
Aberrant DNA Methylation as a Biomarker and a Therapeutic Target of Cholangiocarcinoma

Abstract
: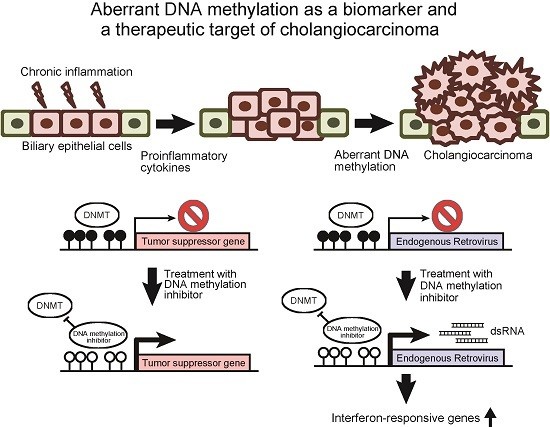
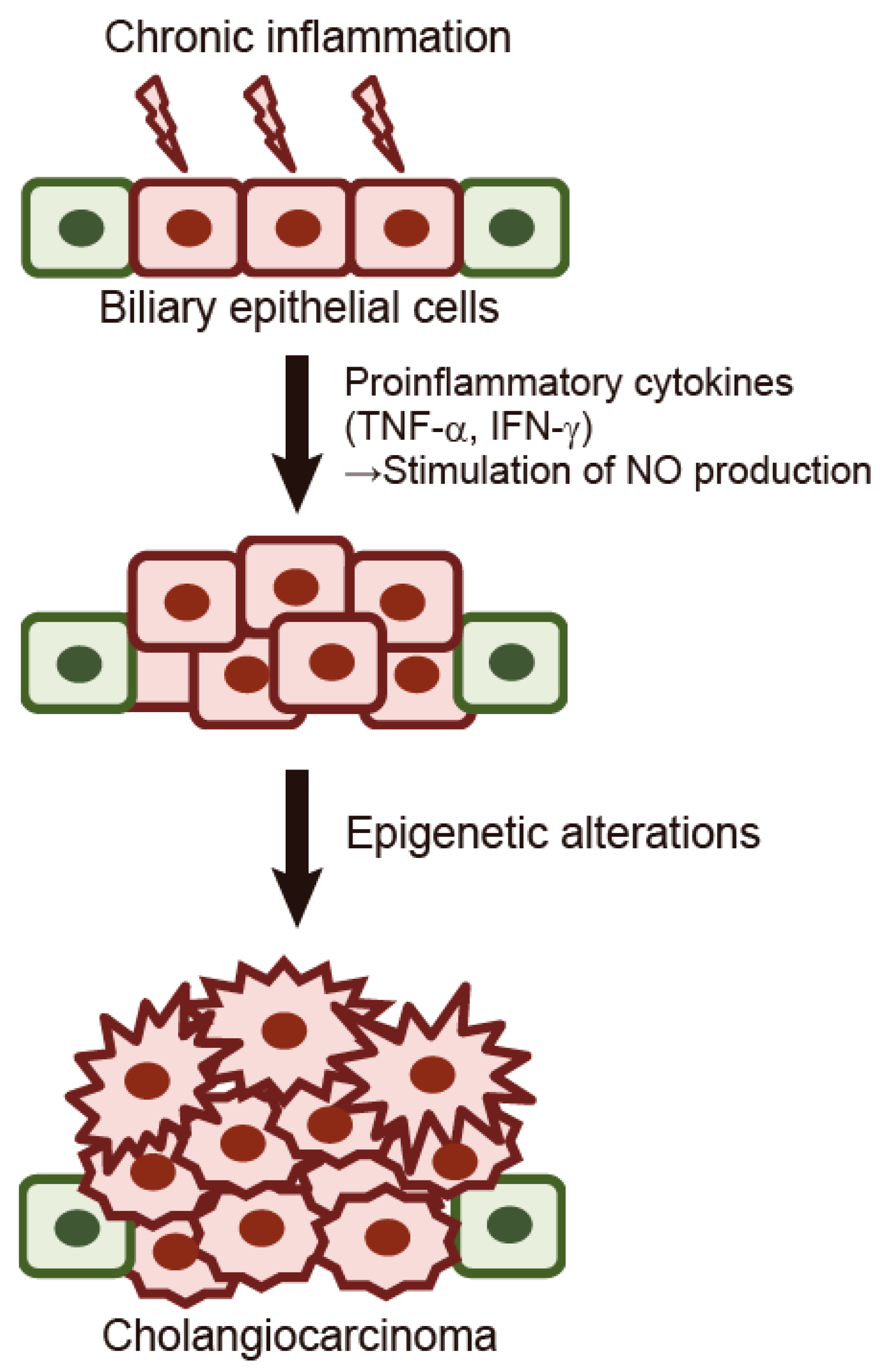
1. Introduction
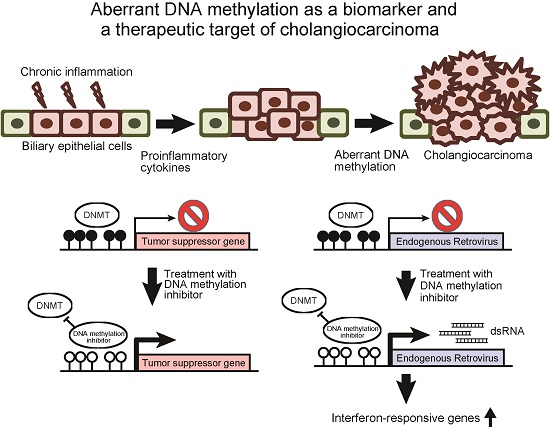
2. Aberrant DNA Methylation as a Biomarker of Cholangiocarcinoma
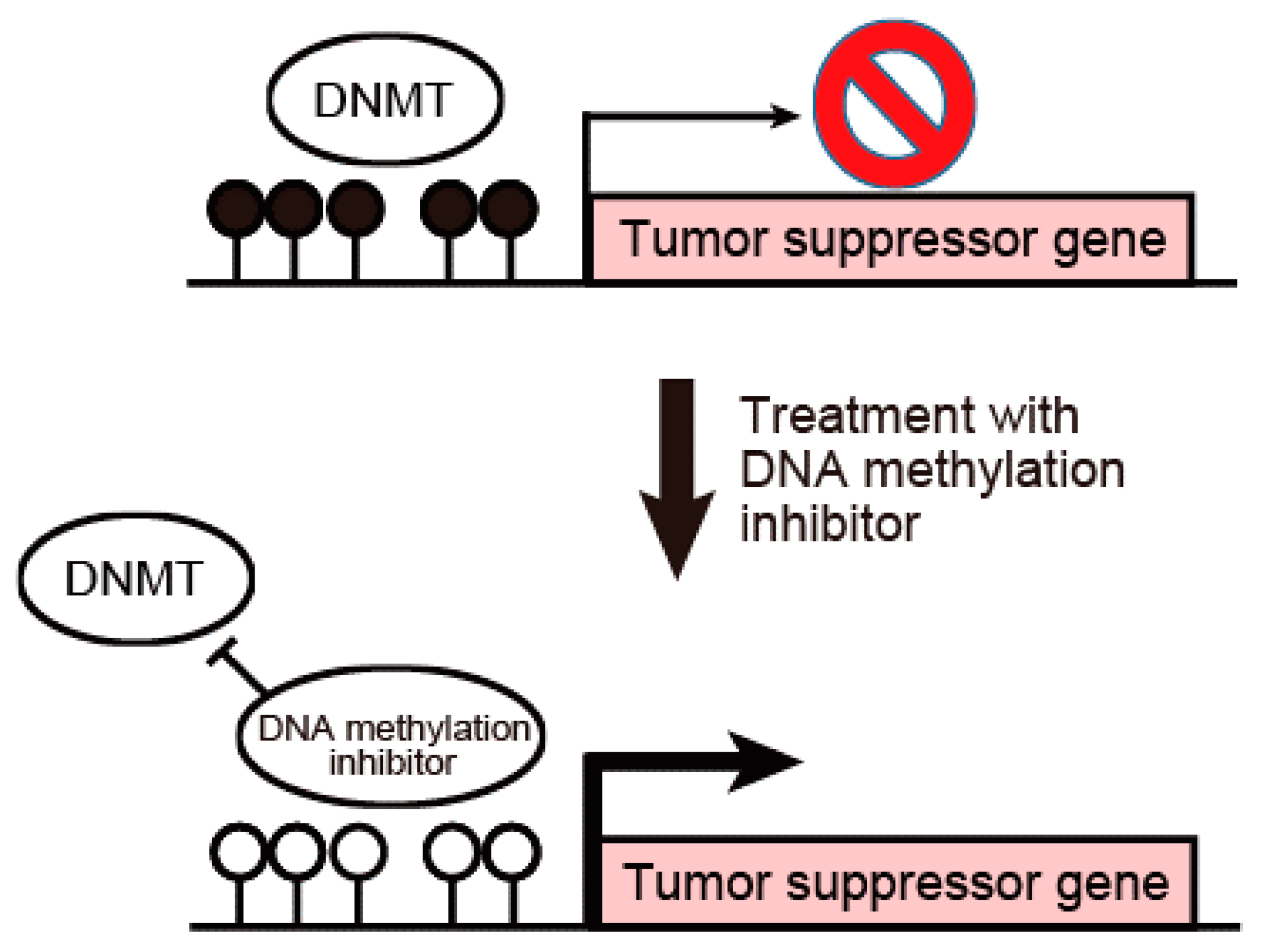
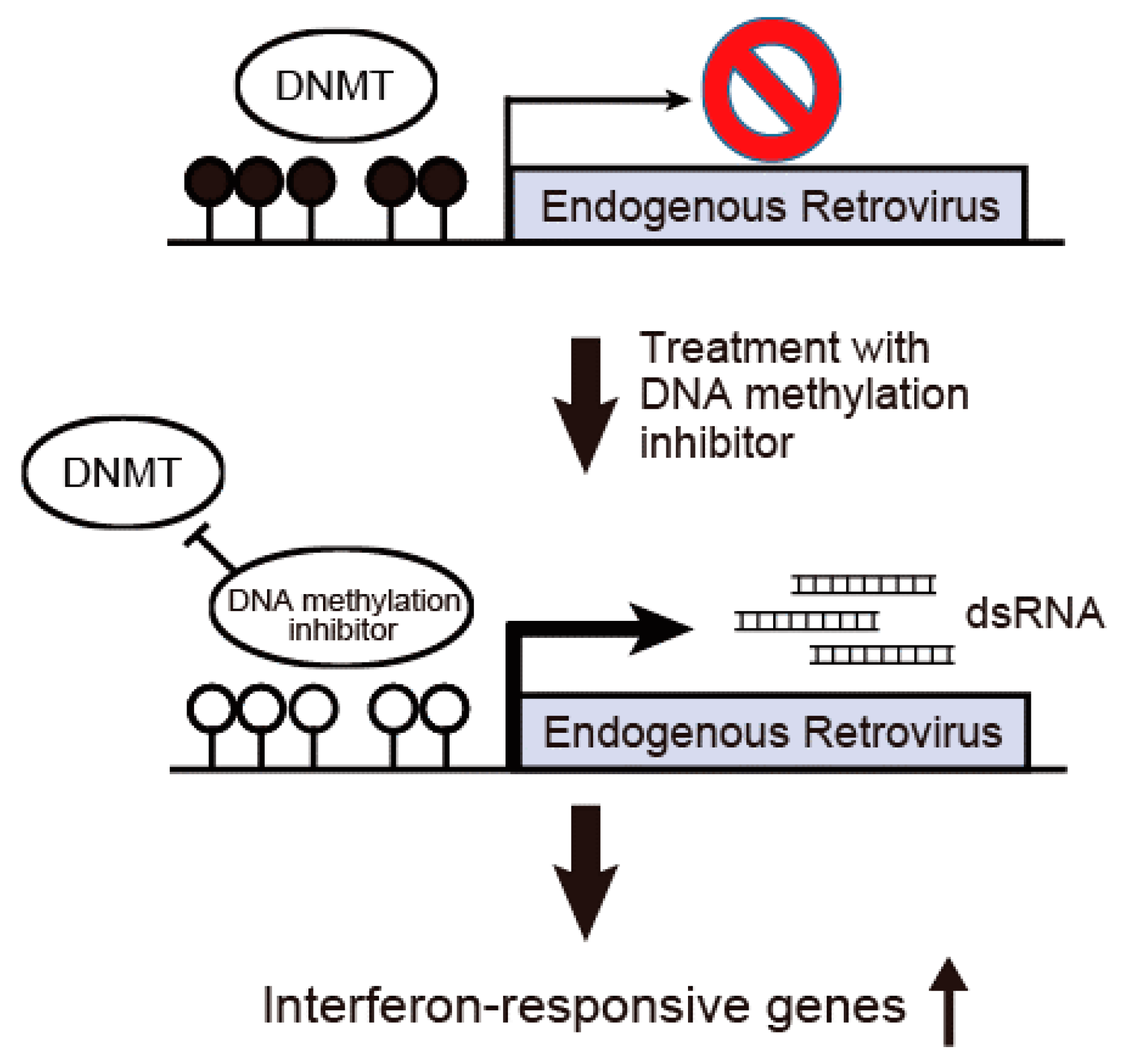
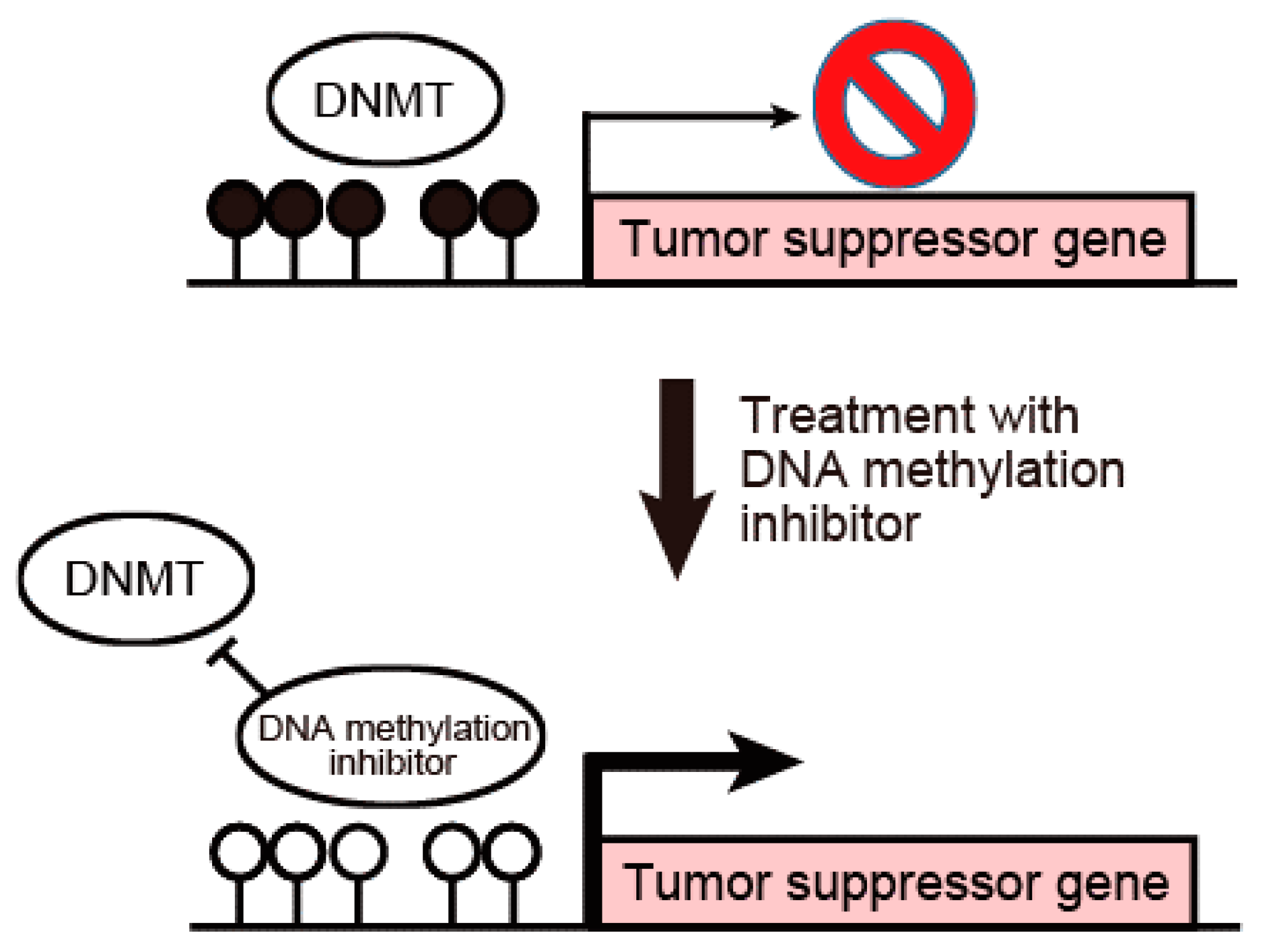
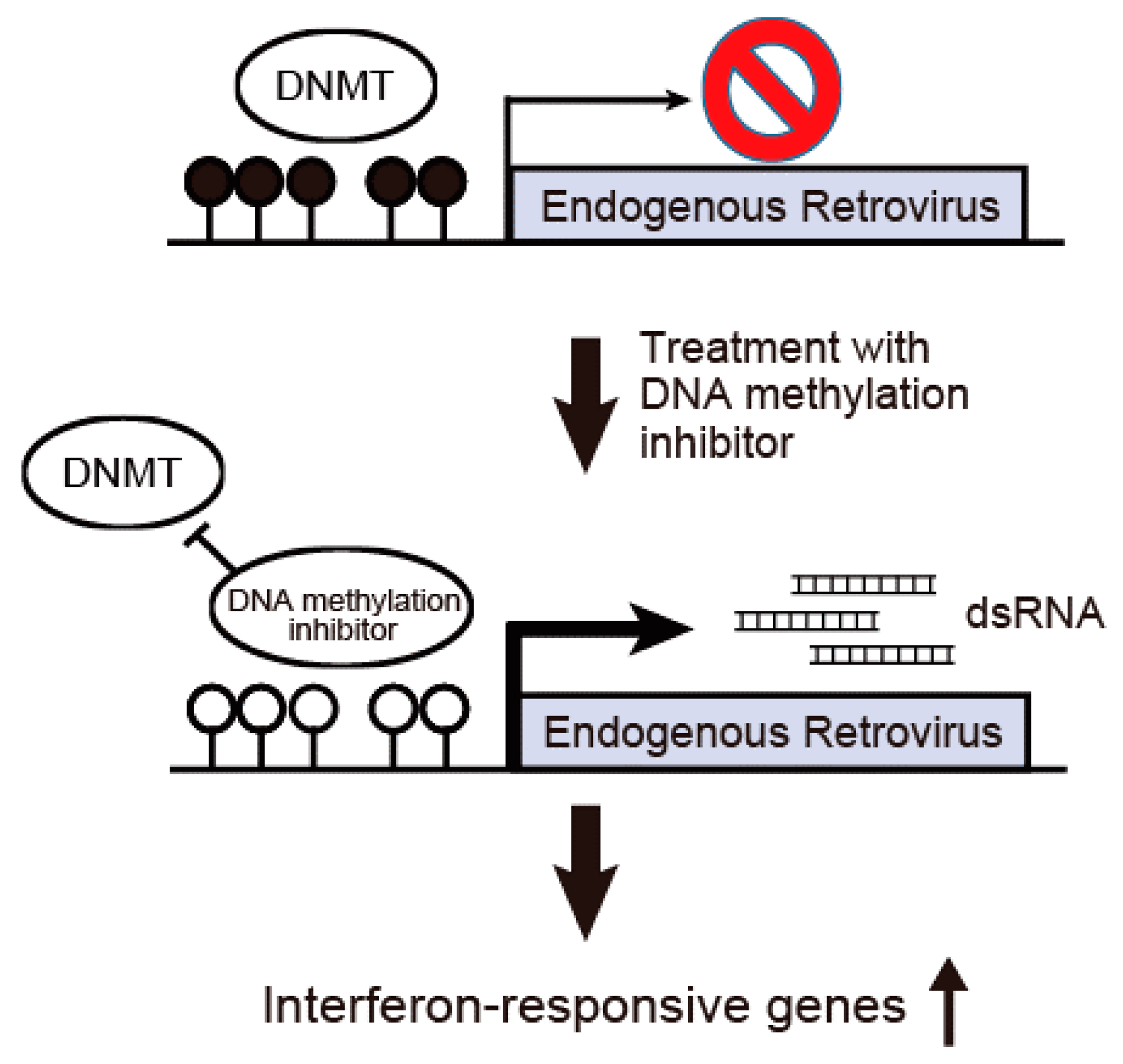
3. DNA Methylation Inhibitors Are Promising Therapeutic Agents against Cholangiocarcinoma
4. Suppression of Tumor Suppressor miRNAs by DNA Methylation in Cholangiocarcinoma
5. Therapeutic Perspectives of DNA Methylation Inhibitors against Cholangiocarcinoma
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Blechacz, B.; Komuta, M.; Roskams, T.; Gores, G.J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; de Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Gal-Yam, E.N.; Saito, Y.; Egger, G.; Jones, P.A. Cancer epigenetics: Modifications, screening, and therapy. Annu. Rev. Med. 2008, 59, 267–280. [Google Scholar] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Hibino, S.; Saito, H. Alterations of epigenetics and microRNA in hepatocellular carcinoma. Hepatol. Res. 2014, 44, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Jones, P.A. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 2006, 5, 2220–2222. [Google Scholar] [CrossRef] [PubMed]
- Takaki, Y.; Saito, Y.; Takasugi, A.; Toshimitsu, K.; Yamada, S.; Muramatsu, T.; Kimura, M.; Sugiyama, K.; Suzuki, H.; Arai, E.; et al. Silencing of microRNA-122 is an early event during hepatocarcinogenesis from non-alcoholic steatohepatitis. Cancer Sci. 2014, 105, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Saito, H.; Liang, G.; Friedman, J.M. Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: A critical review. Clin. Rev. Allergy Immunol. 2014, 47, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Saito, H.; Kaneko, F.; Nakamoto, N.; Tada, S.; Hibi, T. Gene expression associated with the decrease in malignant phenotype of human liver cancer cells following stimulation with a histone deacetylase inhibitor. Int. J. Oncol. 2005, 26, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Saito, Y.; Muramatsu, T.; Otani, A.; Kasai, Y.; Kimura, M.; Saito, H. Inhibitors of enhancer of zeste homolog 2 (EZH2) activate tumor-suppressor microRNAs in human cancer cells. Oncogenesis 2014, 3, e104. [Google Scholar] [CrossRef] [PubMed]
- Limpaiboon, T.; Khaenam, P.; Chinnasri, P.; Soonklang, M.; Jearanaikoon, P.; Sripa, B.; Pairojkul, C.; Bhudhisawasdi, V. Promoter hypermethylation is a major event of hMLH1 gene inactivation in liver fluke related cholangiocarcinoma. Cancer Lett. 2005, 217, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.C.; Lee, J.H.; Boitnott, J.K.; Argani, P.; Furth, E.E.; Wu, T.T. Microsatellite instability in intraductal papillary neoplasms of the biliary tract. Mod. Pathol. 2002, 15, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Vedeld, H.M.; Andresen, K.; Eilertsen, I.A.; Nesbakken, A.; Seruca, R.; Gladhaug, I.P.; Thiis-Evensen, E.; Rognum, T.O.; Boberg, K.M.; Lind, G.E. The novel colorectal cancer biomarkers CDO1, ZSCAN18 and ZNF331 are frequently methylated across gastrointestinal cancers. Int. J. Cancer 2015, 136, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Taniai, M.; Higuchi, H.; Burgart, L.J.; Gores, G.J. p16INK4a promoter mutations are frequent in primary sclerosing cholangitis (PSC) and PSC-associated cholangiocarcinoma. Gastroenterology 2002, 123, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Tannapfel, A.; Busse, C.; Geissler, F.; Witzigmann, H.; Hauss, J.; Wittekind, C. INK4a-ARF alterations in liver cell adenoma. Gut 2002, 51, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Xiaofang, L.; Kun, T.; Shaoping, Y.; Zaiqiu, W.; Hailong, S. Correlation between promoter methylation of p14(ARF), TMS1/ASC, and DAPK, and p53 mutation with prognosis in cholangiocarcinoma. World J. Surg. Oncol. 2012, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Jiang, H.; Zhang, C.S.; Yu, S.P.; Wang, Z.Q.; Su, H.L. Targeted drug regulation on methylation of p53-BAX mitochondrial apoptosis pathway affects the growth of cholangiocarcinoma cells. J. Int. Med. Res. 2012, 40, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Luo, F.; Chen, Y.; Zhu, F.; Wang, J. si-DNMT1 restore tumor suppressor genes expression through the reversal of DNA hypermethylation in cholangiocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Kong, F.M.; Xu, Z.; Yu, S.P.; Sun, F.B.; Zhang, C.S.; Huang, Q.X.; Zhou, X.T.; Song, Z.W. Promoter hypermethylation of death-associated protein kinase gene in cholangiocarcinoma. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 407–411. [Google Scholar] [PubMed]
- Shin, S.H.; Lee, K.; Kim, B.H.; Cho, N.Y.; Jang, J.Y.; Kim, Y.T.; Kim, D.; Jang, J.J.; Kang, G.H. Bile-based detection of extrahepatic cholangiocarcinoma with quantitative DNA methylation markers and its high sensitivity. J. Mol. Diagn. 2012, 14, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, H.; Henson, R.; Meng, F.; Mize-Berge, J.; Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006, 66, 10517–10524. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, K.; Wu, X.; Liu, X.; Li, B.; Zhu, Y.; Yu, Y.; Cheng, Q.; Hu, Z.; Guo, C.; et al. Underexpression of LKB1 tumor suppressor is associated with enhanced Wnt signaling and malignant characteristics of human intrahepatic cholangiocarcinoma. Oncotarget 2015, 6, 18905–18920. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Sriraksa, R.; Zeller, C.; Dai, W.; Siddiq, A.; Walley, A.J.; Limpaiboon, T.; Brown, R. Aberrant DNA methylation at genes associated with a stem cell-like phenotype in cholangiocarcinoma tumors. Cancer Prev. Res. 2013, 6, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Ferrari Junior, A.P.; Lichtenstein, D.R.; Slivka, A.; Chang, C.; Carr-Locke, D.L. Brush cytology during ERCP for the diagnosis of biliary and pancreatic malignancies. Gastrointest. Endosc. 1994, 40, 140–145. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Gores, G.J. Cholangiocarcinoma. Gastroenterology 2005, 128, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Schottenfeld, D.; Beebe-Dimmer, J. Chronic inflammation: A common and important factor in the pathogenesis of neoplasia. CA Cancer J. Clin. 2006, 56, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, E.P.; Wands, J. The molecular pathogenesis of cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Spirli, C.; Fabris, L.; Duner, E.; Fiorotto, R.; Ballardini, G.; Roskams, T.; Larusso, N.F.; Sonzogni, A.; Okolicsanyi, L.; Strazzabosco, M. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology 2003, 124, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Socco, S.; Bovee, R.C.; Palczewski, M.B.; Hickok, J.R.; Thomas, D.D. Epigenetics: The third pillar of nitric oxide signaling. Pharmacol. Res. 2017, 121, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakabayashi, K.; Htet Aung, K.; Aizawa, K.; Hori, N.; Yamauchi, J.; Hata, K.; Tanoue, A. DNA methyltransferase inhibitor zebularine induces human cholangiocarcinoma cell death through alteration of DNA methylation status. PLoS ONE 2015, 10, e0120545. [Google Scholar] [CrossRef] [PubMed]
- Uhm, K.O.; Lee, E.S.; Lee, Y.M.; Kim, H.S.; Park, Y.N.; Park, S.H. Aberrant promoter CpG islands methylation of tumor suppressor genes in cholangiocarcinoma. Oncol. Res. 2008, 17, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Ou, D.S.; Lee, S.B.; Chang, L.H.; Lin, R.K.; Li, Y.S.; Upadhyay, A.K.; Cheng, X.; Wang, Y.C.; Hsu, H.S.; et al. hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J. Clin. Investig. 2010, 120, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Braconi, C.; Huang, N.; Patel, T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010, 51, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Wehbe-Janek, H.; Henson, R.; Smith, H.; Patel, T. Epigenetic regulation of microRNA-370 by interleukin-6 in malignant human cholangiocytes. Oncogene 2008, 27, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, J.; Kikuchi, K.; Mizuguchi, Y.; Kawahigashi, Y.; Yoshida, H.; Uchida, E.; Takizawa, T. MiR-376c down-regulation accelerates EGF-dependent migration by targeting GRB2 in the HuCCT1 human intrahepatic cholangiocarcinoma cell line. PLoS ONE 2013, 8, e69496. [Google Scholar] [CrossRef] [PubMed]
- Hatano, Y.; Semi, K.; Hashimoto, K.; Lee, M.S.; Hirata, A.; Tomita, H.; Kuno, T.; Takamatsu, M.; Aoki, K.; Taketo, M.M.; et al. Reducing DNA methylation suppresses colon carcinogenesis by inducing tumor cell differentiation. Carcinogenesis 2015, 36, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; Makarov, V.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Abiru, S.; Migita, K.; Maeda, Y.; Daikoku, M.; Ito, M.; Ohata, K.; Nagaoka, S.; Matsumoto, T.; Takii, Y.; Kusumoto, K.; et al. Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int. 2006, 26, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Hurst, T.P.; Magiorkinis, G. Activation of the innate immune response by endogenous retroviruses. J. Gen. Virol. 2015, 96, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakaoka, T.; Sakai, K.; Muramatsu, T.; Toshimitsu, K.; Kimura, M.; Kanai, T.; Sato, T.; Saito, H. Inhibition of DNA Methylation Suppresses Intestinal Tumor Organoids by Inducing an Anti-Viral Response. Sci. Rep. 2016, 6, 25311. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Vici, P.; Pizzuti, L.; Natoli, C.; Moscetti, L.; Mentuccia, L.; Vaccaro, A.; Sergi, D.; Di Lauro, L.; Trenta, P.; Seminara, P.; et al. Outcomes of HER2-positive early breast cancer patients in the pre-trastuzumab and trastuzumab eras: A real-world multicenter observational analysis. The RETROHER study. Breast Cancer Res. Treat. 2014, 147, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Raynal, N.J.; Da Costa, E.M.; Lee, J.T.; Gharibyan, V.; Ahmed, S.; Zhang, H.; Sato, T.; Malouf, G.G.; Issa, J.J. Repositioning FDA-Approved Drugs in Combination with Epigenetic Drugs to Reprogram Colon Cancer Epigenome. Mol. Cancer Ther. 2016. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function | Sample | Reference |
|---|---|---|---|
| MLH1 | DNA repair | tissue | [14,15] |
| DCLK1 | stemness | tissue | [16] |
| CDO1 | growth | tissue | [16] |
| ZSCAN18 | unknown | tissue | [16] |
| ZNF331 | growth invasion | tissue | [16] |
| p14 (ARF) | cell cycle regulator | tissue | [17,18,19] |
| p16 (INK4a, CDKN2A) | cell cycle regulator | tissue QBC939 cell line | [17,18,19,20,21] |
| DAPK | apoptosis | tissue QBC939 cell line | [19,20,22] |
| CCND2 | growth | bile fluid | [23] |
| CDH13 | growth invasion | bile fluid | [23] |
| GRIN2B | growth | bile fluid | [23] |
| RUNX3 | growth differentiation | bile fluid | [23] |
| TWIST1 | migration invasion | bile fluid | [23] |
| EGFR | growth | Mz-ChA-1 cell line | [24] |
| LKB1 | growth migration invasion | tissue HuH-28 cell line RBE cell line SSP-25 cell line | [25] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakaoka, T.; Saito, Y.; Saito, H. Aberrant DNA Methylation as a Biomarker and a Therapeutic Target of Cholangiocarcinoma. Int. J. Mol. Sci. 2017, 18, 1111. https://doi.org/10.3390/ijms18061111
Nakaoka T, Saito Y, Saito H. Aberrant DNA Methylation as a Biomarker and a Therapeutic Target of Cholangiocarcinoma. International Journal of Molecular Sciences. 2017; 18(6):1111. https://doi.org/10.3390/ijms18061111
Chicago/Turabian StyleNakaoka, Toshiaki, Yoshimasa Saito, and Hidetsugu Saito. 2017. "Aberrant DNA Methylation as a Biomarker and a Therapeutic Target of Cholangiocarcinoma" International Journal of Molecular Sciences 18, no. 6: 1111. https://doi.org/10.3390/ijms18061111
APA StyleNakaoka, T., Saito, Y., & Saito, H. (2017). Aberrant DNA Methylation as a Biomarker and a Therapeutic Target of Cholangiocarcinoma. International Journal of Molecular Sciences, 18(6), 1111. https://doi.org/10.3390/ijms18061111