. Diabetes. Obes. Metab. 2008, 10 (Suppl. S4), 157–169. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, B.D.; Graham, J.L.; Stanhope, K.L.; Fiehn, O.; Havel, P.J.; Adams, S.H. Plasma amino acid and metabolite signatures tracking diabetes progression in the UCD-T2DM rat model. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E958–E969. [Google Scholar] [CrossRef] [PubMed]
- Wang-Sattler, R.; Yu, Z.; Herder, C.; Messias, A.C.; Floegel, A.; He, Y.; Heim, K.; Campillos, M.; Holzapfel, C.; Thorand, B.; et al. Novel biomarkers for pre-diabetes identified by metabolomics. Mol. Syst. Biol. 2012, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Würtz, P.; Mäkinen, V.-P.; Soininen, P.; Kangas, A.J.; Tukiainen, T.; Kettunen, J.; Savolainen, M.J.; Tammelin, T.; Viikari, J.S.; Rönnemaa, T.; et al. Metabolic signatures of insulin resistance in 7098 young adults. Diabetes 2012, 61, 1372–1380. [Google Scholar] [PubMed]
- Klein, M.S.; Shearer, J. Metabolomics and Type 2 diabetes: Translating basic research into clinical application. J. Diabetes Res. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.S.; Price, L.L.; Phillips, E.M.; Fielding, R.A. Serum glycine is associated with regional body fat and insulin resistance in functionally-limited older adults. PLoS ONE 2013, 8, e84034. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Braun, M. Regulation of Insulin Secretion in Human Pancreatic Islets. Annu. Rev. Physiol. 2013, 75, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.-S.; Jo, S.-H.; Kang, B.-H.; Apostolidis, E.; Lee, M.S.; Jang, H.-D.; Kwon, Y.-I. In vitro and in vivo antihyperglycemic effect of 2 amadori rearrangement compounds, arginyl-fructose and arginyl-fructosyl-glucose. J. Food Sci. 2011, 76, H188–H193. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Ha, K.-S.; Jo, S.-H.; Lee, C.M.; Kim, Y.-C.; Chung, K.-H.; Kwon, Y.-I. Effect of long-term dietary arginyl-fructose (AF) on hyperglycemia and HbA1c in diabetic db/db mice. Int. J. Mol. Sci. 2014, 15, 8352–8359. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Kim, O.-H.; Kwak, J.H.; Lee, K.-H.; Kwon, Y.-I.; Chung, K.H.; Lee, J.H. Antihyperglycemic effect of short-term arginyl-fructose supplementation in subjects with prediabetes and newly diagnosed Type 2 diabetes: Randomized, double-blinded, placebo-controlled trial. Trials 2015, 16, 521. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schünemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and Type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [PubMed]
- Morio, B.; Fardet, A.; Legrand, P.; Lecerf, J.-M. Involvement of dietary saturated fats, from all sources or of dairy origin only, in insulin resistance and Type 2 diabetes. Nutr. Rev. 2016, 74, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, C.; Jia, C. Association of fish and n-3 fatty acid intake with the risk of Type 2 diabetes: A meta-analysis of prospective studies. Br. J. Nutr. 2012, 108, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Lindström, J.; Ilanne-Parikka, P.; Peltonen, M.; Aunola, S.; Eriksson, J.G.; Hemiö, K.; Hämäläinen, H.; Härkönen, P.; Keinänen-Kiukaanniemi, S.; Laakso, M.; et al. Sustained reduction in the incidence of Type 2 diabetes by lifestyle intervention: Follow-up of the Finnish Diabetes Prevention Study. Lancet 2006, 368, 1673–1679. [Google Scholar] [CrossRef]
- Wallin, A.; Di Giuseppe, D.; Orsini, N.; Patel, P.S.; Forouhi, N.G.; Wolk, A. Fish consumption, dietary long-chain n-3 fatty acids, and risk of Type 2 diabetes: Systematic review and meta-analysis of prospective studies. Diabetes Care 2012, 35, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; List, E.O.; Kopchick, J.J. Differentially expressed proteins in the pancreas of diet-induced diabetic mice. Mol. Cell. Proteom. 2005, 4, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Ritz-Laser, B.; Meda, P.; Constant, I.; Klages, N.; Charollais, A.; Morales, A.; Magnan, C.; Ktorza, A.; Philippe, J. Glucose-induced preproinsulin gene expression is inhibited by the free fatty acid palmitate. Endocrinology 1999, 140, 4005–4014. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Hagman, D.; Stein, R.; Artner, I.; Robertson, R.P.; Harmon, J.S. Regulation of the insulin gene by glucose and fatty acids. J. Nutr. 2006, 136, 873–876. [Google Scholar] [PubMed]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Tajiri, Y.; Sako, Y.; Hashimoto, T.; Umeda, F.; Nawata, H. Effects of free fatty acids on β-cell functions: A possible involvement of peroxisome proliferator-activated receptors α or pancreatic/duodenal homeobox. Metabolism 2001, 50, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.C.; Ugas, M.A.; Hagman, D.K.; Parazzoli, S.D.; Poitout, V. Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 2004, 53, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signalling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Ruiz, E.; Varela, L.M.; Luque, A.; Berná, G.; Cahuana, G.; Martinez-Force, E.; Gallego-Durán, R.; Soria, B.; de Roos, B.; Romero Gómez, M.; et al. An extra virgin olive oil rich diet intervention ameliorates the nonalcoholic steatohepatitis induced by a high-fat "Western-type" diet in mice. Mol. Nutr. Food Res. 2017, 61, 1600549. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Imperlini, E.; Nigro, E.; Montagnese, C.; Daniele, A.; Orrù, S.; Buono, P. Biological and Nutritional Properties of Palm Oil and Palmitic Acid: Effects on Health. Molecules 2015, 20, 17339–17361. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Gabler, N.; Walker-Daniels, J.; Spurlock, M. The c-Jun N-terminal kinase mediates the induction of oxidative stress and insulin resistance by palmitate and Toll-like receptor 2 and 4 ligands in 3T3-L1 adipocytes. Horm. Metab. Res. 2009, 41, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Poels, S.; Esser, N.; L’homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/Type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Bugliani, M.; Boggi, U.; Masini, M.; Marselli, L. The pancreatic beta cells in human Type 2 diabetes. Adv. Exp. Med. Biol. 2012, 771, 288–309. [Google Scholar] [PubMed]
- Rosqvist, F.; Iggman, D.; Kullberg, J.; Cedernaes, J.; Johansson, H.-E.; Larsson, A.; Johansson, L.; Ahlström, H.; Arner, P.; Dahlman, I.; et al. Overfeeding polyunsaturated and saturated fat causes distinct effects on liver and visceral fat accumulation in humans. Diabetes 2014, 63, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Vessby, B.; Uusitupa, M.; Hermansen, K.; Riccardi, G.; Rivellese, A.A.; Tapsell, L.C.; Nälsén, C.; Berglund, L.; Louheranta, A.; Rasmussen, B.M.; et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU Study. Diabetologia 2001, 44, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Jebb, S.A.; Lovegrove, J.A.; Griffin, B.A.; Frost, G.S.; Moore, C.S.; Chatfield, M.D.; Bluck, L.J.; Williams, C.M.; Sanders, T.A.; RISCK Study Group. Effect of changing the amount and type of fat and carbohydrate on insulin sensitivity and cardiovascular risk: The RISCK (Reading, Imperial, Surrey, Cambridge, and Kings) trial. Am. J. Clin. Nutr. 2010, 92, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Koulman, A.; Sharp, S.J.; Imamura, F.; Kröger, J.; Schulze, M.B.; Crowe, F.L.; Huerta, J.M.; Guevara, M.; Beulens, J.W.; et al. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident Type 2 diabetes: The EPIC-InterAct case-cohort study. Lancet Diabetes Endocrinol. 2014, 2, 810–818. [Google Scholar] [CrossRef]
- Pérez-Martínez, P.; García-Ríos, A.; Delgado-Lista, J.; Pérez-Jiménez, F.; López-Miranda, J. Mediterranean diet rich in olive oil and obesity, metabolic syndrome and diabetes mellitus. Curr. Pharm. Des. 2011, 17, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, D.; van Leeuwen, M.; Akdogan, D.; Adams, H.; Jansen, H.; Verhoeven, A.J.M. Activation of hepatic lipase expression by oleic acid: Possible involvement of USF1. Nutrients 2009, 1, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, H.; Xu, H.; Halim, V.; Zhang, W.; Wang, H.; Ong, K.T.; Woo, S.-L.; Walzem, R.L.; Mashek, D.G.; et al. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS ONE 2012, 7, e39286. [Google Scholar] [CrossRef] [PubMed]
- Nunes, E.A.; Rafacho, A. Implications of palmitoleic acid (palmitoleate) on glucose homeostasis, insulin resistance and diabetes. Curr. Drug Targets 2015, 18, 619–628. [Google Scholar] [CrossRef]
- Wang, L.; Folsom, A.R.; Zheng, Z.-J.; Pankow, J.S.; Eckfeldt, J.H.; ARIC Study Investigators. Plasma fatty acid composition and incidence of diabetes in middle-aged adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Clin. Nutr. 2003, 78, 91–98. [Google Scholar] [PubMed]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Shi, J.-J.; Li, Y.-M.; Zhang, X.-Y.; Chen, Y.; Calder, P.C.; Tang, L.-J. What is the impact of n-3 PUFAs on inflammation markers in Type 2 diabetic mellitus populations?: A systematic review and meta-analysis of randomized controlled trials. Lipids Health Dis. 2016, 15, 133. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Cicero, A.F.G.; D’Angelo, A.; Borghi, C.; Maffioli, P. Effects of n-3 pufas on fasting plasma glucose and insulin resistance in patients with impaired fasting glucose or impaired glucose tolerance. Biofactors 2016, 42, 316–322. [Google Scholar] [PubMed]
- Chen, C.; Yu, X.; Shao, S. Effects of omega-3 fatty acid supplementation on glucose control and lipid levels in Type 2 diabetes: A meta-analysis. PLoS ONE 2015, 10, e0139565. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Shimano, H.; Yamamoto, T.; Ishikawa, M.; Kumadaki, S.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N.; Nakakuki, M.; Hasty, A.H.; et al. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes 2008, 57, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Pinel, A.; Morio-Liondore, B.; Capel, F. n-3 Polyunsaturated fatty acids modulate metabolism of insulin-sensitive tissues: Implication for the prevention of Type 2 diabetes. J. Physiol. Biochem. 2014, 70, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Risérus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of Type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Hennig, B.; Toborek, M.; Joshi-Barve, S.; Barger, S.W.; Barve, S.; Mattson, M.P.; McClain, C.J. Linoleic acid activates nuclear transcription factor-kappa B (NF-κB) and induces NF-κB-dependent transcription in cultured endothelial cells. Am. J. Clin. Nutr. 1996, 63, 322–328. [Google Scholar] [PubMed]
- Pischon, T.; Hankinson, S.E.; Hotamisligil, G.S.; Rifai, N.; Willett, W.C.; Rimm, E.B. Habitual dietary intake of n-3 and n-6 fatty acids in relation to inflammatory markers among US men and women. Circulation 2003, 108, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Functional roles of fatty acids and their effects on human health. J. Parenter. Enter. Nutr. 2015, 39, 18S–32S. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Kaul, N.; Ali, S. Genes, Genetics, and Environment in Type 2 diabetes: Implication in Personalized Medicine. DNA Cell Biol. 2016, 35, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Hu, F.B. Gene-environment interactions in the development of Type 2 diabetes: Recent progress and continuing challenges. Annu. Rev. Nutr. 2012, 32, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.F.A.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of Type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; McCarthy, M.I. TCF7L2: The biggest story in diabetes genetics since HLA? Diabetologia 2007, 50, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Chandak, G.R.; Janipalli, C.S.; Bhaskar, S.; Kulkarni, S.R.; Mohankrishna, P.; Hattersley, A.T.; Frayling, T.M.; Yajnik, C.S. Common variants in the TCF7L2 gene are strongly associated with Type 2 diabetes mellitus in the Indian population. Diabetologia 2006, 50, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, M.; Hara, K.; Ito, C.; Nagai, R.; Froguel, P.; Kadowaki, T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of Type 2 diabetes in the Japanese population. Diabetologia 2007, 50, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Lehman, D.M.; Hunt, K.J.; Leach, R.J.; Hamlington, J.; Arya, R.; Abboud, H.E.; Duggirala, R.; Blangero, J.; Göring, H.H.H.; Stern, M.P. Haplotypes of transcription factor 7-like 2 (TCF7L2) gene and its upstream region are associated with Type 2 diabetes and age of onset in Mexican Americans. Diabetes 2007, 56, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Ma, E.; Yin, L.; Jin, Y.; Wang, H. The association between gene polymorphism of TCF7L2 and Type 2 diabetes in Chinese Han population: A meta-analysis. PLoS ONE 2013, 8, e59495. [Google Scholar] [CrossRef] [PubMed]
- Yako, Y.Y.; Guewo-Fokeng, M.; Balti, E.V.; Bouatia-Naji, N.; Matsha, T.E.; Sobngwi, E.; Erasmus, R.T.; Echouffo-Tcheugui, J.B.; Kengne, A.P. Genetic risk of Type 2 diabetes in populations of the African continent: A systematic review and meta-analyses. Diabetes Res. Clin. Pract. 2016, 114, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Hindy, G.; Mollet, I.G.; Rukh, G.; Ericson, U.; Orho-Melander, M. Several Type 2 diabetes-associated variants in genes annotated to WNT signalling interact with dietary fibre in relation to incidence of Type 2 diabetes. Genes Nutr. 2016, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Hindy, G.; Sonestedt, E.; Ericson, U.; Jing, X.-J.; Zhou, Y.; Hansson, O.; Renström, E.; Wirfält, E.; Orho-Melander, M. Role of TCF7L2 risk variant and dietary fibre intake on incident Type 2 diabetes. Diabetologia 2012, 55, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- InterAct Consortium. Investigation of gene-diet interactions in the incretin system and risk of Type 2 diabetes: The EPIC-InterAct study. Diabetologia 2016, 59, 2613–2621. [Google Scholar]
- Chimienti, F. Zinc, pancreatic islet cell function and diabetes: New insights into an old story. Nutr. Res. Rev. 2013, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chimienti, F. SLC30A8 mutations in Type 2 diabetes. Diabetologia 2015, 58, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.J.; Chen, R.; Kodama, K.; Ioannidis, J.P.A.; Butte, A.J. Systematic identification of interaction effects between genome- and environment-wide associations in Type 2 diabetes mellitus. Hum. Genet. 2013, 132, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Kanoni, S.; Nettleton, J.A.; Hivert, M.-F.; Ye, Z.; van Rooij, F.J.A.; Shungin, D.; Sonestedt, E.; Ngwa, J.S.; Wojczynski, M.K.; Lemaitre, R.N.; et al. Total zinc intake may modify the glucose-raising effect of a zinc transporter (SLC30A8) variant: A 14-cohort meta-analysis. Diabetes 2011, 60, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- Sonestedt, E.; Lyssenko, V.; Ericson, U.; Gullberg, B.; Wirfält, E.; Groop, L.; Orho-Melander, M. Genetic variation in the glucose-dependent insulinotropic polypeptide receptor modifies the association between carbohydrate and fat intake and risk of Type 2 diabetes in the Malmo Diet and Cancer cohort. J. Clin. Endocrinol. Metab. 2012, 97, E810–E818. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; McKeown, N.M.; Kanoni, S.; Lemaitre, R.N.; Hivert, M.-F.; Ngwa, J.; van Rooij, F.J.A.; Sonestedt, E.; Wojczynski, M.K.; Ye, Z.; et al. Interactions of dietary whole-grain intake with fasting glucose- and insulin-related genetic loci in individuals of European descent: A meta-analysis of 14 cohort studies. Diabetes Care 2010, 33, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Rojo, R.; Delgado-Lista, J.; Lee, Y.-C.; Lai, C.-Q.; Perez-Martinez, P.; Rangel-Zuñiga, O.; Smith, C.E.; Hidalgo, B.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; et al. Interaction of an S100A9 gene variant with saturated fat and carbohydrates to modulate insulin resistance in 3 populations of different ancestries. Am. J. Clin. Nutr. 2016, 104, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Zhang, X.; Lee, N.R.; Jin, H.-S. TRPV1 gene polymorphisms are associated with Type 2 diabetes by their interaction with fat consumption in the Korean genome epidemiology study. J. Nutrigenet. Nutrigenom. 2016, 9, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Hsu, Y.-H.; Niu, T.; Manson, J.E.; Buring, J.E.; Liu, S. Common genetic variants of the ion channel transient receptor potential membrane melastatin 6 and 7 (TRPM6 and TRPM7), magnesium intake, and risk of Type 2 diabetes in women. BMC Med. Genet. 2009, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Chen, C.-H.; Hu, C.; Long, J.; Hee Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for Type 2 diabetes in east Asians. Nat. Genet. 2011, 44, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kaido, T.; Yebra, M.; Cirulli, V.; Rhodes, C.; Diaferia, G.; Montgomery, A.M. Impact of defined matrix interactions on insulin production by cultured human beta-cells: Effect on insulin content, secretion, and gene transcription. Diabetes 2006, 55, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Huang, T.; Li, K.; Chen, Y.; Xie, H.; Xu, D.; Sun, J.; Li, D. Modulation of the association between the PEPD variant and the risk of Type 2 diabetes by n-3 fatty acids in Chinese Hans. J. Nutrigenet. Nutrigenom. 2015, 8, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Lamri, A.; Abi Khalil, C.; Jaziri, R.; Velho, G.; Lantieri, O.; Vol, S.; Froguel, P.; Balkau, B.; Marre, M.; Fumeron, F. Dietary fat intake and polymorphisms at the PPARG locus modulate BMI and Type 2 diabetes risk in the D.E.S.I.R. prospective study. Int. J. Obes. 2012, 36, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Park, J.E.; Choi, Y.J.; Huh, K.B.; Chang, N.; Kim, W.Y. Carbohydrate intake interacts with SNP276G>T polymorphism in the adiponectin gene to affect fasting blood glucose, HbA1C, and HDL cholesterol in Korean patients with Type 2 diabetes. J. Am. Coll. Nutr. 2013, 32, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, A.; Crepostnaia, D.; Maniou, Z.; Lewis, F.J.; Hall, W.L.; Sanders, T.A.B.; O’Dell, S.D.; MARINA study team. Adiponectin gene variant interacts with fish oil supplementation to influence serum adiponectin in older individuals. J. Nutr. 2013, 143, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Arnett, D.K.; Parnell, L.D.; Smith, C.E.; Li, D.; Borecki, I.B.; Tucker, K.L.; Ordovás, J.M.; Lai, C.-Q. Modulation by dietary fat and carbohydrate of IRS1 association with Type 2 diabetes traits in two populations of different ancestries. Diabetes Care 2013, 36, 2621–2627. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Parnell, L.D.; Smith, C.E.; Lee, Y.-C.; Jamal-Allial, A.; Ma, Y.; Li, D.; Tucker, K.L.; Ordovás, J.M.; Lai, C.-Q. Circulating 25-hydroxyvitamin D, IRS1 variant rs2943641, and insulin resistance: Replication of a gene-nutrient interaction in 4 populations of different ancestries. Clin. Chem. 2014, 60, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.; Schreiber, S.; Joost, H.-G.; Boeing, H.; Döring, F. A two-step association study identifies CAV2 rs2270188 single nucleotide polymorphism interaction with fat intake in Type 2 diabetes risk. J. Nutr. 2011, 141, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Azorín, C.; Sorlí, J.V.; Asensio, E.M.; Coltell, O.; Martínez-González, M.; Salas-Salvadó, J.; Covas, M.-I.; Arós, F.; Lapetra, J.; Serra-Majem, L.; et al. Associations of the FTO rs9939609 and the MC4R rs17782313 polymorphisms with Type 2 diabetes are modulated by diet, being higher when adherence to the Mediterranean diet pattern is low. Cardiovasc. Diabetol. 2012, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; Coltell, O.; Sorlí, J.; Estruch, R.; Quiles, L.; Martínez-González, M.; Salas-Salvadó, J.; Castañer, O.; Arós, F.; Ortega-Calvo, M.; et al. Polymorphism of the transcription factor 7-like 2 gene (TCF7L2) interacts with obesity on Type-2 diabetes in the PREDIMED study emphasizing the heterogeneity of genetic variants in type-2 diabetes risk prediction: time for obesity-specific genetic risk scores. Nutrients 2016, 8, 793. [Google Scholar]
- Dashti, H.S.; Smith, C.E.; Lee, Y.-C.; Parnell, L.D.; Lai, C.-Q.; Arnett, D.K.; Ordovás, J.M.; Garaulet, M. CRY1 circadian gene variant interacts with carbohydrate intake for insulin resistance in two independent populations: Mediterranean and North American. Chronobiol. Int. 2014, 31, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Koochakpoor, G.; Hosseini-Esfahani, F.; Daneshpour, M.S.; Hosseini, S.A.; Mirmiran, P. Effect of interactions of polymorphisms in the Melanocortin-4 receptor gene with dietary factors on the risk of obesity and Type 2 diabetes: A systematic review. Diabet. Med. 2016, 33, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; Qi, L.; Tai, E.S.; Deurenberg-Yap, M.; Tan, C.E.; Chew, S.K.; Ordovas, J.M. Perilipin gene variation determines higher susceptibility to insulin resistance in Asian women when consuming a high-saturated fat, low-carbohydrate diet. Diabetes Care 2006, 29, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Yu, D.; Jin, X.; Li, C.; Zhu, F.; Zheng, Z.; Lv, C.; He, X. The association between the FABP2 Ala54Thr variant and the risk of Type 2 diabetes mellitus: A meta-analysis based on 11 case-control studies. Int. J. Clin. Exp. Med. 2015, 8, 5422–5429. [Google Scholar] [PubMed]
- Gouda, H.N.; Sagoo, G.S.; Harding, A.-H.; Yates, J.; Sandhu, M.S.; Higgins, J.P.T. The association between the peroxisome proliferator-activated receptor-gamma2 (PPARG2) Pro12Ala gene variant and Type 2 diabetes mellitus: A HuGE review and meta-analysis. Am. J. Epidemiol. 2010, 171, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, K.; Xu, S.; Chen, G.; Di, H.; Cao, M.; Liu, C. Association between ADIPOQ +45T>G polymorphism and Type 2 diabetes: A systematic review and meta-analysis. Int. J. Mol. Sci. 2014, 16, 704–723. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qiao, Y.; Wang, C.; Zhang, G.; Zhang, X.; Xu, L. Associations between two single-nucleotide polymorphisms (rs1801278 and rs2943641) of insulin receptor substrate 1 gene and Type 2 diabetes susceptibility: A meta-analysis. Endocrine 2016, 51, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, B.; Xia, W.; Yan, J.; Liu, H.-Y.; Hu, L.; Liu, S.-M. FTO genotype and Type 2 diabetes mellitus: spatial analysis and meta-analysis of 62 case-control studies from different regions. Genes 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the CLOCK components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Zhang, C.; Greenberg, A.; Hu, F.B. Common variations in perilipin gene, central obesity, and risk of Type 2 diabetes in US women. Obesity 2008, 16, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, C.; Xie, J.; Xu, G.; Li, Y.; Liu, J.; Chen, B.; Pan, J.; Shen, M.; Yang, L.; Hu, D. Association between three genetic variants of the Perilipin Gene (PLIN) and glucose metabolism: Results from a replication study among Chinese adults and a meta-analysis. Endocr. Res. 2013, 38, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Marín, C.; Pérez-Jiménez, F.; Gómez, P.; Delgado, J.; Paniagua, J.A.; Lozano, A.; Cortés, B.; Jiménez-Gómez, Y.; Gómez, M.J.; López-Miranda, J. The Ala54Thr polymorphism of the fatty acid-binding protein 2 gene is associated with a change in insulin sensitivity after a change in the type of dietary fat. Am. J. Clin. Nutr. 2005, 82, 196–200. [Google Scholar] [PubMed]
- Franks, P.W.; McCarthy, M.I. Exposing the exposures responsible for Type 2 diabetes and obesity. Science 2016, 354, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Sommese, L.; Zullo, A.; Mancini, F.P.; Fabbricini, R.; Soricelli, A.; Napoli, C. Clinical relevance of epigenetics in the onset and management of Type 2 diabetes mellitus. Epigenetics 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S. Diabetes and pancreas size, does it matter? J. Diabetes Investig. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Carrasco, M.; Delgado, I.; Cobo, N.; Tejedo, J.R.; Bedoya, F.J.; Gauthier, B.; Soria, B.; Martín, F. Signaling pathways and transcription factors involved in pancreatic islet development. In Advances in Experimental Medicine and Biology: The Islets of Langerhans 2; Islam, S., Ed.; Springer: Berlin, Germany, 2014; pp. 109–128. [Google Scholar]
- Volkmar, M.; Dedeurwaerder, S.; Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 31, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Georgia, S.; Tschen, S.-I.; Fan, G.; Bhushan, A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell 2011, 20, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Dayeh, T.; Kirkpatrick, C.L.; Wollheim, C.B.; Dekker Nitert, M.; Ling, C. DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med. Genet. 2013, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific control of pancreatic endocrine β- and δ-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2671. [Google Scholar] [CrossRef] [PubMed]
- Tarabra, E.; Pelengaris, S.; Khan, M. A simple matter of life and death-the trials of postnatal beta-cell mass regulation. Int. J. Endocrinol. 2012, 2012, 516718. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gu, X.; Su, I.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Mosley, A.L.; Corbett, J.A.; Ozcan, S. Glucose regulation of insulin gene expression requires the recruitment of p300 by the beta-cell-specific transcription factor Pdx-1. Mol. Endocrinol. 2004, 18, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Evans-Molina, C.; Robbins, R.D.; Kono, T.; Tersey, S.A.; Vestermark, G.L.; Nunemaker, C.S.; Garmey, J.C.; Deering, T.G.; Keller, S.R.; Maier, B.; et al. Peroxisome proliferator-activated receptor gamma activation restores islet function in diabetic mice through reduction of endoplasmic reticulum stress and maintenance of euchromatin structure. Mol. Cell. Biol. 2009, 29, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Deiuliis, J.A. MicroRNAs as regulators of metabolic disease: Pathophysiologic significance and emerging role as biomarkers and therapeutics. Int. J. Obes. 2016, 40, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Q.; Franck, N.; Egan, B.; Sjögren, R.J.O.; Katayama, M.; Duque-Guimaraes, D.; Arner, P.; Zierath, J.R.; Krook, A. Autocrine role of interleukin-13 on skeletal muscle glucose metabolism in type 2 diabetic patients involves microRNA let-7. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1359–E1366. [Google Scholar] [CrossRef] [PubMed]
- Crépin, D.; Benomar, Y.; Riffault, L.; Amine, H.; Gertler, A.; Taouis, M. The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signalling impairment. Mol. Cell. Endocrinol. 2014, 384, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Meng, C.; Guo, X.; Cheruku, P.S.; Shi, L.; Xu, H.; Li, H.; Wang, G.; Evans, A.R.; Safe, S.; et al. A novel regulator of macrophage activation: MiR-223 in obesity-associated adipose tissue inflammation. Circulation 2012, 125, 2892–2903. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Zhu, L.; Gupta, N.; Chang, Y.; Fang, F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol. Endocrinol. 2007, 21, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, C.L.; Peck, B.C.E.; Fannin, E.E.; Beysen, C.; Miao, J.; Landstreet, S.R.; Ding, S.; Turaga, V.; Lund, P.K.; Turner, S.; et al. MicroRNA-29 fine-tunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes 2014, 63, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Hausser, J.; Trajkovski, M.; Braun, M.; Collins, S.; Rorsman, P.; Zavolan, M.; Stoffel, M. miR-375 maintains normal pancreatic α- and beta-cell mass. Proc. Natl. Acad. Sci. USA 2009, 106, 5813–5818. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, M.V.; Joglekar, V.M.; Hardikar, A.A. Expression of islet-specific microRNAs during human pancreatic development. Gene Expr. Patterns 2009, 9, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.T.; Dayeh, T.A.; Kirkpatrick, C.L.; Taneera, J.; Kumar, R.; Groop, L.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia 2011, 54, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renström, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with Type 2 diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Lovis, P.; Roggli, E.; Laybutt, D.R.; Gattesco, S.; Yang, J.-Y.; Widmann, C.; Abderrahmani, A.; Regazzi, R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008, 57, 2728–2736. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. New emerging tasks for microRNAs in the control of β-cell activities. Biochim. Biophys. Acta 2016, 1861, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Akirav, E.M.; Lebastchi, J.; Galvan, E.M.; Henegariu, O.; Akirav, M.; Ablamunits, V.; Lizardi, P.M.; Herold, K.C. Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 19018–19023. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, B.; Irvin, M.R.; Sha, J.; Zhi, D.; Aslibekyan, S.; Absher, D.; Tiwari, H.K.; Kabagambe, E.K.; Ordovas, J.M.; Arnett, D.K. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the genetics of lipid lowering drugs and diet network study. Diabetes 2014, 63, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Xia, Y.; Bell, C.G.; Yet, I.; Ferreira, T.; Ward, K.J.; Gao, F.; Loomis, A.K.; Hyde, C.L.; Wu, H.; et al. An integrated epigenomic analysis for Type 2 diabetes susceptibility loci in monozygotic twins. Nat. Commun. 2014, 5, 5719. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Jansson, P.A.; Perfilyev, A.; Volkov, P.; Pedersen, M.; Svensson, M.K.; Poulsen, P.; Ribel-Madsen, R.; Pedersen, N.L.; Almgren, P.; et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with Type 2 diabetes. Diabetes 2014, 63, 2962–2976. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.; Volkov, P.; Salö, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.L.; Wollheim, C.B.; Eliasson, L.; Rönn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from Type 2 Diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.E.; Liu, S.; Wang, D.; Shen, J.; Hu, Q.; Hong, C.-C.; Newman, V.A.; Zhao, H. Reduction of dietary glycaemic load modifies the expression of microRNA potentially associated with energy balance and cancer pathways in pre-menopausal women. Br. J. Nutr. 2013, 109, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, C.; Iaffaldano, L.; Ferrigno, M.; Labruna, G.; Maruotti, G.M.; Quaglia, F.; Capobianco, V.; Di Noto, R.; Del Vecchio, L.; Martinelli, P.; et al. Characterization and predicted role of the microRNA expression profile in amnion from obese pregnant women. Int. J. Obes. 2014, 38, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.F.; Fazzari, M.J.; Niu, H.; Barzilai, N.; Simmons, R.A.; Greally, J.M. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J. Biol. Chem. 2010, 285, 15111–15118. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of Type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Smith, N.H.; Nitert, M.D.; Ackers-Johnson, M.; Uribe-Lewis, S.; Ito, Y.; Jones, R.H.; Marquez, V.E.; Cairns, W.; Tadayyon, M.; et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 2011, 108, 5449–5454. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Chillaron, J.C.; Isganaitis, E.; Charalambous, M.; Gesta, S.; Pentinat-Pelegrin, T.; Faucette, R.R.; Otis, J.P.; Chow, A.; Diaz, R.; Ferguson-Smith, A.; et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 2009, 58, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Radford, E.J.; Ito, M.; Shi, H.; Corish, J.A.; Yamazawa, K.; Isganaitis, E.; Seisenberger, S.; Hore, T.A.; Reik, W.; Erkek, S.; et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 2014, 345, 1255903. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.A.; Bale, T.L. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology 2009, 150, 4999–5009. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.-L.; Wang, F.-F.; Shu, J.; Tian, S.; Jiang, Y.; Zhang, D.; Wang, N.; Luo, Q.; Zhang, Y.; Jin, F.; et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 2012, 61, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-F.; Lin, R.C.Y.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.A.; Bale, T.L. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 2011, 152, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Fullston, T.; Ohlsson Teague, E.M.C.; Palmer, N.O.; DeBlasio, M.J.; Mitchell, M.; Corbett, M.; Print, C.G.; Owens, J.A.; Lane, M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. 2013, 27, 4226–4243. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.C.; van der Meulen, J.H.; Michels, R.P.; Osmond, C.; Barker, D.J.; Hales, C.N.; Bleker, O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- De Rooij, S.R.; Painter, R.C.; Phillips, D.I.W.; Osmond, C.; Michels, R.P.J.; Godsland, I.F.; Bossuyt, P.M.M.; Bleker, O.P.; Roseboom, T.J. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 2006, 29, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Hillier, T.A.; Pedula, K.L.; Schmidt, M.M.; Mullen, J.A.; Charles, M.-A.; Pettitt, D.J. Childhood obesity and metabolic imprinting: The ongoing effects of maternal hyperglycemia. Diabetes Care 2007, 30, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Finer, S.; Iqbal, M.S.; Lowe, R.; Ogunkolade, B.W.; Pervin, S.; Mathews, C.; Smart, M.; Alam, D.S.; Hitman, G.A. Is famine exposure during developmental life in rural Bangladesh associated with a metabolic and epigenetic signature in young adulthood? A historical cohort study. BMJ Open 2016, 6, e011768. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar] [PubMed]
- Ludwig, D.S.; Currie, J. The association between pregnancy weight gain and birthweight: A within-family comparison. Lancet 2010, 376, 984–990. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Kaati, G.; Bygren, L.O.; Edvinsson, S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. 2002, 10, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Veenendaal, M.; Painter, R.; de Rooij, S.; Bossuyt, P.; van der Post, J.; Gluckman, P.; Hanson, M.; Roseboom, T. Transgenerational effects of prenatal exposure to the 1944–1945 Dutch famine. BJOG Int. J. Obstet. Gynaecol. 2013, 120, 548–554. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
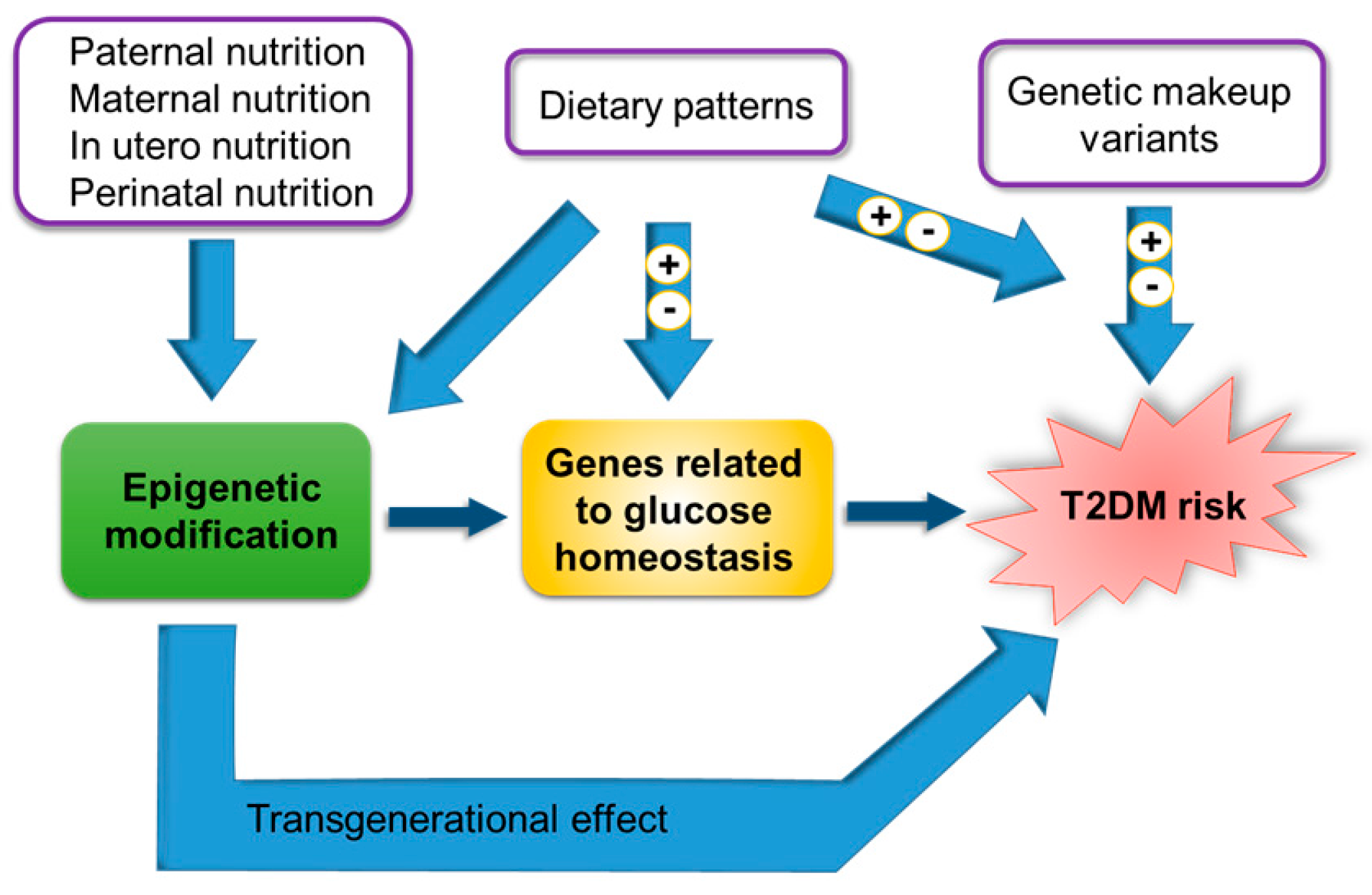{kind=link}
| Flavonoids | Genes and Gene Products Affected | Function | Experimental Model | Reference |
|---|---|---|---|---|
| Flavan-3-ols | ||||
| catechins | ↑ PI3K, ↑ eNOS signalling system | ↓ Hyperglycemia | STZ-diabetic rats | [20] |
| epigallocatechingallate (EGCG) | ↑ Irs2, ↑ Akt, ↑ Foxo1, ↑ Pdx1 | ↑ Viability of β-cell, ↑ Insulin secretion | RIN-m5F cells | [21] |
| ↑ AMPK ↑ PI3K/Akt pathway | ↓ IR | dexamethasone-induced rat L6 cells | [22] | |
| ↑ PI3K phosphorylation | ↑ Glucose uptake | differentiated rat L6 myotubes | [23] | |
| ↑ Bcl-2 | ↓ Apoptosis, ↑ Glucose uptake | RINm5F cells | [24] | |
| ↓ l-cpt-1, ↓ Ddit3, ↓ Ppp1r15a, ↓ Cdkn1a | ↑ Insulin secretion, preserve islet structure | db/db mice | [25] | |
| ↑ Glucokinase, ↑ Acyl CoA oxidase-1 (ACO-1) ↑ Carnitine palmitoyl transferase-1 (CPT-1) ↓ Phosphofructokinase ↓ Glut 1 | ↑ Insulin secretion ↓ Gluconeogenesis ↑ Glycolysis | db/db mice and ZDF rats | [26] | |
| Flavanones | ||||
| naringin or hesperidin | ↑ Gk (liver), ↑ Glut 4, ↑ PPAR-γ | ↓ Hyperglycemia | C57BL/KsJ-db/db mice | [30] |
| naringin | ↑ PPAR-γ, ↑ Hsp | ↓ Hyperglycemia, ↓ Hyperinsulinemia, ↓ IR, ↑ β-cell function | HFD-STZ-induced T2D rats | [31] |
| ↓ PEPCK ↓ G6Pase | ↓ Hyperglycemia, ↓ Hyperinsulinemia, ↓ IR, ↑ β-cell function | HFD-STZ-induced T2D rats | [30] | |
| Flavonols | ||||
| quercetin | ↑ Antioxidant enzyme activity ↓ Nitric oxide (NO) production | ↓ Lipid peroxidation, ↑ β-cell preservation | STZ-induced DM mice | [32] |
| ↑ Nrf-2/HO-1 activation ↓ NF-κB | ↓ High glucose induced apoptosis | Dorsal root ganglion neurons of rats | [33] | |
| ↓ Cdkn1a | ↓ Hyperglycemia, ↑ Insulin plasma levels ↑ Pancreatic cell proliferation | STZ-induced DM mice | [35] | |
| ↓ interferon-γ, ↓ IL-1α, ↓ IL-4 | ↓ Circulating markers of inflammation | C57Bl/6j mice | [36] | |
| ↑ adiponectin, ↓ NOx levels in plasma, ↓ TNF-α | ↓ Dyslipidemia ↓ Hypertension ↓Hyperinsulinemia | obese Zucker rats | [37] | |
| Flavones | ||||
| luteolin, apigenin | ↓ iNos ↓ NF-κB | ↓ Apoptosis ↓ IL-1β- and IFN-γ-mediated inhibition of insulin secretion | RINm5F cells | [38] |
| Isoflavones | ||||
| daidzein and genistein | ↓ PPAR-γ ↓ Glut-2 ↓ SREBP-1 | ↑ Insulin secretion ↓Hyperinsulinemia | rats and pancreatic islets of rats in a HFD | [39] |
| genistein | ↑ cAMP/PKA-dependent ERK1/2 signalling pathway | ↓ Hyperglycemia ↑ Glucose tolerance, ↑ Insulin plasma levels ↑ β-cell preservation | STZ-induced DM mice | [40] |
| Anthocyanins | ||||
| ↑ heme oxygenase-1 (HO-1) gene ↑ ERK1/2 and PI3K/Akt signalling | ↑ Insulin secretion ↓Apoptosis | pancreatic β INS-1 cells and primary islets | [42] | |
| ↑ Glut 4 ↓ retinol binding protein 4 (RBP4) | ↓ Hyperglycemia ↑ Insulin sensitivity | T2D KK-A(y) mice | [43] | |
| ↑ Glut 4 ↑ AMPK, ↑ PPAR-α, ↓ Acetyl-CoA carboxylase ↑ Acyl-CoA oxidase (ACO), ↑ Carnitine palmitoyltransferase-1A-(CPT-1) | ↓ Hyperglycemia ↑ Insulin sensitivity | T2D KK-A(y) mice | [44] | |
| Vitamins | Genes and Gene Products Affected | Function | Experimental Model | References |
|---|---|---|---|---|
| vitamin D | ||||
| ↑ Human insulin receptor gene | ↑ Insulin sensitivity | U-937 human promonocytic cells | [75] | |
| ↓ TNF-α, IL-6, IL-1, and IL-8 | ↓ β-cell cytokine-induced apoptosis | CD14+ monocytes isolated from peripheral blood insulin-treated T2DM patients | [77] | |
| Biotin | ||||
| ↑ Foxa2, ↑ Pdx-1 ↑ Hnf-4α ↑ Ins, ↑ Gk, ↑ Cacna1d, ↑ Acac | ↑ Insulin secretion ↑ Islet function | BALB/cAnN Hsd mice | [98] | |
| ↑ PPAR-γ ↑ IRS-1 ↓ NF-κB | ↑ Antioxidant ↓ Hyperlipidaemia ↓ Inflammation ↓ Hyperglycaemia | T2DM rat model induced by high-fat diet (HFD) and low-dose STZ | [99] | |
| Riboflavin | ||||
| ↓ IL-6 upregulation | ↓ Cytokines-induced p38 phosphorylation | Insulinoma NIT-1 cells and isolated rodent islets | [111] | |
| Nicotinamide | ||||
| ↑ MafA | ↑ Insulin synthesis | INS-1 cells, pancreatic islets | [115] | |
| ↑ Pdx-1 | Pancreatic β-cell differentiation | Mouse embryonic stem cells | [112,113,114] | |
| Genes Related with Stimulus-Secretion Coupling | |||||
| Gene | SNP | T2D Risk | Types of Studies/Dietary Factors | Effect of Interaction between SNP and Dietary Factors | References |
| TCF7L2 | rs12255372 G>T | Allele-T risk increase | MDCS cohort/fibre intake | Higher risk in individuals carrying the allele-T. Smaller risk in individuals homozygous GG | [205] |
| EPIC InterAct study/Coffee Intake | Smaller risk in individuals carrying the allele-T | [207] | |||
| rs7903146 C>T | Allele-T risk increase | MDCS cohort/fibre intake | Higher risk in individuals carrying the allele-T. Smaller risk Individuals homozygous CC | [205] | |
| ZBED3 | rs4457053 G>A | Allele-G risk increase | MDCS cohort/fibre intake | Smaller risk in individuals homozygous for the risk factor (GG) | [205] |
| NOTCH2 | rs10923931 G>T | Allele-T risk increase | MDCS cohort/fibre intake | Smaller risk in individuals carrying the allele-T | [205] |
| SLC30A8 | rs13266634 C>T | Allele-C risk increase | NHANES cohort study/trans-β-carotene intake | Smaller risk in individuals carrying the allele-C | [209,210] |
| NHANES cohort study/γ-tocoferol intake | Higher risk in individuals carrying the allele-C | [209,210] | |||
| rs11558471 A>G | Allele-A increases levels of fasting plasma glucose | 14-Cohort meta-analysis study/zinc intake | Smaller levels of fasting plasma glucose in individuals carrying the allele-A | [211] | |
| GIPR | rs10423928 T>A | Allele-A risk increase | MDCS cohort/fats and carbohydrates intake | Smaller risk in individuals homozygous for de risk factor AA consuming high-fat, low-carbohydrate diets. High-carbohydrate, low-fat diets benefit of the population homozygous for the T-allele | [212] |
| GCKR | rs780094 C>T | Allele-T risk increase | 14-cohort meta-analysis study/whole-grain intake | Smaller risk in individuals carrying the allele-T | [213] |
| S100A9 | rs3014866 C>T | Allele-T risk decrease | Intervention trial in CORDIOPERV, GOLDN and BPRHS/SFAs:carbohydrates ration | Ration high: Individuals with the non-protective variant (CC) had a higher HOMA-IR. Ration low: Individuals with the non-protective variant (CC) had a normal HOMA-IR. | [214] |
| TRPV1 | rs161364 T>C | Allele-C risk decrease | Cohort study/high fat diet intake | Smaller risk in individuals carrying the allele-C Higher risk in individuals carrying the allele-T | [215] |
| rs8065080 T>C | |||||
| TRPM6 | rs2274924 C>T | Allele-T risk increase | Cases-control study/magnesium intake <250 mg/day | Higher risk in women carrying the allele-T | [216] |
| rs3750425 C>T | |||||
| PEPD | rs3786897 G>A | Allele-A risk increase | Cases-control study/High n-3 PUFAs | The risk disappears in individuals carrying the allele-A | [219] |
| Genes Related with Insulin Signaling | |||||
| Gene | SNP | T2D | Dietary Factors | Effect of Interaction between SNP and Dietary Factors | References |
| PPAR-γ | rs180282 C>G | Allele-G risk increase | DESIR cohort study/high fat intake increases | Higher risk in individuals carrying the allele-T | [220] |
| ADIPOQ | rs1501299 G>T | Allele-T risk increase | Cohort study/carbohydrate intake | Higher fasting blood glucose and HbA1C concentrations in individuals carrying allele-T | [221] |
| rs2241766 T>G | Allele-G risk increase | MARINA trial study/hight n-3 PUFAs | Smaller risk in individuals carrying the allele-G | [222] | |
| IRS1 | rs7578326 A>G | Allele-G lower risk of IR and lower fasting insulin | GOLDN and BPRHS cohort studies/low total fat and SFA:carbohydrate ratio intake | Decrease resistance to insulin and plasma insulin | [223] |
| rs2943641 C>T | Allele-T lower risk of IR and lower fasting insulin | GOLDN and BPRHS cohort studies/low fat and SFA:carbohydrate ratio intake | Decrease resistance to insulin and plasma insulin | [223] | |
| Cohort study/vitamin D intake | Smaller risk in individuals carrying the allele-T | [224] | |||
| CAV2 | rs2270188 G>T | Allele-T risk increase | Cases-cohort study/high fat or SFA intake | Higher risk in individuals carrying the allele-T. | [225] |
| Other Genes | |||||
| Gene | SNP | T2D | Dietary Factors | Effect of Interaction between SNP and Dietary Factors | References |
| FTO | rs9939609 T>A | Allele-A risk increase | Cases-control study/adherence to Mediterranean diet | The risk disappears in individuals carrying the allele-A | [226] |
| rs8050136 C>A | Allele-A risk increase | Cases-control study/adherence to Mediterranean diet | The risk disappears in individuals carrying the allele-A | [226] | |
| CLOCK | rs4580704 C>G | Allele-G risk decrease | PREDIMED trial study/high MUFA intake | Lower risk in individuals carrying the allele-G | [227] |
| CRY1 | rs2287161 G>C | Allele-C risk increase | Cohorts Mediterranean and North American study/high carbohydrate intake | Increase in HOMA-IR index, fasting insulin and a decrease in QUICKI | [228] |
| MC4R | rs17782313 T>C | Allele-C risk increase | Systematic review study/adherence to Mediterranean diet | The risk disappears in individuals carrying the allele-C | [226,229] |
| PLIN | rs11482 G>A | Allele-A risk increase | cross-sectional study/SFAs:carbohydrates ration | Ration high: women homozygous for AA increased HOMA-IR | [230] |
| FABP2 | rs1799883 G>A | Allele-A risk increase | 11-cases-control meta-analysis study/MUFA intake | Decrease in HOMA-IR index | [231] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, Á.; Berná, G.; Rojas, A.; Martín, F.; Soria, B. Gene-Diet Interactions in Type 2 Diabetes: The Chicken and Egg Debate. Int. J. Mol. Sci. 2017, 18, 1188. https://doi.org/10.3390/ijms18061188
Ortega Á, Berná G, Rojas A, Martín F, Soria B. Gene-Diet Interactions in Type 2 Diabetes: The Chicken and Egg Debate. International Journal of Molecular Sciences. 2017; 18(6):1188. https://doi.org/10.3390/ijms18061188
Chicago/Turabian StyleOrtega, Ángeles, Genoveva Berná, Anabel Rojas, Franz Martín, and Bernat Soria. 2017. "Gene-Diet Interactions in Type 2 Diabetes: The Chicken and Egg Debate" International Journal of Molecular Sciences 18, no. 6: 1188. https://doi.org/10.3390/ijms18061188
APA StyleOrtega, Á., Berná, G., Rojas, A., Martín, F., & Soria, B. (2017). Gene-Diet Interactions in Type 2 Diabetes: The Chicken and Egg Debate. International Journal of Molecular Sciences, 18(6), 1188. https://doi.org/10.3390/ijms18061188