
AF1q Mediates Tumor Progression in Colorectal Cancer by Regulating AKT Signaling

Abstract
:1. Introduction
2. Results
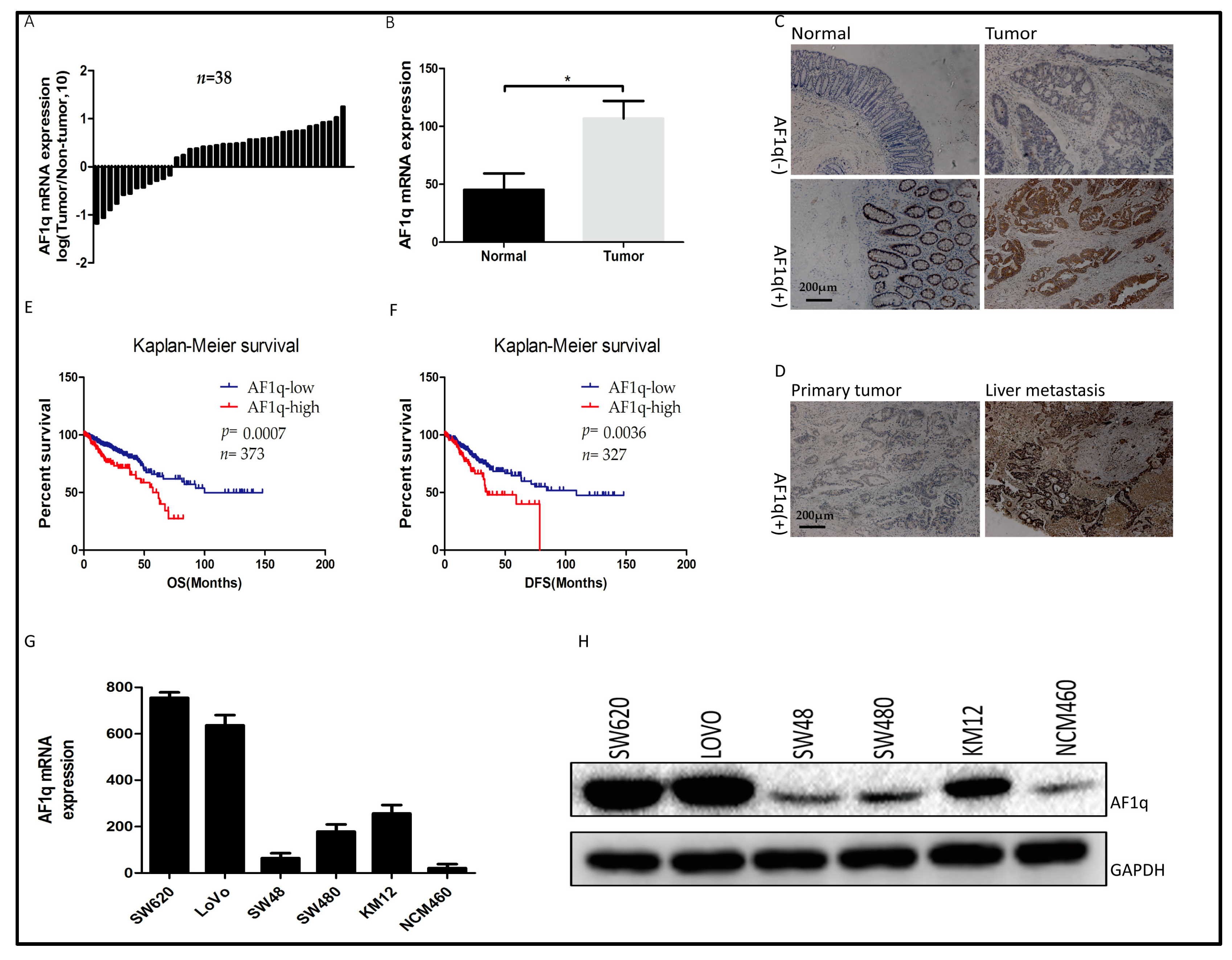
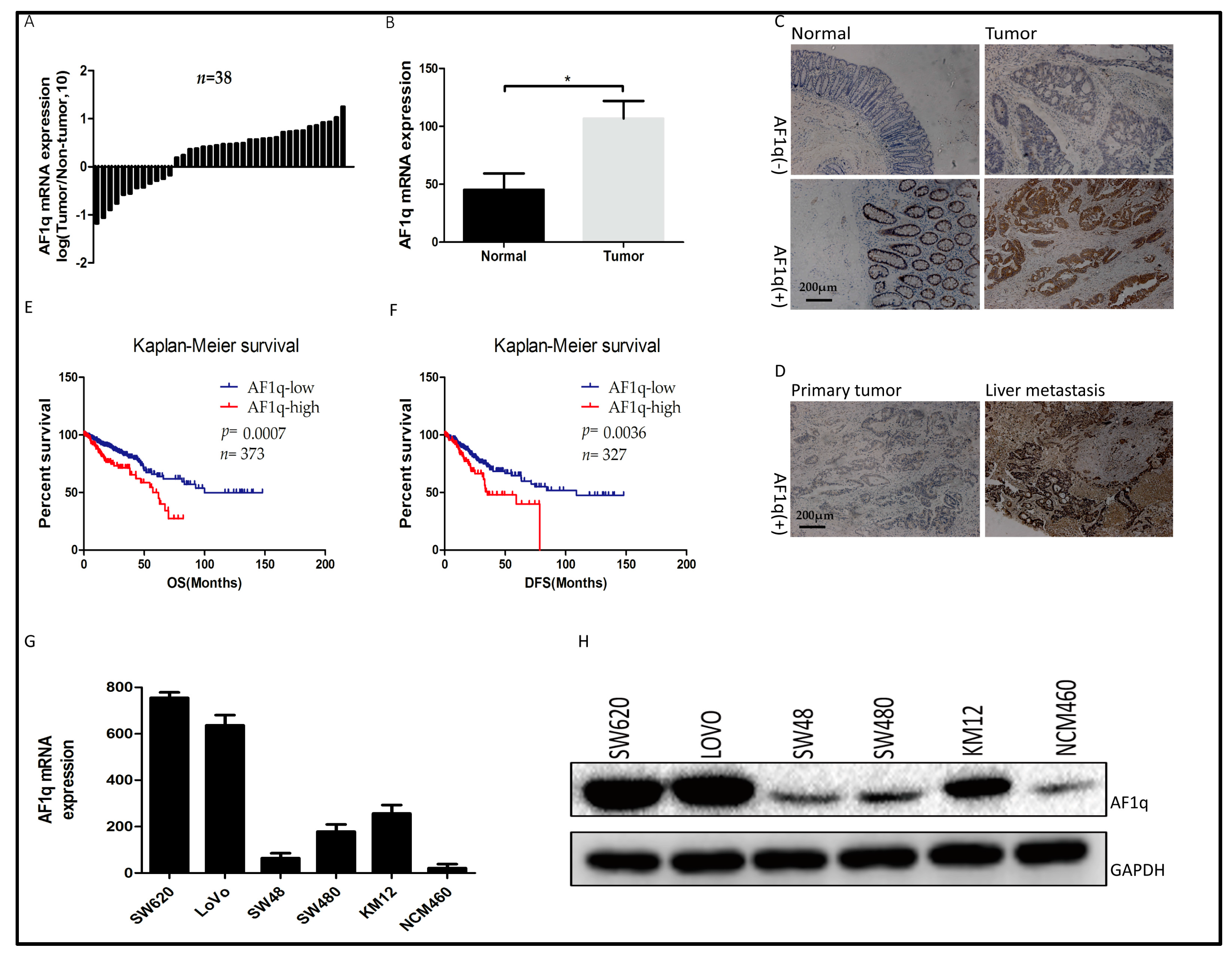
2.1. AF1q Was Upregulated in Human CRC Specimens and Was Indicative of Poor Prognosis
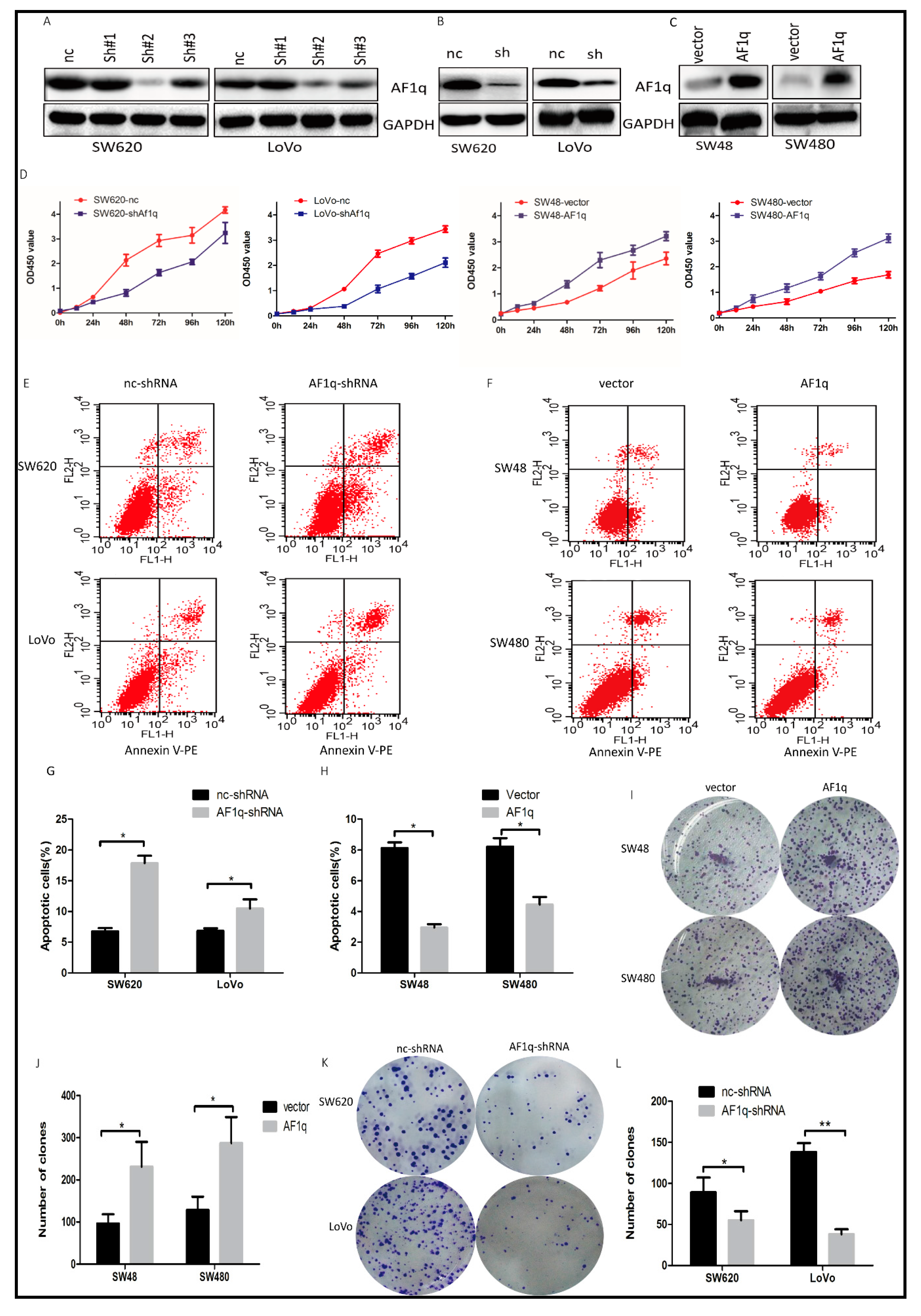
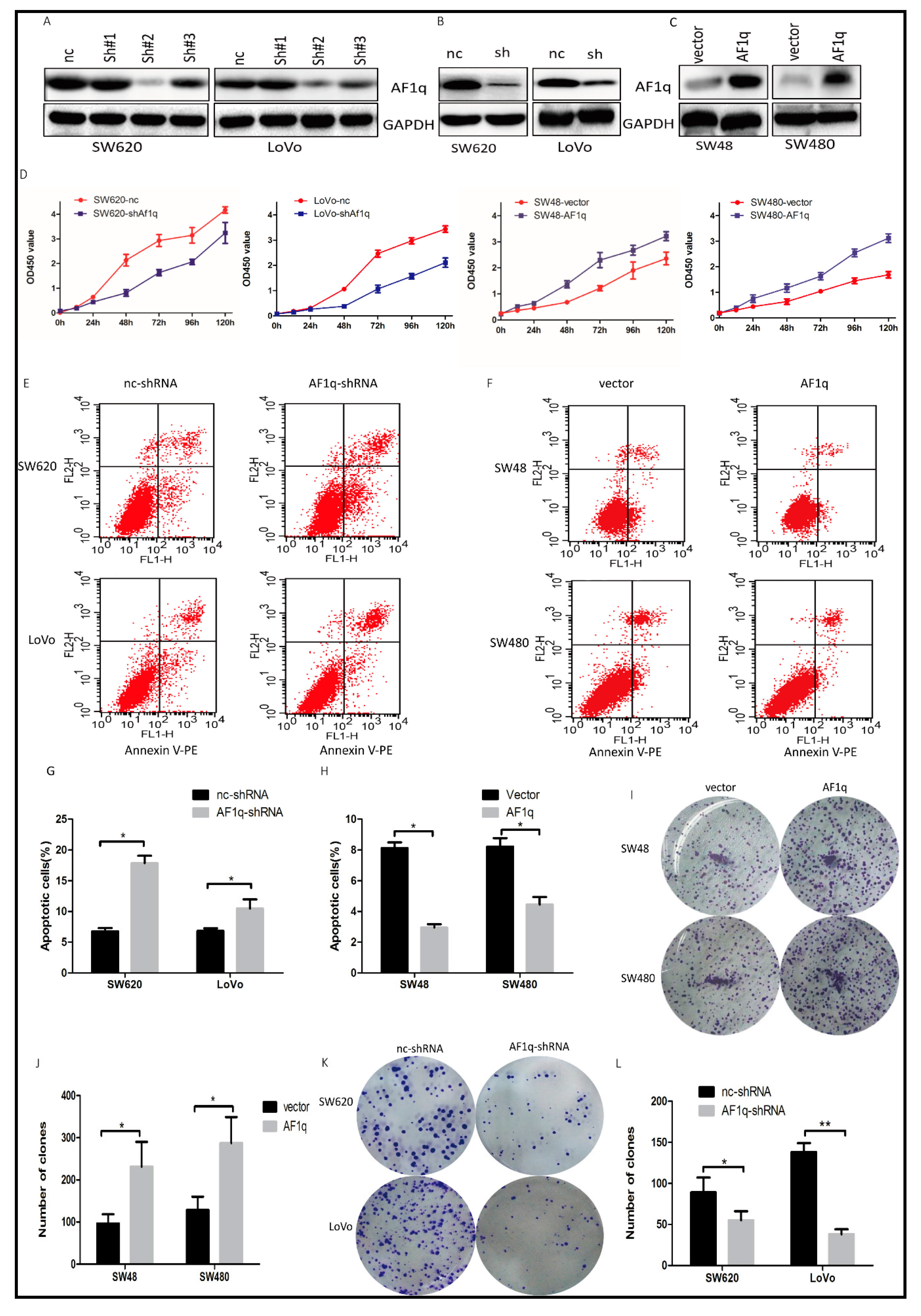
2.2. AF1q Promotes CRC Cell Proliferation In Vitro
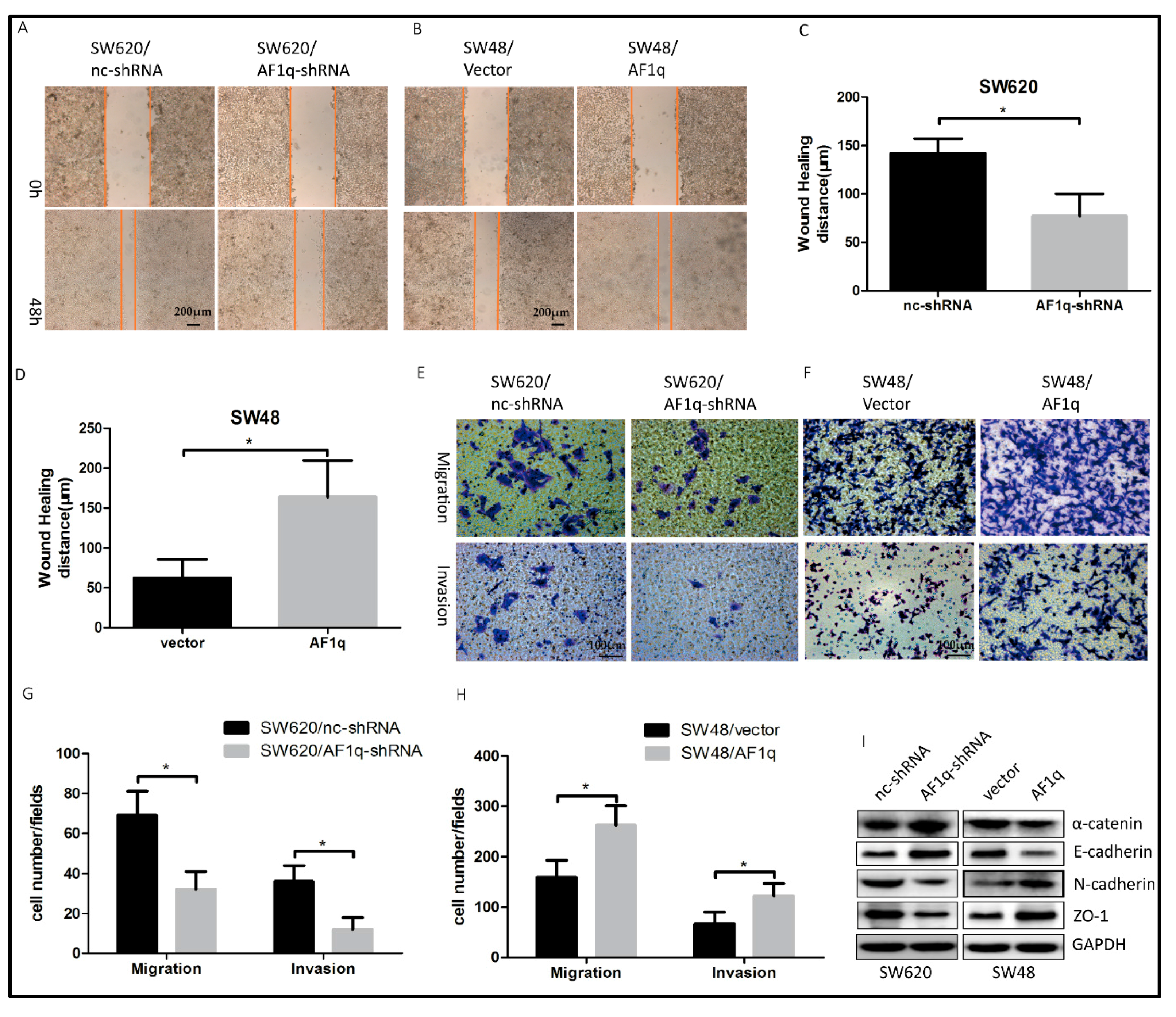
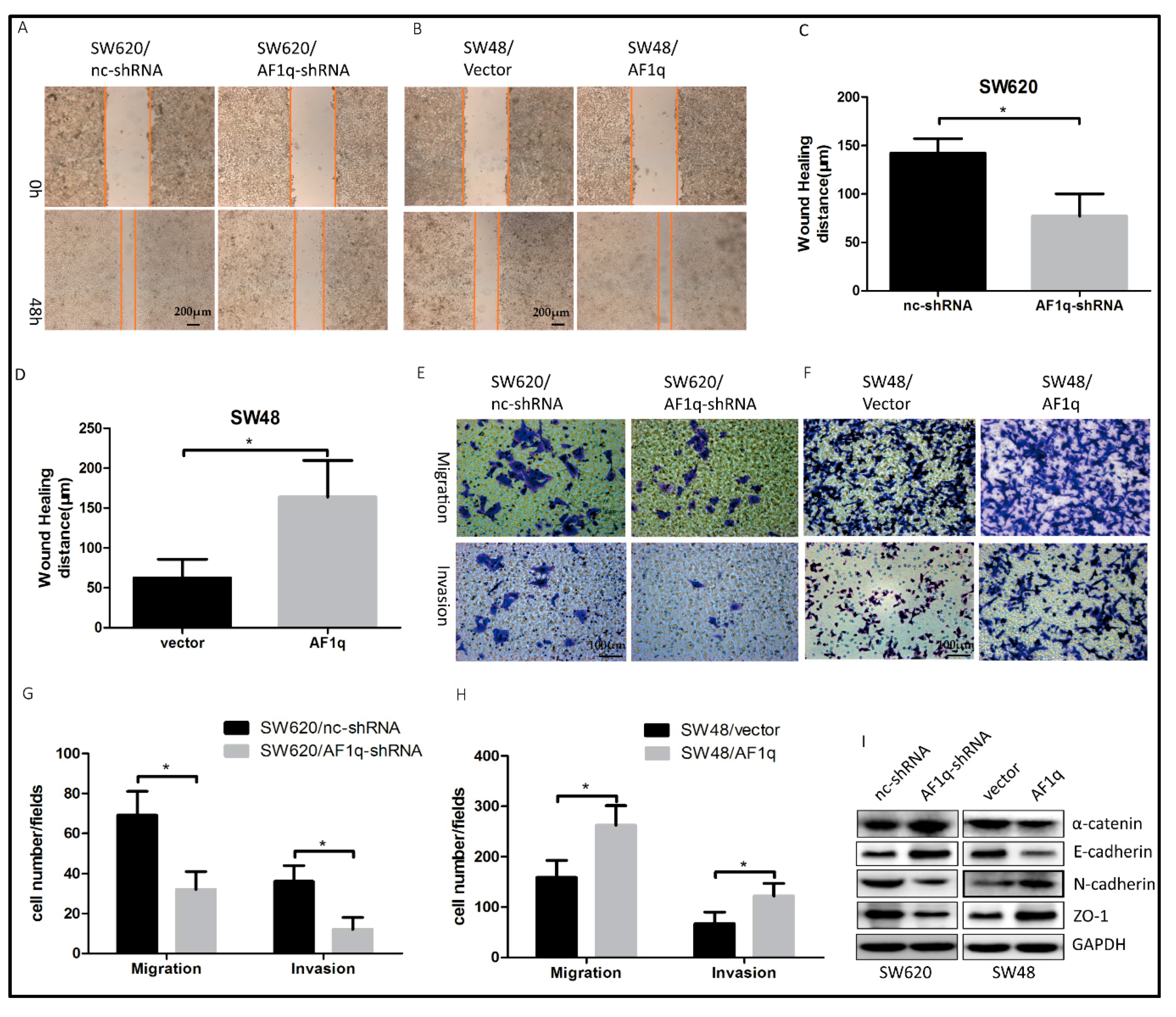
2.3. AF1q Promotes CRC Cell Migration, Invasion, and EMT In Vitro
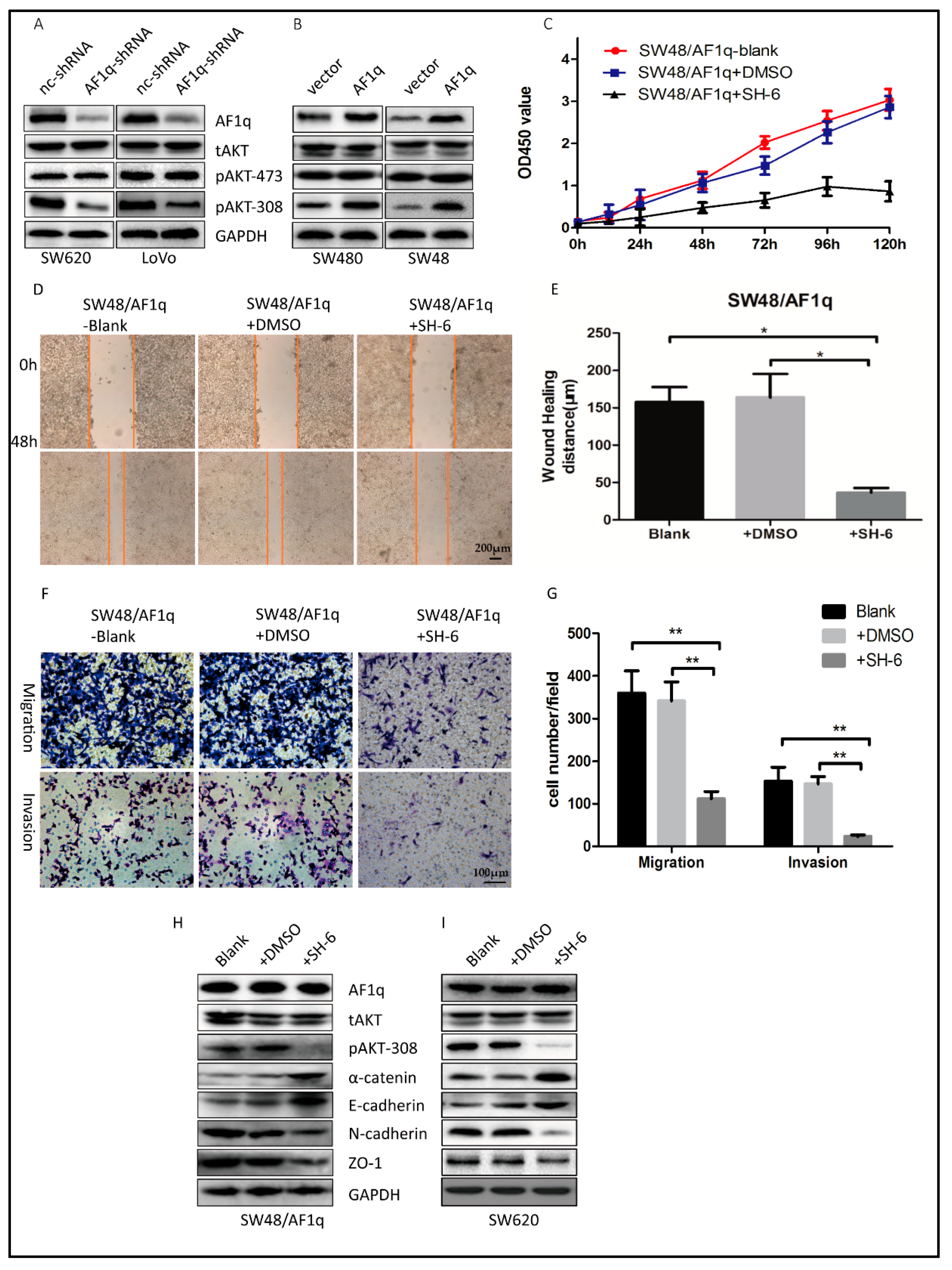
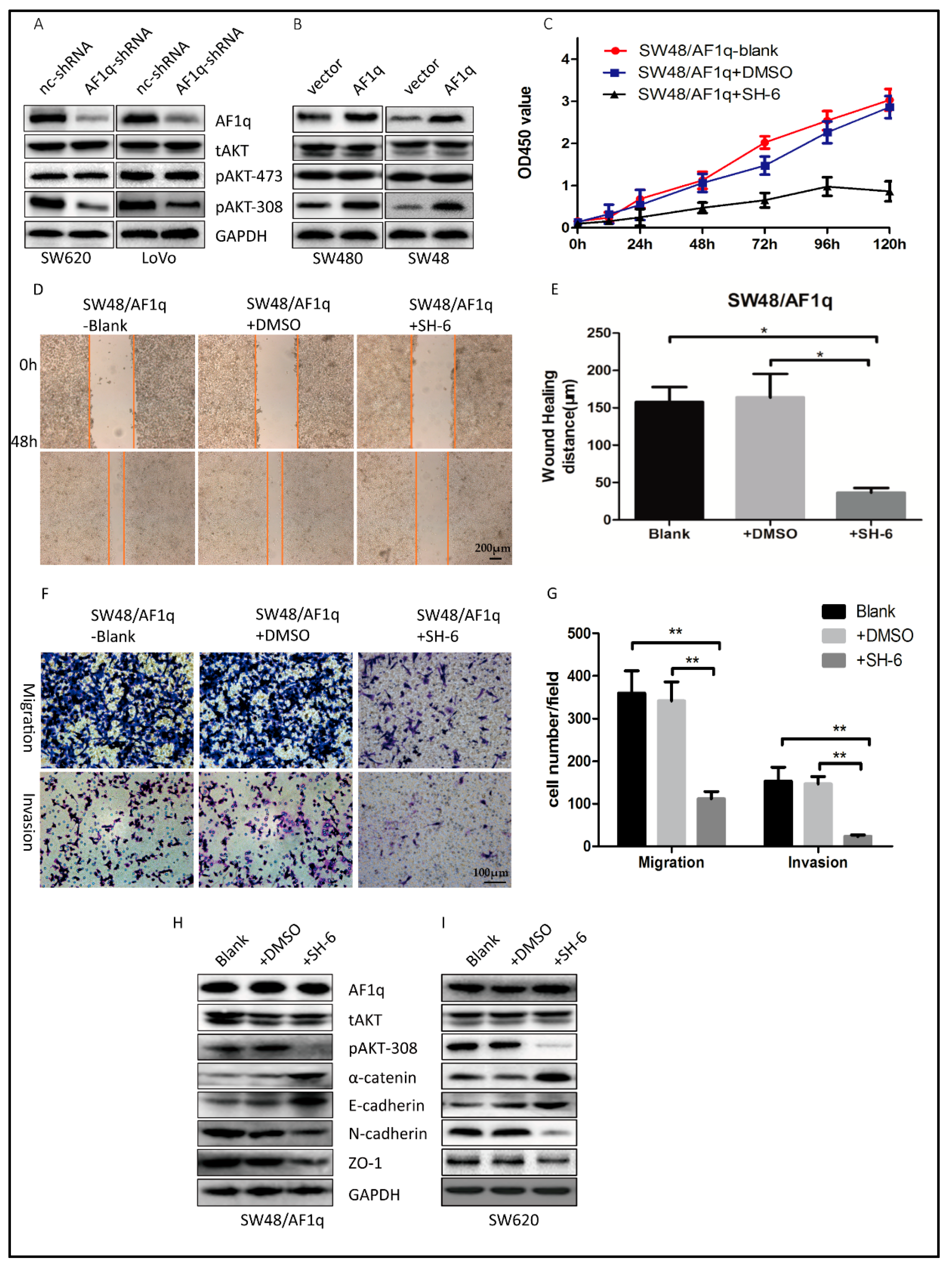
2.4. AF1q-Induced CRC Tumor Promotion Is Mediated by Activation of the AKT Signaling Pathway
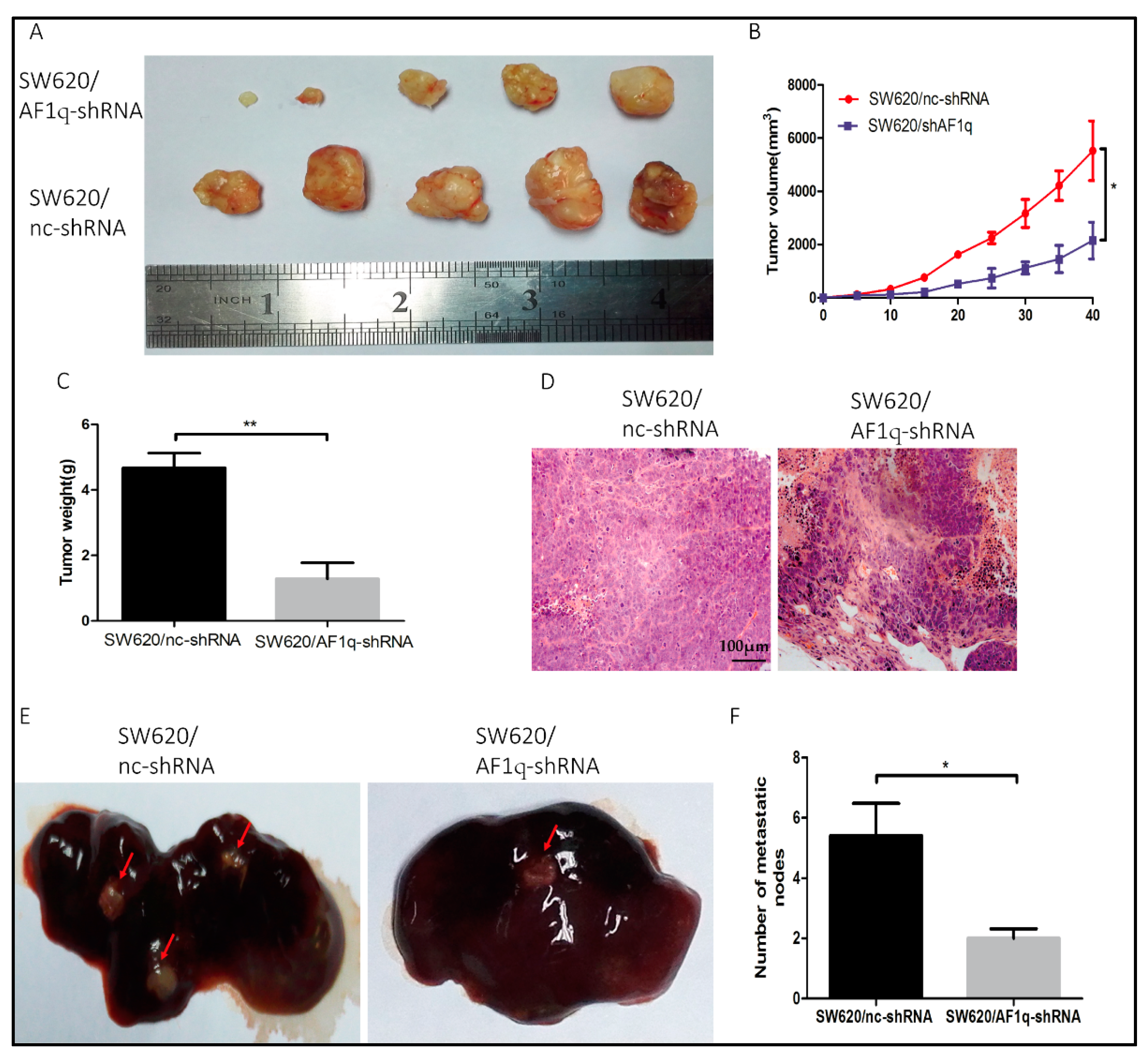
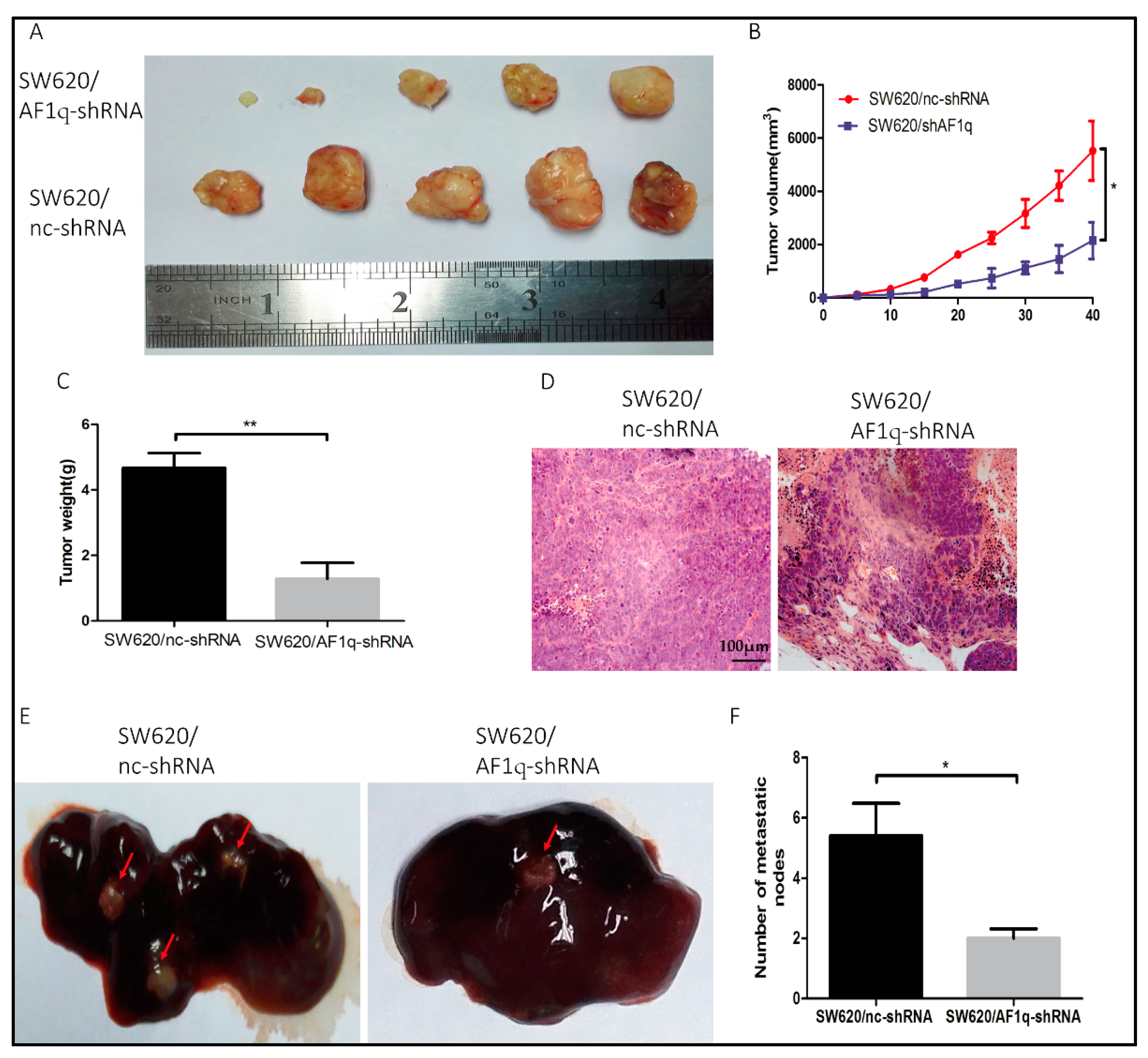
2.5. AF1q Down-Regulation Inhibits CRC Cell Proliferation and Metastasis In Vivo
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Culture
4.3. Tissue Samples and Histological Examination
4.4. Immunohistochemistry
4.5. RNA Extraction and qRT-PCR
4.6. Vector Construction and Transfection
4.7. Cell Proliferation Assay
4.8. Cell Apoptosis Assay
4.9. Western Blot Assay and Antibodies
4.10. Wound-Healing Assays
4.11. Migration and Invasion Assays
4.12. Xenograft Tumor Growth and Metastasis Assays
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AF1q | ALL1-fused gene from chromosome 1q |
| EMT | Epithelial–mesenchymal transition |
| CRC | Colorectal cancer |
| AKT | Protein kinase B |
References
- Gu, Y.; Wang, Q.; Guo, K.; Qin, W.; Liao, W.; Wang, S.; Ding, Y.; Lin, J. TUSC3 promotes colorectal cancer progression and epithelial–mesenchymal transition (EMT) through WNT/β-catenin and MAPK signalling. J. Pathol. 2016, 239, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Aran, V.; Victorino, A.P.; Thuler, L.C.; Ferreira, C.G. Colorectal cancer: Epidemiology, disease mechanisms and interventions to reduce onset and mortality. Clin. Colorectal Cancer 2016, 15, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Tse, W.; Zhu, W.; Chen, H.S.; Cohen, A. A novel gene, AF1q, fused to MLL in t(1;11) (q21;q23), is specifically expressed in leukemic and immature hematopoietic cells. Blood 1995, 85, 650–656. [Google Scholar] [PubMed]
- Tiberio, P.; Cavadini, E.; Callari, M.; Daidone, M.G.; Appierto, V. AF1q: A novel mediator of basal and 4-HPR-induced apoptosis in ovarian cancer cells. PLoS ONE 2012, 7, e39968. [Google Scholar] [CrossRef] [PubMed]
- Co, N.N.; Tsang, W.P.; Wong, T.W.; Cheung, H.H.; Tsang, T.Y.; Kong, S.K.; Kwok, T.T. Oncogene AF1q enhances doxorubicin-induced apoptosis through BAD-mediated mitochondrial apoptotic pathway. Mol. Cancer Ther. 2008, 7, 3160–3168. [Google Scholar] [CrossRef] [PubMed]
- Co, N.N.; Tsang, W.P.; Tsang, T.Y.; Yeung, C.L.; Yau, P.L.; Kong, S.K.; Kwok, T.T. AF1q enhancement of γ irradiation-induced apoptosis by up-regulation of BAD expression via NF-κB in human squamous carcinoma A431 cells. Oncol. Rep. 2010, 24, 547–554. [Google Scholar] [PubMed]
- Park, J.; Schlederer, M.; Schreiber, M.; Ice, R.; Merkel, O.; Bilban, M.; Hofbauer, S.; Kim, S.; Addison, J.; Zou, J.; et al. AF1q is a novel TCF7 co-factor which activates CD44 and promotes breast cancer metastasis. Oncotarget 2015, 6, 20697–20710. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Bohlken, A.; Kuljaca, S.; Lee, M.; Nguyen, T.; Smith, S.; Cheung, B.; Norris, M.D.; Haber, M.; Holloway, A.J.; Bowtell, D.D.; et al. The retinoid anticancer signal: Mechanisms of target gene regulation. Br. J. Cancer 2005, 93, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Shaffer, K.M.; Sun, Z.; Jay, G.; He, W.W.; Ma, W. AF1q, a differentially expressed gene during neuronal differentiation, transforms HEK cells into neuron-like cells. Brain Res. Mol. Brain Res. 2004, 131, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Li, Z.; Ji, M.; Tan, A.C.; Bemis, J.; Tse, J.V.; Huang, G.; Park, J.; Ji, C.; Chen, J.; et al. MIR29B regulates expression of MLLT11 (AF1Q), an MLL fusion partner, and low MIR29B expression associates with adverse cytogenetics and poor overall survival in AML. Br. J. Haematol. 2011, 153, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Pirone, D.M.; Oberst, M.D.; Stylianou, D.; Burbelo, P.D. The genomic structure of the human SPEC1 gene reveals complex splicing and close promoter proximity to the AF1q translocation gene. Gene 2001, 273, 295–303. [Google Scholar] [CrossRef]
- So, C.W.; Ma, S.K.; Wan, T.S.; Chan, G.C.; Ha, S.Y.; Chan, L.C. Analysis of MLL-derived transcripts in infant acute monocytic leukemia with a complex translocation (1;11;4)(q21;q23;p16). Cancer Genet. Cytogenet. 2000, 117, 24–27. [Google Scholar] [CrossRef]
- Cao, T.T.; Gao, L.; Zhou, M.H.; Guo, Y.L.; Yan, Z.; Zhang, S.S.; Xu, Y.Y.; Ding, Y.; Wang, L.L.; Yu, L. Application of multiplex nested RT-PCR to detecting 10 fusion genes related with MLL gene in myelodysplastic syndrome. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2012, 20, 933–936. [Google Scholar] [PubMed]
- Chang, X.Z.; Li, D.Q.; Hou, Y.F.; Wu, J.; Lu, J.S.; Di, G.H.; Jin, W.; Ou, Z.L.; Shen, Z.Z.; Shao, Z.M. Identification of the functional role of AF1Q in the progression of breast cancer. Breast Cancer Res. Treat. 2008, 111, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Dong, Q.; Yao, M.; Shi, M.; Ye, J.; Zhao, L.; Su, J.; Gu, W.; Xie, W.; Wang, K.; et al. Establishment of an experimental human lung adenocarcinoma cell line SPC-A-1BM with high bone metastases potency by (99m)Tc-MDP bone scintigraphy. Nucl. Med. Biol. 2009, 36, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, S.; Joh, J.; Remick, S.C.; Miller, D.M.; Yan, J.; Kanaan, Z.; Chao, J.H.; Krem, M.M.; Basu, S.K.; et al. MLLT11/AF1q boosts oncogenic STAT3 activity through Src-PDGFR tyrosine kinase signaling. Oncotarget 2016, 7, 43960–43973. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Craft, B.; Swatloski, T.; Goldman, M.; Ma, S.; Haussler, D.; Zhu, J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci. Rep. 2013, 3, 2652. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, X.; Li, C.; Cao, X.; Luo, X.; Hu, J. PIK3R3 induces epithelial-to-mesenchymal transition and promotes metastasis in colorectal cancer. Mol. Cancer Ther. 2014, 13, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, J.D.; Yan, L.J.; Zhao, X.P. PDGF-D/PDGFRβ promotes tongue squamous carcinoma cell (TSCC) progression via activating p38/AKT/ERK/EMT signal pathway. Biochem. Biophys. Res. Commun. 2016, 478, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fu, Y.; Yang, X.; Luo, X.; Wang, J.; Gong, J.; Hu, J. Brg-1 targeting of novel miR550a-5p/RNF43/Wnt signaling axis regulates colorectal cancer metastasis. Oncogene 2016, 35, 651–661. [Google Scholar] [CrossRef] [PubMed]
- McNutt, N.S.; Mak, L.L.; Kim, Y.S. Comparison of cell peripheries in the human colonic adenocarcinoma cell lines SW480 and SW620 grown in floating chamber culture, cover slip culture, athymic (nude) mice, and BALB/c mice. Lab. Investig. 1981, 44, 309–323. [Google Scholar] [PubMed]
- Stragand, J.J.; Bergerat, J.P.; White, R.A.; Hokanson, J.; Drewinko, B. Biological and cell kinetic properties of a human colonic adenocarcinoma (LoVo) grown in athymic mice. Cancer Res. 1980, 40, 2846–2852. [Google Scholar] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Salas, N.; Dominguez, G.; Barderas, R.; Mendiola, M.; García-Albéniz, X.; Maurel, J.; Batlle, J.F. Clinical relevance of colorectal cancer molecular subtypes. Crit. Rev. Oncol. Hematol. 2017, 109, 9–19. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, L.; Palmans, S.; Andel, D.; Govaere, O.; Boeckx, B.; Smeets, D.; Galle, E.; Wouters, J.; Barras, D.; Suffiotti, M.; et al. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br. J. Cancer 2017, 116, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Silveanu, C.; Fetyko, A.; Butiurca, V.; Kovacs, Z.; Jung, I. Systematic review of the old and new concepts in the epithelial-mesenchymal transition of colorectal cancer. World J. Gastroenterol. 2016, 22, 6764–6775. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Das, T.P.; Sirimulla, S.; Alatassi, H.; Ankem, M.K.; Damodaran, C. Withaferin-A suppress AKT induced tumor growth in colorectal cancer cells. Oncotarget 2016, 7, 13854–13864. [Google Scholar] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Nemcová, L.; Nagyová, E.; Petlach, M.; Tománek, M.; Procházka, R. Molecular mechanisms of insulin-like growth factor 1 promoted synthesis and retention of hyaluronic acid in porcine oocyte-cumulus complexes. Biol. Reprod. 2007, 76, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Rana, C.; Piplani, H.; Vaish, V.; Nehru, B.; Sanyal, S.N. Downregulation of PI3-K/Akt/PTEN pathway and activation of mitochondrial intrinsic apoptosis by Diclofenac and Curcumin in colon cancer. Mol. Cell. Biochem. 2015, 402, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fisher, R.C.; Signs, S.; Molina, L.A.; Shenoy, A.K.; Lopez, M.C.; Baker, H.V.; Koomen, J.M.; Chen, Y.; Gittleman, H.; et al. Inhibition of PI3K/Akt/mTOR signaling in PI3KR2-overexpressing colon cancer stem cells reduces tumor growth due to apoptosis. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [PubMed]
- Vincent, E.E.; Elder, D.J.; Thomas, E.C.; Phillips, L.; Morgan, C.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavaré, J.M. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Gallay, N.; Dos, S.C.; Cuzin, L.; Bousquet, M.; Simmonet, G.V.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Récher, C. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009, 23, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Niit, M.; Hoskin, V.; Carefoot, E.; Geletu, M.; Arulanandam, R.; Elliott, B.; Raptis, L. Cell–cell and cell-matrix adhesion in survival and metastasis: Stat3 versus Akt. Biomol. Concepts 2015, 6, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Steder, M.; Alla, V.; Meier, C.; Spitschak, A.; Pahnke, J.; Fürst, K.; Kowtharapu, B.S.; Engelmann, D.; Petigk, J.; Egberts, F.; et al. DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell 2013, 24, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Castillo, S.S.; Zhang, C.; Petukhov, P.A.; Memmott, R.M.; Hollingshead, M.; Warfel, N.; Han, J.; Kozikowski, A.P.; Dennis, P.A. Phosphatidylinositol ether lipid analogues that inhibit AKT also independently activate the stress kinase, p38α, through MKK3/6-independent and -dependent mechanisms. J. Biol. Chem. 2007, 282, 27020–27029. [Google Scholar] [CrossRef] [PubMed]
- Castillo, S.S.; Brognard, J.; Petukhov, P.A.; Zhang, C.; Tsurutani, J.; Granville, C.A.; Li, M.; Jung, M.; West, K.A.; Gills, J.G.; et al. Preferential inhibition of Akt and killing of Akt-dependent cancer cells by rationally designed phosphatidylinositol ether lipid analogues. Cancer Res. 2004, 64, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Sun, H.; Brognard, J.; Dennis, P.A. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J. Am. Chem. Soc. 2003, 125, 1144–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Chen, L.; Liang, H.F.; Li, G.Z.; Zhang, B.X.; Chen, X.P. Smad3 sensitizes hepatocelluar carcinoma cells to cisplatin by repressing phosphorylation of AKT. Int. J. Mol. Sci. 2016, 17, 610. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Yan, L.; Vitfell-Rasmussen, J.; Zernhelt, A.M.; Sullivan, D.M.; Cassier, P.A.; Chen, E.; Biondo, A.; Tetteh, E.; Siu, L.L.; et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J. Hematol. Oncol. 2014, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Chang, T.; Wilson, T.W.; Wu, L. Methylglyoxal mediates adipocyte proliferation by increasing phosphorylation of Akt1. PLoS ONE 2012, 7, e36610. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathologic Parameters | AF1q Staining | ||
|---|---|---|---|
| Low (%) (n = 42) | High (%) (n = 54) | p-Value | |
| Gender | |||
| Male | 22 (22.9) | 31 (32.3) | 0.623 |
| Female | 20 (20.8) | 23 (24.0) | |
| Age (years) | |||
| <50 | 6 (6.3) | 7 (7.3) | 0.851 |
| ≥50 | 36 (37.5) | 47 (48.9) | |
| T | |||
| T1, T2 | 9 (9.4) | 11 (11.5) | 0.899 |
| T3, T4 | 33 (34.4) | 43 (44.8) | |
| Tumor grade | |||
| G1, G2 | 34 (35.4) | 40 (41.7) | 0.426 |
| G3, G4 | 8 (8.3) | 14 (14.6) | |
| Lymph node metastasis | |||
| Negative | 20 (32.3) | 13 (13.5) | 0.0159 |
| Positive | 22 (20.8) | 41 (42.7) | |
| Distant metastasis | |||
| Negative | 40 (41.7) | 45 (46.9) | 0.135 |
| Positive | 2 (2.1) | 9 (9.4) | |
| TNM Stage | |||
| I, II | 17 (17.7) | 10 (10.4) | 0.018 |
| III, IV | 25 (26.0) | 44 (45.8) | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Li, G.; Liu, L.; Wang, Y.; Li, X.; Gong, J. AF1q Mediates Tumor Progression in Colorectal Cancer by Regulating AKT Signaling. Int. J. Mol. Sci. 2017, 18, 987. https://doi.org/10.3390/ijms18050987
Hu J, Li G, Liu L, Wang Y, Li X, Gong J. AF1q Mediates Tumor Progression in Colorectal Cancer by Regulating AKT Signaling. International Journal of Molecular Sciences. 2017; 18(5):987. https://doi.org/10.3390/ijms18050987
Chicago/Turabian StyleHu, Jingwei, Guodong Li, Liang Liu, Yatao Wang, Xiaolan Li, and Jianping Gong. 2017. "AF1q Mediates Tumor Progression in Colorectal Cancer by Regulating AKT Signaling" International Journal of Molecular Sciences 18, no. 5: 987. https://doi.org/10.3390/ijms18050987
APA StyleHu, J., Li, G., Liu, L., Wang, Y., Li, X., & Gong, J. (2017). AF1q Mediates Tumor Progression in Colorectal Cancer by Regulating AKT Signaling. International Journal of Molecular Sciences, 18(5), 987. https://doi.org/10.3390/ijms18050987